
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycosylated polyplex micelles from oppositely charged block copolymers†
Mokun
Chen
ab,
Théophile
Pelras
a,
Joël
Benninga
ab,
Vincent S. D.
Voet
b,
Rudy
Folkersma
b and
Katja
Loos
 *a
*a
aMacromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 3, 9747 AG, The Netherlands. E-mail: k.u.loos@rug.nl
bCircular Plastics, Academy Tech & Design, NHL Stenden University of Applied Sciences, 7811 KL Emmen, The Netherlands
First published on 24th March 2025
Abstract
Glycosylated nanoparticles hold significant promise for applications in biomedicine, because of their ability to mimic complex carbohydrate interactions. Herein, we report the synthesis of block copolymers featuring both a glycosylated segment and a charged block via RAFT polymerization and postpolymerization modifications. Additionally, we prepared glycosylated polyplex micelles by mixing oppositely charged glycosylated block copolymers in aqueous media. Electrostatic interactions between the charged segments occur, triggering the formation of glycosylated nanoparticles with a polyplex core. The resulting nanoparticles were characterized via dynamic light scattering (DLS), ζ-potential measurements and transmission electron microscopy (TEM), which confirmed their nonspherical morphology. Furthermore, we expanded this strategy by incorporating oppositely charged homopolymers, highlighting the versatility of our approach. These findings demonstrate a robust and modular platform for the design of glycosylated nanoparticles, paving the way for future exploration of their dynamic properties and potential use as responsive carriers for drug delivery.
Introduction
Carbohydrates are ubiquitous in nature, and play crucial roles as energy storage1 (e.g., glycogen and starch) and as structural material2–4 (e.g., cellulose, chitin and collagen). Most importantly, carbohydrates are involved in numerous biomolecular recognition events, including the inflammatory response and microbial or viral infections, due to their ability to selectively and specifically bind to proteins located on the surface of cells. Although the monovalent interaction between a monosaccharide and its receptor is relatively weak, the multivalent interactions offered by carbohydrates, known as the ‘cluster glycoside effect’,5 significantly increase the overall binding strength. This multivalency leads to greater biological activity, which can be leveraged for applications in recognition processes6 and drug delivery systems.7 Polysaccharides, as naturally occurring and highly abundant carbohydrates, possess inherent advantages for use as materials.8–11 However, their use in biomedical applications can be hampered by challenges such as poor structural control, limited solubility and inherent fragility.Nature remains a vast source of inspiration, particularly in the field of biomedicine, where mimicking biological behaviors with tailor-made molecules is essential for achieving significant breakthroughs. Glycopolymers, are synthetic analogues of polysaccharides, and feature pendant carbohydrate moieties instead of sugar motifs within the polymer backbone. Glycopolymers have been developed as laboratory-made alternatives that can be more easily produced and tailored. They can be synthesized directly from glycomonomers via various polymerization techniques, or sugar moieties can later be attached onto a functional polymer backbone.12 This enables the facile production of glycopolymers with tailored properties, such as chain length, dispersity or end-group functionality. This degree of control becomes even more important when the synthesis of sequence-controlled glycopolymers, such as glycosylated block copolymers, is sought.
Various glycosylated block copolymers have already been utilized for the production of glycosylated nanoparticles, which can be tailored for a wide range of applications.13–16 Stenzel et al. not only demonstrated the formation of polymer micelles featuring a hydrophobic core and a glycosylated shell, but also investigated their applications for drug delivery. While a hydrophobic drug can be encapsulated and subsequently released from the micellar core,17 smart macromolecular design can also bring shape-changing capability18,19 or enzymatic degradability20 of the nanoparticles. These examples of glycosylated nanoparticles rely on solvophobic interactions for self-assembly, where the hydrophobic block collapses and is stabilized by the hydrophilic glycosylated segment. Additionally, a plethora of alternative self-assembly methods, such as direct hydration,21–23 polymerization-induced self-assembly,24,25 sugar deprotection self-assembly26–28 and temperature induced self-assembly,29,30 have been reported for the preparation of glycosylated nanoparticles in aqueous media.
Despite its potential, interpolyelectrolyte complexation has been absent from the toolkit for preparing glycosylated nanoparticles. The utilization of electrostatic interactions between oppositely charged polymers is a well-established strategy for the creation of soft materials, particularly in aqueous environments, where the hydrophilicity of charged polymers enhances their functional performance. Complexation between oppositely charged polymers triggers the formation of a largely dehydrated coacervate. Owing to the pseudo-hydrophobic31 nature of the polyplex domains, particularly when charge-neutrality is achieved, instability occurs and the coacervate tends to precipitate or aggregate. Therefore, the attachment of water-soluble yet charged neutral chains onto polyelectrolytes is necessary to stabilize the particles in water. Before the advent of carbohydrate-decorated polymers, poly(ethylene glycol) was widely used for this purpose,32,33 even offering a certain degree of biocompatibility, but concerns regarding its immune response,34,35 as well as the desire to move toward biosourced14,36 and biodegradable37 alternatives are opening doors toward the formation of glycosylated polyplex micelles.
In this study, an efficient approach for the construction of glycosylated polyplex micelles via electrostatic interactions was demonstrated (Scheme 1). Two block copolymers, each comprising a glycosylated block and a charged segment, were synthesized via a combination of sequential reversible addition–fragmentation chain-transfer (RAFT) polymerization and postpolymerization modification techniques. Through stoichiometric mixing of the block copolymer solutions, nanoparticles with a polyplex core (i.e., a complex formed between the charged segments) and stabilized by a glycosylated corona were successfully formed. The resulting nanoparticles were characterized via dynamic light scattering (DLS), ζ-potential measurements and transmission electron microscopy (TEM), which confirmed their size and morphology.
 | ||
| Scheme 1 Synthesis of charged glycosylated block copolymers. | ||
Results and discussion
The overall synthesis strategy involves the use of RAFT polymerization to produce a glycopolymer as the macromolecular chain-transfer agent (macro-CTA), which is then chain-extended to incorporate poly(2-(dimethylamino)ethyl methacrylate), followed by deprotection to expose the glucose units, and quaternization to yield a glycopolymer containing a strong polycationic block. An alternative pathway involves chain extension with poly(tert-butyl methacrylate), followed by simultaneous deprotection to obtain a glycopolymer featuring a weak polyanionic segment.Although glycosylated polymers can be directly synthesized via the polymerization of hydrophilic sugar monomers,38–40 an alternative approach was employed to convert glucose into a hydrophobic protected glycomonomer. This strategy not only facilitates its polymerization and subsequent chain extension reactions but also enables more straightforward characterization of the various polymer intermediates.41–43 The protected glycomonomer, acetonide-protected glucose methacrylate (PrGlcMA), was synthesized through a two-step reaction: (i) the introduction of two acetonide protective groups onto α-D-glucose, followed by (ii) the addition of a methacrylic moiety to the remaining hydroxyl group. This method enhances both the reactivity and manageability of the monomer during the polymerization process (details are in ESI†). After flash column chromatography, PrGlcMA was obtained at high purity, as confirmed by 1H and 13C nuclear magnetic resonance (1H NMR and 13C NMR, respectively, Fig. S1, ESI†), as well as high-resolution mass spectrometry (Fig. S2, PrGlcMA + Natheo = 351.14142, PrGlcMA + Naexp = 351.14103, ESI†).
The synthesis of glycosylated block copolymers was initiated with the polymerization of PrGlcMA to poly(acetonide protected glucose methacrylate) (PPrGlcMA). RAFT polymerization, which is mediated by 2-cyanopropan-2-yl propyl trithiocarbonate as the CTA, was employed because of its ability to produce polymers with low dispersity, high chain-end fidelity, and compatibility with a wide range of monomers. This particular CTA was selected for its enhanced stability, as the trithiocarbonate group is less susceptible to hydrolysis44 and shows greater resistance to strong nucleophiles, which is crucial for subsequent deprotection steps. Additionally, its short C3 alkane chain is less hydrophobic than that of commercially available C12 CTAs, which is particularly relevant for our hydrophilic diblock copolymers.
The successful synthesis of the glycosylated homopolymer was verified via1H NMR (Fig. 1A1), which allowed for the calculation of the polymer's chain length on the basis of monomer conversion (conv. = 95%, DPPPrGlcMA = 72, Mn,NMR = 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 Da), while size exclusion chromatography (SEC) was used to monitor the dispersity of the polymers (Đ = 1.56, Fig. 1B). Although featuring a high dispersity, the polymer elugram retains a Gaussian distribution (i.e., absence of chain-chain termination or tailing), suggesting a potential interaction with the column material. Most importantly, an excellent ratio was found between the acetonide signals (12H, 1.33–1.49 ppm) and the glucose ring (7H, 3.68–5.81 ppm), confirming the absence of unwanted deprotection.
800 Da), while size exclusion chromatography (SEC) was used to monitor the dispersity of the polymers (Đ = 1.56, Fig. 1B). Although featuring a high dispersity, the polymer elugram retains a Gaussian distribution (i.e., absence of chain-chain termination or tailing), suggesting a potential interaction with the column material. Most importantly, an excellent ratio was found between the acetonide signals (12H, 1.33–1.49 ppm) and the glucose ring (7H, 3.68–5.81 ppm), confirming the absence of unwanted deprotection.
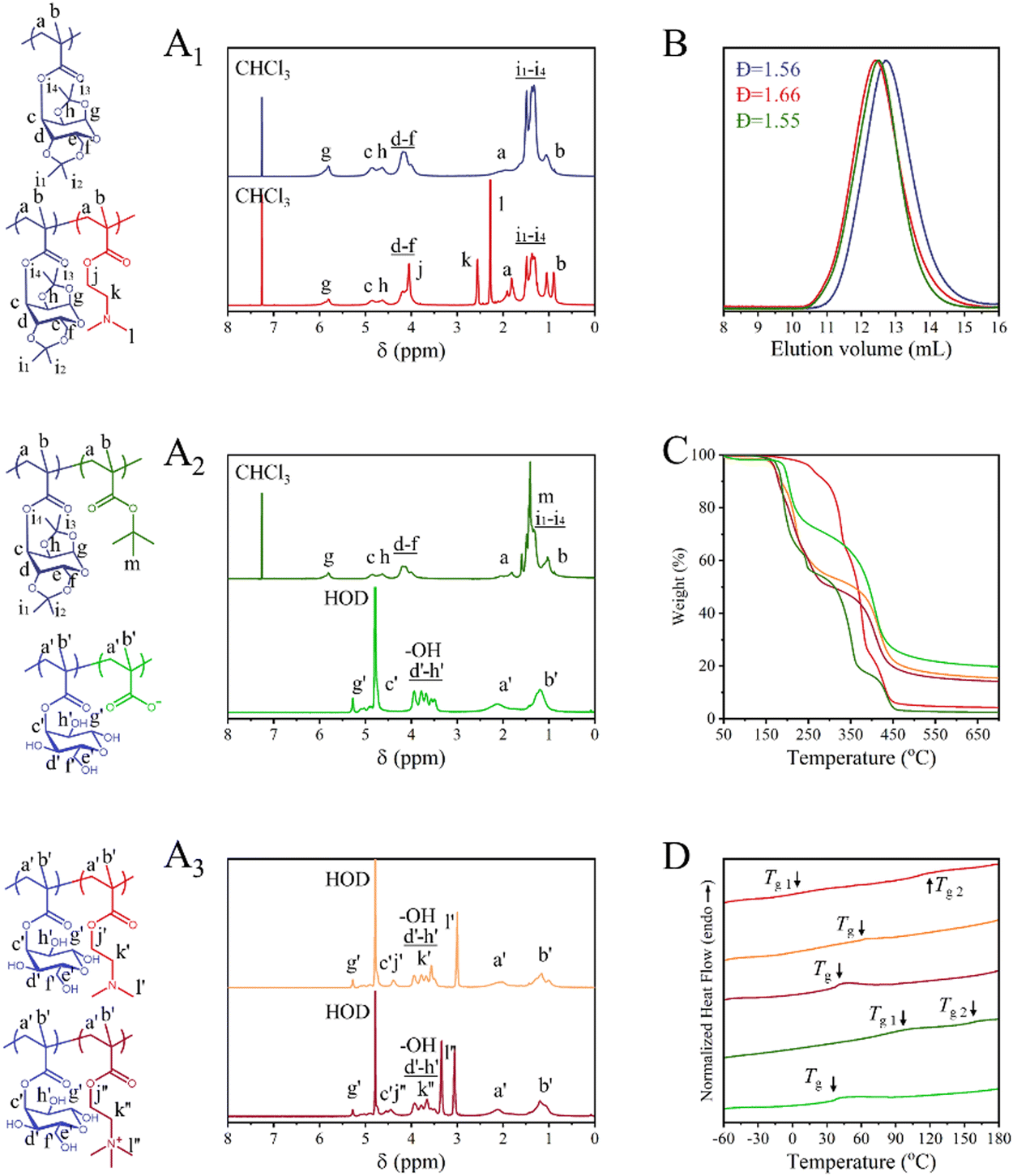 | ||
| Fig. 1 Characterization of oppositely charged glycosylated block copolymers and their precursors. (A1)–(A3) 1H NMR spectra of the polymers: PPrGlcMA72 (dark blue), PPrGlcMA72-b-PDMAEMA71 (red), and PPrGlcMA72-b-PtBMA85 (dark green), PGlcMA72-b-PMAA85 (light green), PGlcMA72-b-PDMAEMA71 (orange), PGlcMA72-b-PMETAI71 (dark red). (B) SEC elugrams of PPrGlcMA72 (dark blue), PPrGlcMA72-b-PDMAEMA71 (red) and PPrGlcMA72-b-PtBMA85 (dark green). (C) TGA and (D) DSC analyses of PPrGlcMA72-b-PDMAEMA71 (red), PGlcMA72-b-PDMAEMA71 (orange), PGlcMA72-b-PMETAI71 (dark red), PPrGlcMA72-b-PtBMA85 (dark green) and PGlcMA72-b-PMAA85 (light green). | ||
Subsequently, chain extension reactions via RAFT polymerization were performed to produce different block copolymers. The PPrGlcMA-block-poly(2-(dimethylamino)ethyl methacrylate) (PPrGlcMA72-b-PDMAEMA71) block copolymer was synthesized using PPrGlcMA as the macro-CTA. 1H NMR confirmed the introduction of the new block, as evidenced by the new proton signals (CH2, 2H, 4.05 ppm; CH2 2H, 2.56 ppm; CH3, 6H, 2.28 ppm, Fig. 1A1), and enabled the calculation of the degree of polymerization of the second block through monomer conversion (conv. = 71%, DPPDMAEMA = 71, Mn,NMR = 35000 Da). SEC confirmed the chain extension, as indicated by a complete shift of the peak toward a lower retention time and the absence of chain–chain termination, while preserving a reasonable dispersity (Đ = 1.66). The PPrGlcMA-block-poly(tert-butyl methacrylate) (PPrGlcMA72-b-PtBMA85, Fig. 1A2) block was similarly synthesized using PPrGlcMA as the macro-CTA. 1H NMR again confirmed the introduction of the new block, as shown by the appearance of new proton signals (CH3, 9H, 1.41 ppm, Fig. 1A2), and was used to calculate the degree of polymerization of the second block on the basis of the monomer conversion (conv. = 81%, DPPtBMA = 85, Mn,NMR = 35900 Da). SEC further confirmed the chain extension by complete shift of the peak toward a lower retention time and reasonable dispersity (Đ = 1.55).
The two diblock copolymers, which are currently in their protected and hydrophobic state, require postpolymerization modifications to achieve their desired functionality. While hexafluoroisopronanol/hydrochloric acid has been previously reported as an efficient method for the removal of tert-butyl protective groups,45 trifluoroacetic acid (TFA) has demonstrated the ability to simultaneously remove both acetonide20,46 and tert-butyl groups.47,48 Preliminary tests on PPrGlcMA and PtBMA homopolymers (details are in ESI†) revealed that both routes are viable, with the TFA route yielding slightly better results, as shown by clearer signals of the glucose ring in the 1H NMR spectra (Fig. S4-1, ESI†). This method was first applied to PPrGlcMA72-b-PtBMA85 to produce a poly(glucose methacrylate)-block-poly(methacrylic acid) (PGlcMA72-b-PMAA85) through a facile two-in-one deprotection step. PGlcMA72-b-PMAA85, now soluble in water, was analyzed by 1H NMR, which confirmed the complete removal of both protective groups, with the disappearance of the acetonide and tert-butyl signals (1.18–1.64 ppm, respectively, Fig. 1A2). Fourier-transform infrared spectroscopy (FT-IR, Fig. S5-2, ESI†) also evidenced the loss of the tert-butyl protective groups and the appearance of O–H stretching at 3000–3600 cm−1. The same protocol was used to expose the glucose units on PPrGlcMA72-b-PDMAEMA71, yielding water-soluble PGlcMA72-b-PDMAEMA71, which is now soluble in water (Fig. 1A3). To ensure the PDMAEMA segment would remain soluble at any pH, a quaternization step was performed following a previously reported procedure.47,491H NMR confirmed the modification by the shift of the CH2 and CH3 proton signals (from 3.05 to 3.34 ppm), yielding a PGlcMA-block-poly(2-(methacryloyloxy)ethyl trimethylammonium iodide) (PGlcMA72-b-PMETAI71) diblock copolymer featuring a strong polyelectrolyte segment.
Thermal characterization was conducted on the various block copolymers and their precursors, beginning with thermogravimetric analysis (TGA, Fig. 1C and Fig. S6, ESI†). The results indicate that none of the protected block copolymers exhibit significant degradation below 180 °C, highlighting the relatively high thermal stability of the acetonide and tert-butyl protective groups. PPrGlcMA72-b-PDMAEMA71 demonstrates greater thermal stability, with the initial weight loss occurring at approximately 190 °C, corresponding to the degradation of the acetonide protective groups. PGlcMA72-b-PDMAEMA71 rapidly decomposes above 190 °C because of the removal of protective acetonide groups, and the weight loss is attributed primarily to the degradation of the glucose rings. PGlcMA72-b-PMETAI71 follows a similar decomposition trend, even after quaternization. While PPrGlcMA72-b-PtBMA85 demonstrates the earliest onset of degradation, it experiences a rapid weight loss of approximately 40% at approximately 250 °C. This degradation is primarily due to the early decomposition of the tert-butyl groups,50 followed by the subsequent thermolysis or hydrolysis of the remaining polymer. In contrast, after deprotection, PGlcMA72-b-PMAA85 shows improved thermal stability and higher residual mass.
Differential scanning calorimetry (DSC) was performed to assess the thermal behaviour of the block copolymers and their precursors (Fig. 1D and Fig. S6, ESI†). PPrGlcMA72-b-PDMAEMA71 has two distinct glass transition temperatures (TgPPrGlcMA-b-PDMAEMA1 = 119 °C and TgPPrGlcMA-b-PDMAEMA2 = 5 °C, Table S1, ESI†), which suggests that the blocks do not mix. The higher Tg is attributed to the rigid PPrGlcMA segment (TgPPrGlcMA = 150 °C, Table S2, ESI†), whereas the lower Tg corresponds to the softer PDMAEMA segment (TgPDMAEMA = 6 °C, Table S2, ESI†). The Tg values of TgPPrGlcMA-b-PDMAEMA1 and TgPPrGlcMA-b-PDMAEMA2 are lower than the Tg values of individual PPrGlcMA and PDMAEMA, presumably due to block mixing near the interface,51,52 which increases the mobility of the polymer chains. After deprotection, the PGlcMA segment becomes more flexible (TgPGlcMA = 61 °C, Table S2, ESI†), and PGlcMA72-b-PDMAEMA71 features a unique Tg at 62 °C. This suggests either that deprotection enables mixing of the two blocks or that the thermal behavior of the PrGlcMA block prevails over that of the PDMAEMA. Following quaternization, the PMETAI segment becomes more rigid because of stronger interactions between the charged polymer chains (TgPMETAI = 48 °C, Table S2, ESI†). PGlcMA72-b-PMETAI71 exhibited a single thermal transition (TgPGlcMA-b-PMETAI = 40 °C, Table S1, ESI†), possibly due to the mixing of the blocks. The thermogram of PPrGlcMA72-b-PtBMA85 shows two distinct transitions (TgPPrGlcMA-b-PtBMA1 =153 °C and TgPPrGlcMA-b-PtBMA2 = 83 °C, Table S1, ESI†), corresponding to those of the PPrGlcMA (Tg PPrGlcMA = 150 °C, Table S2, ESI†) and PtBMA (TgPtBMA = 120 °C, Table S2, ESI†) homopolymers. After deprotection, both segments of PGlcMA72-b-PMAA85 become softer, and polymer exhibits a broad thermal transition near the Tg of the individual polymers (TgPGlcMA-b-PMAA = 38 °C, Table S1 (ESI†); TgPGlcMA = 61 °C and TgPMAA12= 45 °C, Table S2, ESI†).
The formation of glycosylated polyplex micelles (GPM) was subsequently investigated through electrostatic complexation between oppositely charged PMETAI and PMAA segments (Fig. 2A). Solutions of each block copolymer were prepared at a concentration of 1 g L−1 in 10 mM KNO3 and a pH of 7.2, which permits the PMAA85 block to be charged (pKaPMAA= 4.8–5.5),53 while the charge of PMETAI71 is independent of pH. When aqueous solutions of PGlcMA72-b-PMAA85 and PGlcMA72-b-PMETAI71 (GPM1) were mixed, micelles formed almost instantaneously. A stoichiometric ratio (1:1) between the positively charged PMETAI and the negatively charged PMAA segments was used to balance the charges so that a charge-neutral and pseudo hydrophobic domain formed, i.e., the core of the micelles. Dynamic light scattering (DLS) experiments were conducted to measure the effective hydrodynamic radii of the particles. Using this technique, we confirmed the presence of small particles with a mean core diameter of DhGPM1 = 14.6 nm and a rather low polydispersity index (PDI = 0.149) (Fig. 2B, Fig. S7-1, ESI†). The small diameter may be attributed to the glycosylated corona around the core, as PGlcMA segments not only provide stability and solubility in water but also prevent the micelles from continuously growing into larger particles.54 Additionally, the low salt concentration of the solution leads to stronger charge repulsion between polyelectrolytes and results in slower diffusion of the individual polymer molecules in the solution, which inhibits polymer aggregation into larger clusters.55ζ-Potential measurements (ζGPM1 = 5.0 mV, Table 1) confirmed the quasi neutral nature of the particles and the near-compensation of all charges. Visualization of the nanoparticles was conducted via transmission electron microscopy (TEM) on negatively stained samples (Fig. 2C). Uranyl acetate was used to stain the polyplex domains at the particle cores, resulting in an inverted contrast (i.e., ‘positive staining’).56 The particles observed in most samples were elliptical. Polyplex micelles in solution with a nonspherical morphology are not common, and elliptical shape is theoretically predicted as possible intermediates in the transition from spheres to lamellae.57 Elliptical overall shape and disc-like core formed from the complexation of oppositely charged block copolymers have been reported previously,58,59 with the proposed mechanism of coronal microphase separation of the stabilizing segments from different block copolymers. For GPM1, elliptical micelles with constant dimensions were observed on a large scale, suggesting that the PGlcMA corona plays an important role in stabilizing the nonspherical morphology. Additionally, the mean core diameter (ØGPM1 = 21.5 nm) of GPM1 was obtained from statistical analyses of several TEM images (Fig. 2D). The glycosylated polyplex micelles exhibited a good stability due to their rigid complex core and stabilizing segments. No dissociation or aggregation was observed during measurements.
 | ||
| Fig. 2 Formation of glycosylated polyplex micelles through electrostatic complexation between oppositely charged glycosylated block copolymers. (A) Schematic representation of the self-assembly process. (B) DLS (173°) intensity plot (bars) and correlation coefficient (line) of glycosylated nanoparticles in a 10 mM KNO3 solution. (C) TEM images of uranyl acetate-stained glycosylated nanoparticles. (D) Statistical analysis of glycosylated nanoparticles (data extracted from 300 particles in several images). | ||
| GPM | Polyanion (x)a | Polycation (y)a | D h (nm) | PDIb | ζ (mV) | Øc (nm) |
|---|---|---|---|---|---|---|
| a Stoichiometric ratio between the polyanion and the polycation. b Determined by DLS/ζ-potential measurements at 25 °C on 1 g L−1 solutions in 10 mM KNO3 and measured in triplicate. c Determined from TEM images and measurements of 300 particles. | ||||||
| GPM1 | PGlcMA72-b-PMAA85 (1) | PGlcMA72-b-PMETAI71 (1) | 14.6 | 0.149 | 5.0 | 21.5 |
| GPM2 | PGlcMA72-b-PMAA85 (1) | PMETAI41 (1) | 63.2 | 0.142 | 1.1 | 50.5 |
| GPM3 | PMAA74 (1) | PGlcMA72-b-PMETAI71 (1) | 69.5 | 0.079 | 6.9 | 70.9 |
| GPM4 | PSPMA-Na114 (1) | PGlcMA72-b-PMETAI71 (1) | 89.3 | 0.092 | −18.0 | 90.4 |
| GPM5 | PSPMA-Na114 (0.5) | PGlcMA72-b-PMETAI71 (1) | 114.0 | 0.081 | −5.0 | 96.2 |
Since the concept of glycosylated polyplex micelles (GPM) with block copolymers was demonstrated, different polymers have been explored for charge compensation, potentially eliminating the need for glycosylated block copolymers. Three charged homopolymers, PMETAI41, PMAA74 and poly(sulfopropyl methacrylate) sodium salt (PSPMA-Na114), were produced via a combination of RAFT polymerization and postpolymerization modification (details are in ESI†). Two of these polymers, namely, PMETAI41 and PSPMA-Na119, are strong polyelectrolytes, i.e., their charge density is pH-independent in the medium, which have significant rigidity and hydrodrynamic volume in water.
The preparation of GPM was conducted as previously described, with the aim of nearly compensating for the charge in 10 mM KNO3. However, here we used a pair consisting of a glycosylated block copolymer and one complementary homopolymer. Aqueous solutions of PMETAI41 and PGlcMA72-b-PMAA85 (GPM2) were mixed at a 1:1 stoichiometric ratio between positively charged PMETAI and negatively charged PMAA segments (Fig. 3A1). This led to the formation of stable and spherical micellar-like polyplexes. ζ-Potential measurements (ζGPM2 = 1.1 mV, Table 1) confirmed the quasi neutrality of the particles’ core and the near-compensation of all charges. The DLS results (Fig. 3B1 and Fig. S7-2, ESI†) revealed the presence of glycosylated polyplex micelles with hydrodynamic radius of DhGPM2 = 63.2 nm, whereas the TEM images (Fig. 3C1) confirmed their spherical morphology with mean core diameters of ØGPM2 = 50.5 nm (Fig. S7-1, ESI†). The hydrodynamic radius from (DhGPM2) DLS is smaller than the mean core diameter (ØGPM2), possibly due to the hydration layer in DLS or/and particle shrinkage in TEM of the micelles. Interestingly, while the components of GPM2 are similar to those of GPM1, the resulting polyplex micelle morphologies are markedly different. The possible reason is that both GPM1 and GPM2 initially underwent a transient phase in which metastable large-scale aggregates formed via spontaneous complexation after mixing.60 However, owing to the less stabilizing corona in GPM2 than in GPM1, the charge-neutral clusters of GPM2 underwent thermodynamic equilibration.61 This process involves fusion-fission or expulsion-insertion events, ultimately yielding spherical polyplex micelles.
 | ||
| Fig. 3 Formation of glycosylated polyplex micelles using one charged glycosylated block copolymer and one oppositely charged homopolymer. Depiction of the electrostatic interaction between (A1) PGlcMA72-b-PMAA85 and PMETAI41 (GMP2), (A2) PGlcMA72-b-PMETAI71 and PMAA74 (GMP3) or (A3) PGlcMA72-b-PMETAI71 and PSPMA-Na114 (GMP4) yielding glycosylated nanoparticles. (B1)–(B3) Corresponding DLS (173°) intensity plots (bars), correlation coefficient (lines) and zeta potential values. (C1)–(C3) Corresponding TEM images of uranyl acetate-stained glycosylated nanoparticles. | ||
We prepared another spherical core-corona polyplex micelle by mixing PMAA74 with PGlcMA72-b-PMETAI71 (GPM3, Fig. 3A2). Like GPM2, GPM3 comprises a core consisting of complexed PMETAI and PMAA encapsulated by a PGlcMA corona. The DLS results (Fig. 3B2 and Fig. S7-3, ESI†) and TEM images (Fig. 3C2) confirmed the near charge neutrality (ζGPM3 = 6.9 mV, Table 1) and spherical morphology of GPM3, with a mean core diameter of ØGPM3 = 70.9 nm (Fig. S8-2, ESI†). Notably, the hydrodynamic radius of GPM3 (DhGPM3 = 69.5 nm, Table 1) is larger than that of GPM2 primarily because of the increased length of the core-forming block.62,63 The glycosylated polyplex micelles formed by the complexation of PSPMA-Na114/PGlcMA72-b-PMETAI71 (GPM4, Fig. 3A3) exhibited a relative large hydrodynamic radius (Dh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) GPM4 = 89.3 nm, Table 1), as confirmed by the DLS results (Fig. 3B3 and Fig. S7-4, ESI†). The increase in size is attributed to the larger volume and higher molar mass of PSPMA-Na than of the PMAA segment. Despite a 1:1 stoichiometric ratio, the ζ-potential (ζGPM4 = −18.0 mV, Table 1) of GPM4 remained negative, indicating incomplete charge compensation of PSPMA-Na units. This could be due to the larger hydrodynamic volume and stiffer nature of the PSPMA-Na chains than those of PMAA, as well as the broad dispersity of the block copolymer contributes to increased heterogeneity during micelle formation, leading to a wider size distribution an uneven ζ-potential among the nanoparticles. Although the PGlcMA corona provides stabilization, the negatively charged GPM4 remains susceptible to aggregation, as confirmed by the TEM images (Fig. 3C3 and Fig. S8-3, ESI†). To achieve complete charge compensation of the PSPMA-Na units, ζ-potential titration across various charge ratios was performed (Fig. S9, ESI†). Therefore, we used a ratio of 0.5:1 between PSPMA-Na114 and PGlcMA72-b-PMETAI71 (GPM5), and the ζ-potential (ζGPM5 = −5.0 mV, Table 1) of GPM5 confirmed the formation of a nearly charge-neutral complex core. TEM images (Fig. 4B) revealed the spherical morphology of GPM5, with variable dimensions of the particles but less aggregation. Both the hydrodynamic radius (DhGPM5 = 114.0 nm, Table 1) and mean core diameter (ØGPM5 = 96.2 nm) increased compared with those of GPM4, likely due to the incorporation of additional PGlcMA72-b-PMETAI71 chains into the complex core to fully compensate for the charges of PSPMA-Na114.64 The larger hydrodynamic radius compared with the mean core diameter is attributed to the intensity-weighted averaging in DLS measurements, which is more sensitive to larger particles.65
GPM4 = 89.3 nm, Table 1), as confirmed by the DLS results (Fig. 3B3 and Fig. S7-4, ESI†). The increase in size is attributed to the larger volume and higher molar mass of PSPMA-Na than of the PMAA segment. Despite a 1:1 stoichiometric ratio, the ζ-potential (ζGPM4 = −18.0 mV, Table 1) of GPM4 remained negative, indicating incomplete charge compensation of PSPMA-Na units. This could be due to the larger hydrodynamic volume and stiffer nature of the PSPMA-Na chains than those of PMAA, as well as the broad dispersity of the block copolymer contributes to increased heterogeneity during micelle formation, leading to a wider size distribution an uneven ζ-potential among the nanoparticles. Although the PGlcMA corona provides stabilization, the negatively charged GPM4 remains susceptible to aggregation, as confirmed by the TEM images (Fig. 3C3 and Fig. S8-3, ESI†). To achieve complete charge compensation of the PSPMA-Na units, ζ-potential titration across various charge ratios was performed (Fig. S9, ESI†). Therefore, we used a ratio of 0.5:1 between PSPMA-Na114 and PGlcMA72-b-PMETAI71 (GPM5), and the ζ-potential (ζGPM5 = −5.0 mV, Table 1) of GPM5 confirmed the formation of a nearly charge-neutral complex core. TEM images (Fig. 4B) revealed the spherical morphology of GPM5, with variable dimensions of the particles but less aggregation. Both the hydrodynamic radius (DhGPM5 = 114.0 nm, Table 1) and mean core diameter (ØGPM5 = 96.2 nm) increased compared with those of GPM4, likely due to the incorporation of additional PGlcMA72-b-PMETAI71 chains into the complex core to fully compensate for the charges of PSPMA-Na114.64 The larger hydrodynamic radius compared with the mean core diameter is attributed to the intensity-weighted averaging in DLS measurements, which is more sensitive to larger particles.65
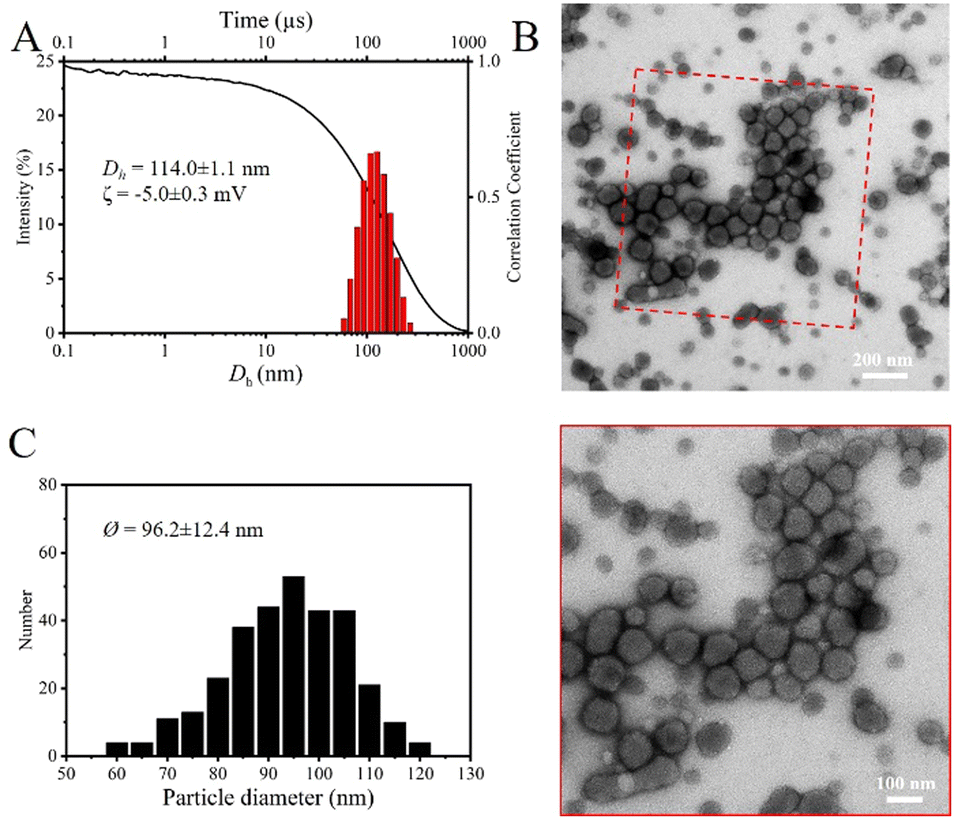 | ||
| Fig. 4 Morphology analyses of GPM5. (A) DLS (173°) intensity plots (bars), correlation coefficient (line) and zeta potential value. (B) TEM images of uranyl acetate-stained glycosylated nanoparticles. (C) Statistical analysis of glycosylated nanoparticles (data extracted from 300 particles in several images). | ||
Conclusions
This study successfully demonstrated the potential to fabricate glycosylated polyplex micelles through electrostatic interactions. Oppositely charged block glycosylated copolymers were produced in a straightforward fashion, via a combination of RAFT polymerization and postpolymerization modification. Upon mixing oppositely charged block copolymers, stable nonspherical nanoparticles were formed. Their morphology was confirmed by dynamic light scattering and transmission electron microscopy, underscoring the precision of this method in the preparation of glycosylated nanoparticles. The versatility of this system was further explored by employing oppositely charged homopolymers, broadening the potential applications of glycosylated nanoparticles. This approach offers a promising pathway toward more intricate nanoparticle designs, with potential for use in responsive delivery systems and other advanced biomedical applications. The insights gained from this work pave the way for future research into the responsive characteristics of these nanoparticles, facilitating their evolution into more sophisticated carriers for therapeutic cargo.Author contributions
The manuscript was written through the contributions of all authors. All the authors approved the final version of the manuscript. Conceptualization, M. C., T. P., J. B., V. S. D. V., R. F. and K. L.; methodology, M. C., T. P. and J. B.; writing – reviewing & editing, M. C., T. P., J. B., V. S. D. V., R. F. and K. L.; funding acquisition, V. S. D. V., R. F. and K. L.; resources, V. S. D. V., R. F. and K. L.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
The authors warmly thank Jur van Dijken, Dr Marc C. A. Stuart, Renze Sneep, Albert Woortman and Eleni Thomou for their technical assistance with thermogravimetric analyses, electron microscopy, HRMS and GPC, respectively. M. C. thanks Kai Lu for his assistance in analysing the morphologies of the glycosylated polyplex micelles. This research received funding from the Dutch Research Council (NWO) in the framework of the ENW PPP Fund for the top sectors and from the Ministry of Economic Affairs in the framework of the ‘PPS-Toeslagregeling’. M. C. thanks financial support from a Chinese Scholarship Council grant (no. 202006240066) and University of Groningen scholarship top-up.References
- R. C. Saxena, D. K. Adhikari and H. B. Goyal, Renewable Sustainable Energy Rev., 2009, 13, 167–178 Search PubMed
.
- D. J. Cosgrove, Curr. Opin. Plant Biol., 2018, 46, 77–86 CAS
- A. Domard, Carbohydr. Polym., 2011, 84, 696–703 CAS
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CAS
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 Search PubMed
- C. S. Mahon, G. C. Wildsmith, D. Haksar, E. de Poel, J. M. Beekman, R. J. Pieters, M. E. Webb and W. B. Turnbull, Faraday Discuss., 2019, 219, 112–127 CAS
- W. Stuart-Walker and C. S. Mahon, Adv. Drug Delivery Rev., 2021, 171, 77–93 CrossRef CAS
- J. Ciric and K. Loos, Carbohydr. Polym., 2013, 93, 31–37 CrossRef CAS
- L. Boetje, X. Lan, F. Silvianti, J. van Dijken, M. Polhuis and K. Loos, Carbohydr. Polym., 2022, 292, 119649 CrossRef CAS
- X. Lan, W. Li, C. Ye, L. Boetje, T. Pelras, F. Silvianti, Q. Chen, Y. Pei and K. Loos, ACS Appl. Mater. Interfaces, 2023, 15, 4398–4407 CrossRef CAS PubMed
- K. Loos, G. Jonas and R. Stadler, Macromol. Chem. Phys., 2001, 202, 3210–3218 Search PubMed
- T. Pelras and K. Loos, Prog. Polym. Sci., 2021, 117, 101393 CrossRef CAS
- A. Adharis, T. Ketelaar, A. G. Komarudin and K. Loos, Biomacromolecules, 2019, 20, 1325–1333 CrossRef CAS PubMed
- A. Adharis, D. Vesper, N. Koning and K. Loos, Green Chem., 2018, 20, 476–484 RSC
- A. Adharis and K. Loos, Macromol. Chem. Phys., 2019, 220, 1900219 CrossRef CAS
- W. M. J. Kloosterman, D. Jovanovic, S. G. M. Brouwer and K. Loos, Green Chem., 2014, 16, 203–210 RSC
- C. Cao, J. Zhao, M. Lu, C. J. Garvey and M. H. Stenzel, Biomacromolecules, 2019, 20, 1545–1554 CrossRef CAS
- E. Prochazkova, C. Cao, A. Rawal, M. Dracinsky, S. Bhattacharyya, I. Cisarova, J. M. Hook and M. H. Stenzel, ACS Appl. Mater. Interfaces, 2019, 11, 28278–28288 CrossRef CAS
- C. Cao, J. Zhao, F. Chen, M. Lu, Y. Y. Khine, A. Macmillan, C. J. Garvey and M. H. Stenzel, Chem. Mater., 2018, 30, 5227–5236 CrossRef CAS
- S. Ganda, Y. Jiang, D. S. Thomas, J. Eliezar and M. H. Stenzel, Macromolecules, 2016, 49, 4136–4146 CrossRef CAS
- H. Schlaad, L. You, R. Sigel, B. Smarsly, M. Heydenreich, A. Mantion and A. Masic, Chem. Commun., 2009, 1478–1480 RSC
- Z. Hordyjewcz-Baran, L. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901–3903 CrossRef
- L. You and H. Schlaad, J. Am. Chem. Soc., 2006, 128, 13336–13337 CrossRef CAS
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS
- L. Qiu, H. Zhang, T. Bick, J. Martin, P. Wendler, A. Boker, U. Glebe and C. Xing, Angew. Chem., Int. Ed., 2021, 60, 11098–11103 CrossRef CAS PubMed
- W. Qi, Y. Zhang, J. Wang, G. Tao, L. Wu, Z. Kochovski, H. Gao, G. Chen and M. Jiang, J. Am. Chem. Soc., 2018, 140, 8851–8857 CrossRef CAS
- X. Wu, L. Su, G. Chen and M. Jiang, Macromolecules, 2015, 48, 3705–3712 Search PubMed
- L. Su, C. Wang, F. Polzer, Y. Lu, G. Chen and M. Jiang, ACS Macro Lett., 2014, 3, 534–539 Search PubMed
- L. Su, Y. Zhao, G. Chen and M. Jiang, Polym. Chem., 2012, 3, 1560–1566 Search PubMed
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 120, 4925–4928 Search PubMed
- D. V. Pergushov, E. V. Remizova, J. Feldthusen, A. B. Zezin, A. H. E. Müller and V. A. Kabanov, J. Phys. Chem. B, 2003, 107, 8093–8096 CrossRef CAS
- Y. Li, T. K. Bronich, P. S. Chelushkin and A. V. Kabanov, Macromolecules, 2008, 41, 5863–5868 CrossRef CAS
- J. Sun and Z. Li, Macromolecules, 2020, 53, 8737–8740 CAS
- M. Ibrahim, E. Ramadan, N. E. Elsadek, S. E. Emam, T. Shimizu, H. Ando, Y. Ishima, O. H. Elgarhy, H. A. Sarhan, A. K. Hussein and T. Ishida, J. Controlled Release, 2022, 351, 215–230 CAS
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug. Delivery, 2012, 9, 1319–1323 CAS
- W. M. J. Kloosterman, S. Roest, S. R. Priatna, E. Stavila and K. Loos, Green Chem., 2014, 16, 1837–1846 CAS
- E. F. Fiandra, L. Shaw, M. Starck, C. J. McGurk and C. S. Mahon, Chem. Soc. Rev., 2023, 52, 8085–8105 CAS
- A. Monaco, V. P. Beyer, R. Napier and C. R. Becer, Biomacromolecules, 2020, 21, 3736–3744 CAS
- T. Tanaka, A. Matsuura, Y. Aso and H. Ohara, Chem. Commun., 2020, 56, 10321–10324 CAS
- E. L. Sahkulubey Kahveci, M. U. Kahveci, A. Celebi, T. Avsar and S. Derman, Biomacromolecules, 2022, 23, 4896–4908 CAS
- J. Zhao, H. Lu, Y. Yao, S. Ganda and M. H. Stenzel, J. Mater. Chem. B, 2018, 6, 4223–4231 CAS
- S. Ganda, M. Dulle, M. Drechsler, B. Förster, S. Förster and M. H. Stenzel, Macromolecules, 2017, 50, 8544–8553 CrossRef CAS
- M. Lu, F. Chen, C. Cao, C. J. Garvey, N. L. Fletcher, Z. H. Houston, H. Lu, M. S. Lord, K. J. Thurecht and M. H. Stenzel, Macromolecules, 2019, 52, 477–486 CrossRef CAS
- A. H. Hofman, R. Fokkink and M. Kamperman, Polym. Chem., 2019, 10, 6109–6115 RSC
- A. D. Filippov, I. A. van Hees, R. Fokkink, I. K. Voets and M. Kamperman, Macromolecules, 2018, 51, 8316–8323 CrossRef CAS PubMed
- A. Dag, M. Callari, H. Lu and M. H. Stenzel, Polym. Chem., 2016, 7, 1031–1036 RSC
- T. Pelras, C. S. Mahon Nonappa, O. Ikkala, A. H. Groschel and M. Mullner, J. Am. Chem. Soc., 2018, 140, 12736–12740 CrossRef CAS
- T. Pelras Nonappa, C. S. Mahon and M. Müllner, Macromol. Rapid Commun., 2020, 42, 2000401 Search PubMed
- M. Vamvakaki, G.-F. Unali, V. Bütün, S. Boucher, K. L. Robinson, N. C. Billingham and S. P. Armes, Macromolecules, 2001, 34, 6839–6841 CrossRef CAS
- J. H. Lai, Macromolecules, 1984, 17, 1010–1012 CrossRef CAS
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS
- A. L. Patterson, S. P. O. Danielsen, B. Yu, E. C. Davidson, G. H. Fredrickson and R. A. Segalman, Macromolecules, 2019, 52, 1277–1286 CrossRef CAS
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC
- D. V. Pergushov, A. H. Müller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC
- M. Sedlák, J. Chem. Phys., 2002, 116, 5256–5262 CrossRef
- C. A. Scarff, M. J. G. Fuller, R. F. Thompson and M. G. Iadanza, J. Visualized Exp., 2018, 6, 57199 Search PubMed
- J. C. Eriksson and S. Ljunggren, Langmuir, 2000, 6, 895–904 CrossRef
- I. K. Voets, A. de Keizer, P. de Waard, P. M. Frederik, P. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. Leermakers and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2006, 45, 6673–6676 CAS
- I. K. Voets, R. Fokkink, T. Hellweg, S. M. King, P. de Waard, A. de Keizer and M. A. C. Stuar, Soft Matter, 2009, 5, 999–1005 RSC
- M. Amann, J. S. Diget, J. Lyngsø, J. S. Pedersen, T. Narayanan and R. Lund, Macromolecules, 2019, 52, 8227–8237 CrossRef CAS
- C. C. M. Sproncken, J. R. Magana and I. K. Voets, ACS Macro Lett., 2021, 10, 167–179 CrossRef CAS PubMed
- S. V. D. Burgh, A. D. Keizer and M. A. C. Stuart, Langmuir, 2004,(20), 1073–1084 CrossRef
- H. M. van der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. van der Gucht, Langmuir, 2012, 28, 14180–14191 CrossRef CAS
- H. E. Cingil, E. B. Boz, J. Wang, M. A. C. Stuart and J. Sprakel, Adv. Funct. Mater., 2016, 26, 1420–1427 CrossRef CAS
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb02760d |
| This journal is © The Royal Society of Chemistry 2025 |