
rC14: engineering a new dual-function carbon allotrope for sustainable energy technologies under conventional and micro-strain conditions†
Yaru
Wei‡
abc,
Baocheng
Yang‡
b,
Shouren
Zhang
 b and
Houyang
Chen
*ac
b and
Houyang
Chen
*ac
aChongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, Chongqing 400714, P. R. China. E-mail: chenhouyang@cigit.ac.cn
bHenan Provincial Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, P. R. China
cChongqing School, University of Chinese Academy of Sciences, Chongqing 400714, P. R. China
First published on 14th April 2025
Abstract
The depletion of fossil fuels and increasing greenhouse gas emissions underscore the urgent need for sustainable energy technologies. Herein, we present rC14, a novel two-dimensional (2D) carbon allotrope engineered by reconfiguring graphene through carbon–carbon bond rotation and defect introduction, which exhibits remarkable multifunctionality for energy storage and conversion applications under conventional and micro-strain conditions. Theoretical calculations indicate that rC14 exhibits excellent dynamical, thermal and mechanical stability. Its high porosity facilitates a theoretical Na-ion storage capacity of 1115 m Ah g−1, approximately three times higher than that of conventional graphite, and this high capacity is retained under small compressive strains. It possesses a low ion diffusion barrier (0.22–0.54 eV) that promises rapid charge/discharge kinetics. Furthermore, the non-uniform charge distribution across rC14, particularly at C1 and C2 sites, renders it an effective metal-free catalyst for the hydrogen evolution reaction (HER). The application of small biaxial compressive strains further refines the electronic structure and adsorption properties of the metal-free catalyst, yielding near-optimal ΔGH* values (|ΔGH*| around 0.05 eV) that surpass the values of conventional catalysts such as the Pt(111) surface (−0.09 eV). This work establishes rC14 as a promising dual-function metal-free material for next-generation energy storage and conversion systems under conventional and micro-strain conditions and demonstrates the effectiveness of the designed strategies in advancing material performance.
1. Introduction
The development of electrochemical energy storage and conversion technologies is essential for achieving a sustainable and clean energy supply, especially in light of the rapid depletion of nonrenewable fossil fuels and the significant emission of greenhouse gases.1–3 Secondary batteries and effective catalysts for hydrogen evolution reactions (HERs) have attracted considerable attention as potential solutions to the energy crisis and environmental pollution. Among secondary batteries, sodium ion batteries (SIBs) are considered one of the most promising alternatives to commercially available lithium ion batteries (LIBs) due to their low cost and natural abundance of sodium resources.4 Despite recent advancements in SIB technology, identifying novel anode materials capable of accommodating large Na-ions remains a significant challenge.5,6 In parallel, hydrogen (H2) produced via water electrolysis offers an attractive solution for high-efficiency energy conversion, where highly efficient HER catalysts play a crucial role.7 Although platinum (Pt)-based catalysts exhibit admirable performance in the HER, their scarcity and high cost severely limit their large-scale practical applications.8 Consequently, developing cost-effective, highly efficient, and long-life electrode materials for both SIBs and HERs is necessary for overcoming the existing challenges.Recently, two-dimensional (2D) anode materials with large surface areas have attracted great interest, which can provide abundant active sites for electrocatalytic reactions.9,10 Pristine graphene, however, is not an ideal choice for these energy-related applications because the high delocalization of its π electrons renders the structure chemically inert despite its stability. This limitation has driven the scientific community to explore new carbon allotropes and develop innovative synthesis methods.11 Notably, carbon structures containing non-six-membered ring units have demonstrated excellent physicochemical and electronic properties, making them promising candidates for electrodes in energy conversion and storage.12 For instance, in 2021, biphenylene was synthesized on the surface of Au(111) single crystals under ultra-high vacuum conditions and then applied it as a metal-free catalyst for water splitting in OERs.13,14 Li et al. identified monolayer biphenylene as a promising high-performance anode for sodium-ion batteries (SIBs).15 Wang et al. has demonstrated a new 2D carbon allotrope (net-τ) as a new high-performance metallic carbon anode material for lithium-ion batteries.16 Wu et al. investigated graphdiyne (GDY) and graphyne (an allotrope of GDY) as high activity catalysts for CO oxidation and the oxygen reduction reaction (ORR),17,18 owing to the positively charged sites within their frameworks, although the HER performance of pristine GDY remained suboptimal.19 More research has focused on two-dimensional carbon materials as single energy storage or catalytic electrodes, while little research has focused on multifunctional materials. In 2022, Yang et al. proposed graphene composed of four- and seven-membered rings as an efficient bifunctional electrocatalyst for both the HER and OER, as well as a promising anode material for potassium-ion batteries.20 These studies underscore the growing interest in multifunctional materials that can serve dual or more purposes while reducing overall costs.
In this work, based on first-principles calculations, we have constructed this novel 2D carbon allotrope, which consists of three-, five-, six- and fourteen-membered rings, and named it rC14. rC14 could be constructed by rotation of the carbon–carbon bond and introduction of defects (Fig. S1†), and these methods have experimental feasibility.21–23 The energy calculations reveal that while rC14 is metastable compared to graphene, it is energetically more favorable than graphdiyne24 (Table S1†). Moreover, our results showed that rC14 is a promising anode material for Na-ion batteries (SIBs), as evidenced by its high Na-ion storage capacity, low diffusion energy barrier and modest average open circuit voltage. Meanwhile, the fully exposed active sites in rC14 enhance its catalytic activity for the HER, with applied strain playing a critical role in modulating the performance. We further analyzed its electronic characteristics and composition that underpin both its energy storage capacity and catalytic activity. The computational strategies and methods used in this study provide a reference for future search of new 2D materials with dual-function applications.
2. Computational methods
Density functional theory calculations were performed in the Vienna Ab initio Simulation Package (VASP).25 The interactions between electrons were described by a projector-augmented-wave (PAW)26 method. The electron exchange-correlation was processed by the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional,27 and the Heyd−Scuseria−Ernzerhof (HSE06)28 functional was adopted to acquire more accurate electronic structures. The plane-wave cut-off was set to 500 eV. All structural relaxation until the residual force on each atom was less than 0.001 eVÅ−1, and the convergence of total energy was set to 10−6 eV. Van der Waals (vdW) corrections with the DFT-D229 method were considered to describe interactions between the Na atom and rC14 monolayer. The Brillouin zone sampled using the 7 × 7 × 1 k-point grid was adopted for geometry relaxation and a 14 × 14 × 1 k-point grid was used for density of states (DOS) calculations. In addition, a vacuum layer of 20 Å was adopted to avoid periodic image interactions. The dynamical stability of rC14 was estimated using the phonopy code with density functional perturbation theory (DFPT).30 The climbing-image nudged elastic band (CI-NEB) method31,32 was applied to explore the diffusion barriers of Na atoms on the surface of RC14. Ab initio molecular dynamics (AIMD) simulations33–35 were performed to examine the thermal stability with the canonical (NVT) ensemble.3. Results and discussion
3.1. Structure and stability
Fig. 1a shows the 3 × 3 supercell optimized configuration of the rC14 monolayer. Its unit cell involves 14 carbon atoms with optimized lattice parameters of a = 5.74 Å and b = 8.45 Å. Its space group is Pmmm. Symmetry analysis demonstrates that atoms in the unit cell can be categorized into five inequivalent sets, labelled C1 through C5. As listed in Table S1,† all bond lengths (1.38–1.44 A) are close to that of graphene (1.42 A), revealing sp2 hybridization. | ||
| Fig. 1 (a) The crystal structure of rC14 in the form of a 3 × 2 supercell, from top and side views. (b) Fluctuations of the total potential energy of rC14 at 300 K performed by DS-PAW. (c) Polar diagrams for plane Young's modulus C and (d) Poisson's ratio ѵ for rC14. | ||
Furthermore, the structural stability of the proposed structure is confirmed. The total energy of rC14 is −8.59 eV per atom, underscoring its energetic stability. The detailed structural information of rC14 and other 2D carbon allotropes is given in Table S1 and Section S1 of the ESI.† The phonon spectrum (Fig. S2†) shows no imaginary phonon mode throughout the entire Brillouin zone, indicating that rC14 is dynamically stable. To evaluate the thermal stability of rC14, AIMD simulations were performed at 300 K and 1200 K. Each simulation was conducted for 5 ps with a time step of 1 fs to ensure accuracy and reliability. The total energy of rC14 possesses slight fluctuations in total energy without any structural changes (Fig. 1b and S3†), signifying outstanding thermal stability even at high temperatures around 1200 K. The elastic constants are C11 = 182 GPa, C22 = 228 GPa, C12 = 51 GPa and C44 = 36 GPa. The elastic constants Cij satisfy the Born–Huang criteria,36 C11C22– C122 > 0 and C44 > 0, implying the mechanical stability of rC14. Furthermore, Young's modulus C and Poisson's ratio ѵ estimated the in-plane stiffness from the linear elastic constants. The polar diagrams of C(θ) and ѵ(θ) in Fig. 1c and d show that the Young's modulus and Poisson's ratios are almost isotropic. Specifically, the plane Young's modulus is 224 GPa nm along the a-axis (Ca) and 178 GPa nm along the b-axis (Cb), implying high in-plane stiffness. The corresponding Poisson's ratios in the a-axis and b-axis directions are 0.22 and 0.28, respectively, which indicate that rC14 is malleable in the axial direction.
3.2. Electronic properties
The electronic properties of rC14 were studied through band structures and their corresponding density of states (DOS) based on PBE/HSE06 functionals (Fig. 2a and b). No band gap is observed between the valence and conduction bands crossing the Fermi level, corresponding to the nonzero density of states (DOS) at the Fermi level, indicating metallic conductivity of rC14. Furthermore, the total DOS around the Fermi level is predominantly contributed by pz orbitals. The electron localized function (ELF) can provide an insight into the gain of the bonding nature, and the value of ELF is between 0 and 1, where 0, 0.5, and 1 correspond to nearly no electrons, fully delocalized electrons and perfect localization, respectively. The ELF result (Fig. 2c) suggests strong localized electrons (ELF close to 1.0), indicating the strong C–C covalent bonding. Almost no electron is found in the large vacancy in the center, which facilitates fast ion loading and large metal ion filling. | ||
| Fig. 2 (a) Band structures by PBE and HSE06 functionals. (b) Projected density of states of rC14 obtained by the PBE functional. The Fermi level is set to zero. (c) The ELF map of rC14. | ||
3.3. Promising application as an anode for Na-ion batteries
To explore the potential application, the adsorption of a single Na atom on the rC14 monolayer (2 × 2 × 1) was investigated to determine the optimized Na absorption site(s). Fig. 3a shows all possible adsorption sites, i.e., three hollow sites (H1–H3), seven bridge sites (B1–B7), and five top sites (T1–T5). Upon full relaxation, Na atoms preferentially locate at sites H1–H3 and B1, with Na atoms initially placed at other sites spontaneously migrating to either a hollow or the B1 site. The H1 site shows the in-plane adsorption, while the other three stable sites present the out-of-plane adsorption. The adsorption energy is defined as: Eads = (EnM−rC14 − ErC14 − nEM)/n, where EnM−rC14 and ErC14 refer to total energies of rC14 with and without Na insertion, respectively; EM represents the total energy per Na atom in bulk metallic sodium, and n is the number of adsorbed Na atoms. By adopting the single atom adsorption method, the calculated adsorption energies for the H1, H2, H3 and B1 sites are −1.05, −0.68, −0.69 and −0.66 eV, respectively. The H1 site shows in-plane adsorption (site H1, Fig. S4a†), implying that H1 sites are the most stable adsorption sites. The strong adsorption of Na in the in-plane H1 sites is unfavorable for ion migration in (stretchable) batteries, e.g., the diffusion energy barrier of Path1 is 4.55 eV (Fig. S4c†). Thus, we further adopt the atom pair adsorption approach to investigate the adsorption behavior of low concentration Na atoms on the surface of rC14.37–39 Compared with the single Na atom adsorption scenario, three stable adsorption sites (out-of-plane sites H2, H3 and B1) remained, and two additional stable sites (B7 and F) emerged, as shown in Fig. 3a. Their adsorption energies are −0.46, −0.40, −0.39, −0.34 and −0.36 eV for H2, H3, B1, B7 and F sites, respectively, which means that the H2 site is the most energetically favorable (Fig. 3b).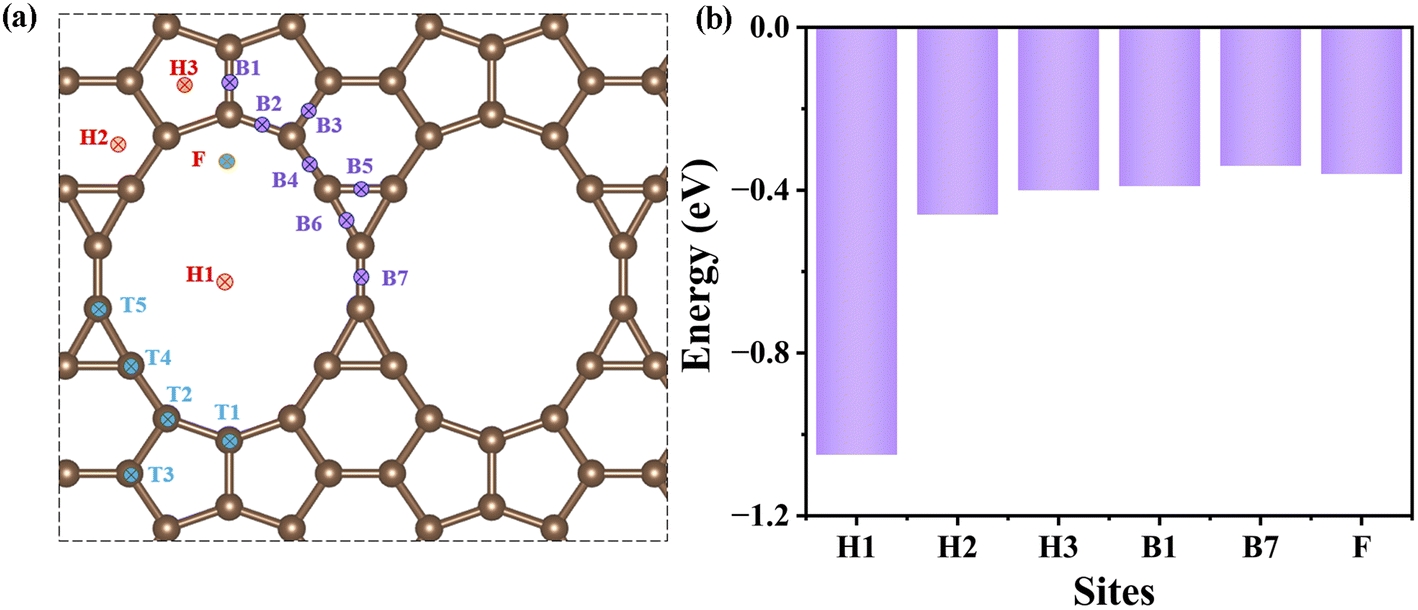 | ||
| Fig. 3 (a) The possible Na adsorption sites on rC14. (b) The adsorption energies of the Na-adsorbed rC14 monolayer at different sites by the atom pair adsorption approach. | ||
Subsequently, a series of Na ions of increasing concentration were adopted to test the storage capacity of the rC14 anode. Their energetically favorable configurations are provided in Fig. S5a–e,† and the computed adsorption energies for Na4C56, Na8C56, Na12C56, Na20C56 and Na28C56 are −0.80, −0.36, −0.16, −0.12 and −0.18 eV, respectively, and our AIMD result shows that Na32C56 is not thermally stable (Fig. S5f†). It implies the maximum stoichiometric ratio is NaC2. Our AIMD result shows that Na28C56 is thermally stable (Fig. S6†), and the corresponding theoretical maximum specific capacity of 1115.78 mA h g−1, which is much higher than those of 2D anode materials such as graphite (237 mA h g−1),40,41 phosphene (433 mA h g−1),42 Ti3C2 (352 mA h g−1),43 phagraphene (557.87 mA h g−1)44 and net-C18 (403 mA h g−1).45 To further assess the performance of the anode for the stretchable SIBs, the mechanochemical approach was employed to check their specific capacities under biaxial strains, as shown in Fig. S7a–d.† At a compressive strain of 0.5–2%, rC14 undergoes a reversible phase transition while maintaining a capacity of approximately 1115 mA h g−1. Moreover, after the metal ions are removed, rC14 fully recovers to its original configuration (Fig. S7e†). This indicates that small compressive strains retain the high capacity of the anode.
To understand the microscale mechanism of the adsorption in-depth, we calculated the charge density differences of most stable adsorption structures at various Na concentrations. The electrons mainly accumulated primarily around the C atoms, and the Na atoms lost a significant portion of their electrons (Fig. S8†), which demonstrates that the charges are transferred from metal atoms to the rC14 monolayer. Bader charge population analysis shows that at low Na concentrations (Na4C56), the average electron loss for each Na atom is 0.90 e, indicating that Na atoms are near-complete ionization. While at high concentrations ((Na28C56)), the average electron transfer per Na atom decreases to 0.51e. Furthermore, electron localization function (ELF) results provide further insight. At lower ionic concentrations (x = 2), only a sparse electron cloud surrounds the Na atoms (ELF values close to 0), indicating the highly delocalized electron density and the formation of strong ionic bonds with neighboring carbon atoms. With the adsorption of two layers of Na atoms on both sides of rC14, the electron-deficient region at the center of rC14 gradually decreases. Partial ionization of Na atoms occurs, while others contribute to delocalized electron density, which accumulates between Na ions to neutralize the repulsion among the Na+ cations and stabilize the adsorbed layer. For the maximum capacity of Na atom adsorption (Fig. S9e†), the electron cloud mainly filled the interstices between the two Na layers, implying the formation of conductive channels that facilitate efficient electron transfer.
To assess the kinetics of the transport of Na ions, we further investigated the charge/discharge rate, which is one of the key indicators for rechargeable batteries and depends on the electronic conductivity and diffusion barrier. The charging/discharging rate is affected by the electronic properties of anodes. To better understand the electronic properties of rC14 with Na ion loading, the density of states (DOS) of the most stable configurations is computed (Fig. 4). With the loading of Na ions, the electron states of the Na atom gradually near the Fermi level, and the total DOS shifts towards the low energy level. Projected density of states (PDOS) shows that there is a clear overlap between the Na s orbital and the carbon p orbital at the Fermi level, indicating significant s-p hybridization and strong interactions between Na atoms and the rC14 monolayer. In addition, the rC14 monolayer retains its metallic character with outstanding electronic conductivity during the Na insertion and extraction processes, which is essential for fast charge/discharge kinetics.
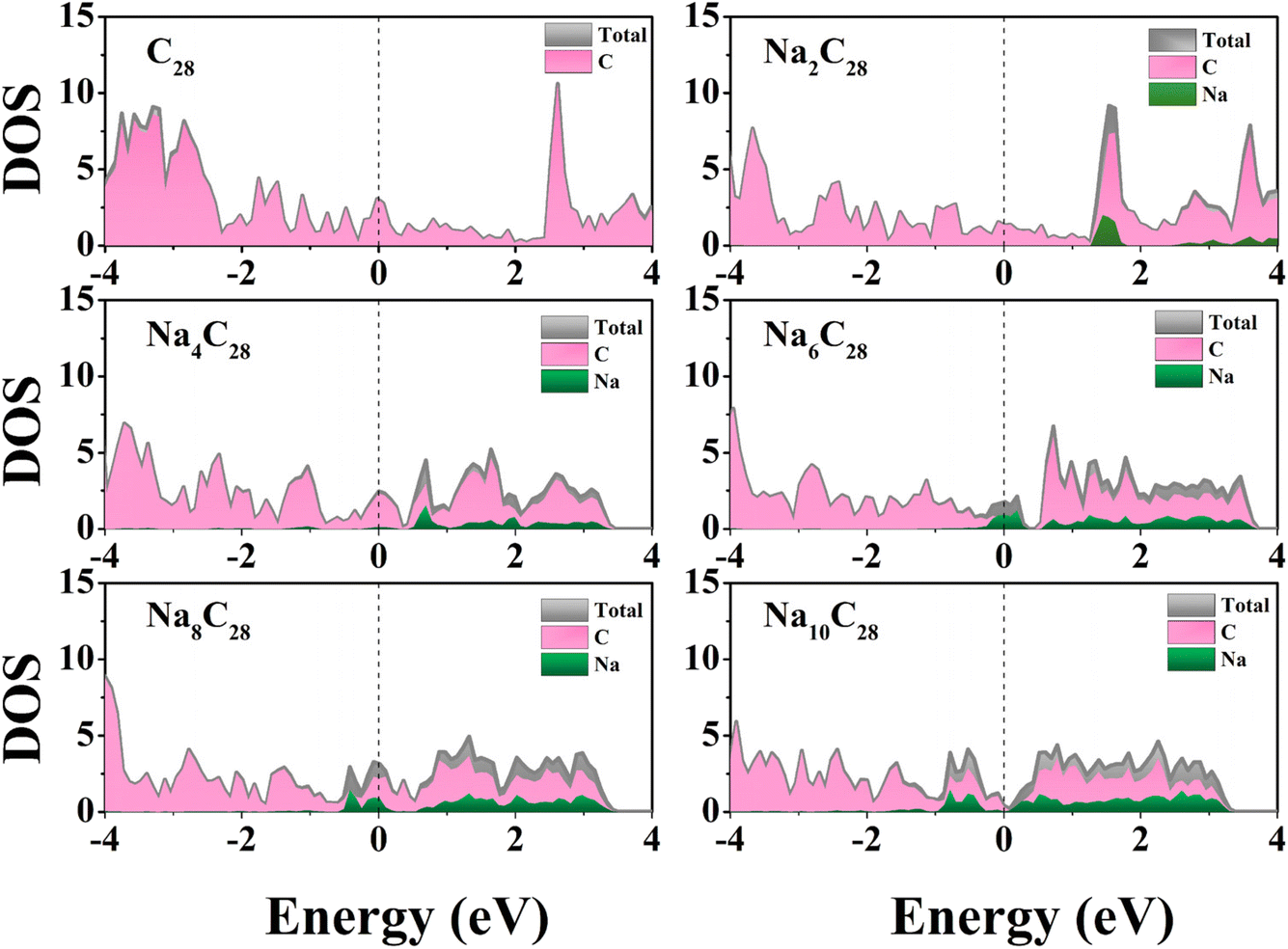 | ||
| Fig. 4 Density of states for the rC14 monolayer with Na atom adsorption. The Fermi level was set as 0 eV and indicated by the black vertical dashed lines. | ||
Next, the mobility of Na atoms on the rC14 monolayer was performed by the climbing-image nudged elastic band (CI-NEB) method. We first investigated the diffusion of one of the metal ions on the rC14 monolayer by considering three possible migration pathways from one energetically favorable H1 site to another along different directions (Fig. S4b†). From Fig. S4c,† the diffusion energy barrier of Path1 is 4.55 eV, indicating that Na-ions are not likely to diffuse along this route. In contrast, both Path2 and Path3 exhibit a migration barrier of 0.51 eV (Fig. S4d and e†), making these pathways more feasible for ion migration. To further understand Na-ion diffusion at low concentrations, we studied the diffusion behavior of Na-ions by adopting a “particle (atom) pair diffusion” model.35 We selected three possible diffusion pathways between the two most stable adsorption sites (H2 sites), which were found in the atom pair adsorption model, marked as Path1, Path2 and Path3 in Fig. 5a. The diffusion energy barrier profiles for these pathways were calculated (Fig. 5b). The corresponding diffusion barriers are 0.31, 0.54 and 0.22 eV for Paths1–3, respectively, which are much lower than that the mobility of single Na atoms. The migration barriers of Na atoms in rC14 are smaller than those observed for the commercial graphite anode (∼0.6 eV).41,46,47 The low-energy migration barrier of Na diffusion indicates its potential for achieving high charge–discharge rates as an anode material.
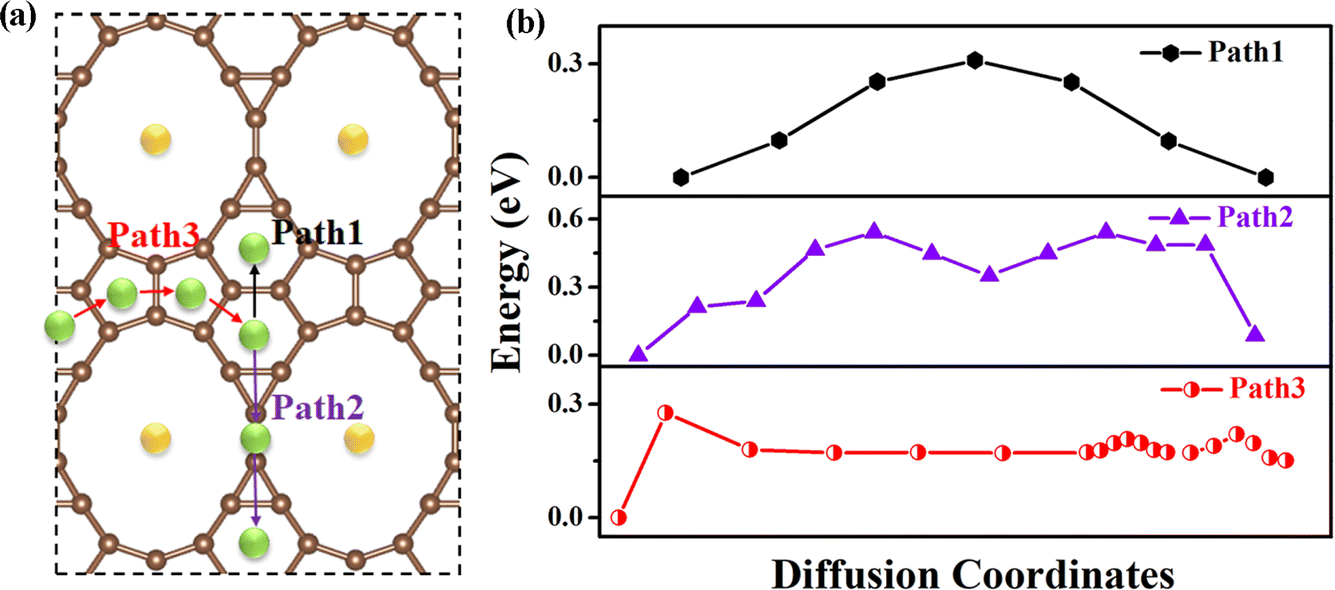 | ||
| Fig. 5 (a) The three possible migration paths and (b) the migration barrier of low concentration Na atom diffusion on rC14. | ||
Finally, the average open-circuit voltage (OCV) was computed by using the following equation:
| OCV = (EM + xENa − EM+Na)/xe | (1) |
3.4. Promising metal-free catalyst for the hydrogen evolution reaction
Given its fully exposed active sites, rC14 is a promising candidate for catalyzing the hydrogen evolution reaction (HER). To investigate its catalytic activity, the initial step is the identification of catalytically active sites on rC14. After systematically examining all hydrogen adsorption sites, the sites C1 and C2 are found to most effectively stabilize atomic hydrogen with the lowest energy. The catalytic activity of the HER is strongly influenced by the Gibbs free energy change of hydrogen adsorption (ΔGH*). Specifically, a smaller absolute value of ΔGH leads to enhanced catalytic activity.52 The Gibbs free energy is defined as follows:| ΔGH = ΔEH + ΔEZPE + TΔSH | (2) |
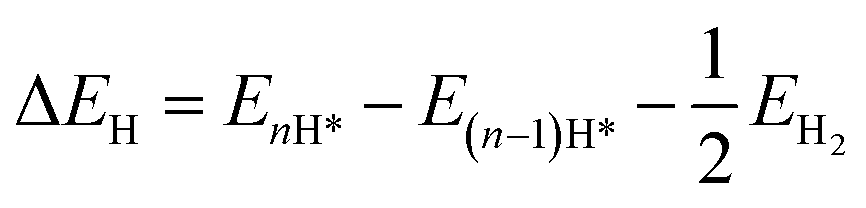 | (3) |
 | (4) |
 and
and  are the corresponding zero-point energies. EH2 and
are the corresponding zero-point energies. EH2 and  are the total energy and zero-point energy of a hydrogen molecule in the gas phase, respectively. The calculated ΔGH* for one hydrogen atom adsorbed on the top site of C1/C2 atoms is 0.26/0.19 eV, which is much lower than those on the top site of C3/C4/C5 (0.91/0.96/0.56 eV), as presented in Fig. 6a. Notably, the ΔGH* of C2 sites is smaller than absolute values of N,P-graphene (0.29 eV),53 Th-graphene (0.24 eV),20 2D B3S nanosheet (−0.30 eV)54 and M-N@graphene (−0.2–0.3 eV),55 indicating that the top site of the C2 atom is particularly well-suited as an active catalytic site for the HER.
are the total energy and zero-point energy of a hydrogen molecule in the gas phase, respectively. The calculated ΔGH* for one hydrogen atom adsorbed on the top site of C1/C2 atoms is 0.26/0.19 eV, which is much lower than those on the top site of C3/C4/C5 (0.91/0.96/0.56 eV), as presented in Fig. 6a. Notably, the ΔGH* of C2 sites is smaller than absolute values of N,P-graphene (0.29 eV),53 Th-graphene (0.24 eV),20 2D B3S nanosheet (−0.30 eV)54 and M-N@graphene (−0.2–0.3 eV),55 indicating that the top site of the C2 atom is particularly well-suited as an active catalytic site for the HER.
 | ||
| Fig. 6 (a) The computed free energy profiles for the HER. (b) The p-band center (εp) of the active C sites of rC14. | ||
To further understand the origin of the enhanced catalytic activity, we performed the charge density and Bader charge population analysis. The electrons are redistributed on the surface of the 2D rC14 monolayer, compared to graphene (Fig. S10†). Although rC14 is entirely composed of carbon atoms, subtle charge transfers occur among the different carbon sites. For example, the net charges are approximately −0.04|e| for the C1 atom and +0.13 |e| for the C2 atom, with similar variations observed for the other carbon atoms. The coexistence of unpaired electrons and empty states is believed to be beneficial for the HER. Additionally, the DOS of various C sites in rC14 is presented in Fig. S11.† The projected DOS reveals that pz orbitals of the carbon atoms play a decisive role in the formation of C–H bonds, as evidenced by the significant overlap with the s orbital of hydrogen. To elucidate the remarkable difference between the adsorption strength of the H* species on different C atoms, we explored the electronic properties of the p-orbital in the C sites based on the amendatory p band-center (εp) model proposed by Wang's group.56–58 Our results (Fig. 6b) indicate that the positions of εp of C1 (−3.71 eV) and C2 (−3.48 eV) are closer to the Fermi level than those of other carbon atoms, implying that C1 and C2 atoms can interact more strongly with reaction intermediates compared to other C atoms of rC14. This strong interaction further supports the role of these sites as the primary active centers for the HER on the rC14 monolayer.
In fact, graphene is not perfectly flat, and the adsorption on the flat surface leads to deformation and an associated increase in energy. However, if the flat surface is properly curved, this energy penalty can be mitigated, thereby enhancing the catalytic activity. To systematically investigate the effect of strain on the HER performance, we applied a series of small external biaxial compressive strains ranging from 0.5% to 2% with intervals of 0.5%. One can see that compression strains would regulate the Gibbs free energy and thus affect the performance of the HER (Fig. 7a). Under 1% and 2% strains of the rC14 system, the C1 and C3 sites exhibit considerable catalytic activity for the HER, and the ΔGH* values are −0.049 and 0.053 eV, respectively. These values are close to the optimal HER catalyst (ΔGH* = 0 eV) and are even lower than those of the well-established Pt(111) surface (−0.09 eV)59 and non-noble metal-based catalysts anchored on GDY.60,61 Although there is no clear trend in the curvature effect on the C–H bond distance, probably due to out-of-plane deformation induced by hydrogen adsorption, the overall adsorption ability increases with increasing rC14 curvature. This observation suggests that the curvature, introduced by strain, can effectively influence the adsorption of reaction intermediates (Table S2†).
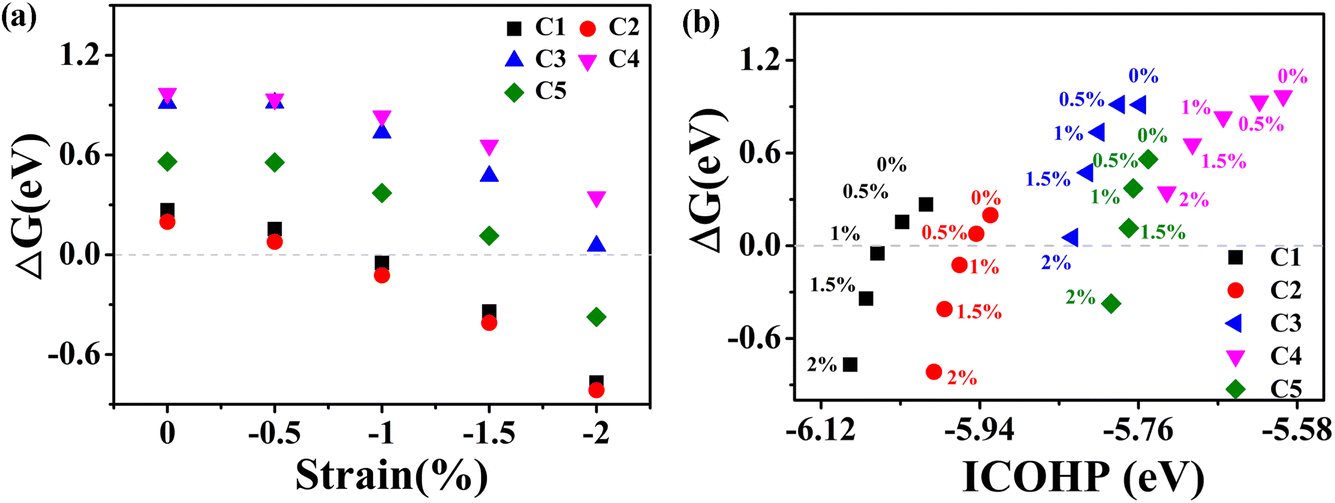 | ||
| Fig. 7 (a) The HER free energy of the active C sites of rC14 with compression ratios of 0 to 2%. (b) The relation between ICOHP and HER free energy. | ||
Furthermore, we analyzed the bonding relationship between the C site of rC14 under strain and adsorbed H* by implementing COHP, as shown in Fig. 7b. The curvature has a significant impact on the bonding strength between C atoms and H atoms. In COHP analysis, a more negative integrated COHP (ICOHP) value corresponds to a stronger bonding interaction. The bonding strength between the active C atoms and H increase with the increase of curvature. For the originally active C1 and C2 atoms, applying a slight strain (0.5–1%) yields an optimal balance interaction between C and H atoms, resulting in adsorption that is neither too strong nor too weak and thus enhances the catalyst activity. However, if the strain is increased further, the adsorption becomes too strong, which hinders the desorption of the hydrogen atom. In summary, the excellent catalytic performance of the C1 and C2 sites can be attributed to the moderate ICOHP values achieved under compressive strains of 1% and 0.5%, respectively. For the less active C3, C4, and C5 atoms, increasing the strain enhances the adsorption interaction between carbon and hydrogen, thereby tuning their HER activity. Notably, for the C3 and C5 atoms, applying strains of 2% and 1.5% results in ΔGH* values of 0.054 and 0.115 eV, respectively, values close to the ideal near-zero condition for optimal HER performance.
To further elucidate the origin of the enhanced catalytic activity in strained rC14, we analyzed the PDOS for rC14 under biaxial compressive strains ranging from 0.5% to 2% (Fig. 8a). The total DOS around the Fermi level (−2 to 5 eV) is primarily contributed by electrons of pz orbitals. With increasing strain, a noticeable downward shift of the Fermi level is observed. Additionally, small but noticeable changes near the Fermi level caused by the out of plane deformation are evident (Fig. 8b). For a compression strain of 0.5%, the total DOS closely resembles that of the flat rC14, indicating that slight out-of-plane deformation has a minimal impact on the electron states around the Fermi level. In contrast, for strains exceeding 1% (where the deformation is more pronounced), the electron states near the Fermi level decrease with increasing deformation. At 1% strain, the electronic state at the Fermi level falls between the values observed at lower and higher strains. Therefore, one could conclude that slight out-of-plane deformation induced by compressive strain regulates the electron states near the Fermi level in rC14 and changes its chemical activity. This modulation may enhance the adsorption and dissociation of hydrogen molecules, thereby improving the overall catalytic performance of rC14 for the hydrogen evolution reaction.
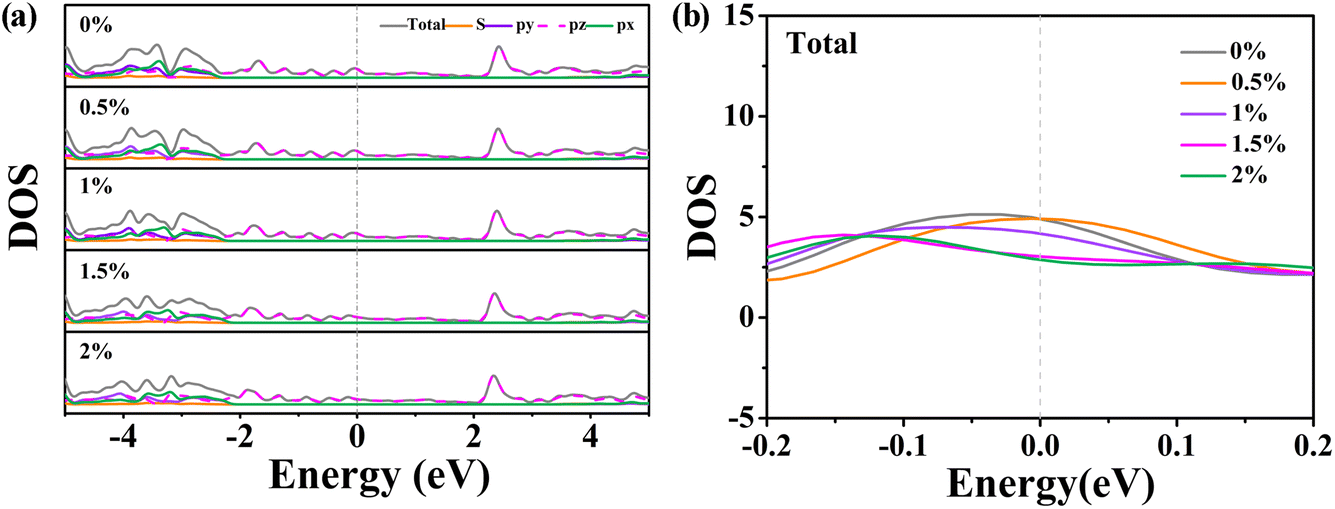 | ||
| Fig. 8 (a) Calculated electronic density of rC14 at compression ratios of 0 to 2%. (b) The total DOS around the Fermi level. | ||
4. Conclusion
In summary, inspired by the remarkable electronic and mechanical properties of graphene, we reconfigured its structure to propose rC14 as a potential metal-free multifunctional material for use as an SIB anode and an HER catalyst, based on density functional theory calculations. Constructed through a combination of carbon–carbon bond rotation and defect engineering, rC14 is dynamically, thermally, and mechanically stable. Our systematic study reveals that the high porosity of the rC14 monolayer yields a high theoretical capacity of 1115.78 m Ah g−1 for Na-ion storage under conventional and micro-strain conditions, while its low diffusion barrier (0.22–0.54 eV) suggests its potential for ultrafast charging/discharging. Furthermore, the non-uniform charge distribution across the carbon atoms in rC14 results in the C1 and C2 sites emerging as the primary active centers for the HER. The application of biaxial compressive strain further enhances the HER catalytic activity by modulating the adsorption state of H* and altering the electron states near the Fermi level. In the 1% and 2% strained rC14 systems, the optimized ΔGH* values (|ΔGH*| around 0.05 eV) approach the ideal condition for HER catalysis, outperforming the well-established Pt(111) surface (−0.09 eV). The fundamental design considerations of this 2D material highlight its potential not only as a high-performance anode for sodium-ion batteries but also as an effective metal-free electrocatalyst for hydrogen production. This work provides valuable insights that will guide the future exploration and development of multifunctional materials to overcome the multifaceted challenges for sustainable energy storage and conversion applications.Data availability
Data will be made available on request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Postdoctoral Program of Natural Science Foundation of Chongqing (CSTB2023NSCQ-BHX0231), Natural Science Foundation of Chongqing (CSTB2023NSCQ-MSX0045), the Startup Foundation of Chongqing Institute of Green and Intelligent Technology, and Chinese Academy of Sciences. The authors acknowledge hzwtech (https://www.hzwtech.com) for providing HPC resources that have contributed to the research results reported within this paper.References
- C. Zhu, R. E. Usiskin, Y. Yu and J. Maier, Science, 2017, 358, 6369 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 6321 CrossRef.
- L. Fan and X. Li, Nano Energy, 2018, 53, 630–642 CrossRef CAS.
- T. Liu, Y. Zhang, Z. Jiang, X. Zeng, J. Ji, Z. Li, X. Gao, M. Sun, Z. Lin, M. Ling, J. Zheng and C. Liang, Energy Environ. Sci., 2019, 12, 1512–1533 RSC.
- H. Yang, R. Xu, Y. Yao, S. Ye, X. Zhou and Y. Yu, Adv. Funct. Mater., 2019, 29, 1809195 CrossRef.
- Y. Dong, J. Xu, M. Chen, Y. Guo, G. Zhou, N. Li, S. Zhou and C. P. Wong, Nano Energy, 2020, 68, 104357 CrossRef CAS.
- X. Chia, A. Y. S. Eng, A. Ambrosi, S. M. Tan and M. Pumera, Chem. Rev., 2015, 115, 11941–11966 CrossRef CAS PubMed.
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC.
- Y. Chang, H. Lin, L. Li and H. Chen, Mater. Today Adv., 2020, 6, 100054 CrossRef.
- K. Chen, I. Balla, N. S. Luu and M. C. Hersam, ACS Energy Lett., 2017, 2, 2026–2034 CrossRef CAS.
- A. Rajkamal and R. Thapa, Adv. Mater. Technol., 2019, 4, 1900307 CrossRef CAS.
- J. Wang, H. Shi, W. Wang, Z. Xu, C. Hong, Y. Xue and F. Tian, Chem. Eng. J., 2022, 432, 133617 CrossRef CAS.
- Q. Fan, L. Yan, M. W. Tripp, O. Krejčí, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Liljeroth and J. M. Gottfried, Science, 2021, 372, 852–856 CrossRef CAS PubMed.
- T. Liu, Y. Jing and Y. Li, J. Phys. Chem. Lett., 2021, 12, 12230–12234 CrossRef CAS.
- T. Han, Y. Liu, X. Lv and F. Li, Phys. Chem. Chem. Phys., 2022, 24, 10712–10716 RSC.
- X. Wang, Z. Feng, J. Rong, Y. Zhang, Y. Zhong, J. Feng, X. Yu and Z. Zhan, Carbon, 2019, 142, 438–444 CrossRef CAS.
- P. Wu, P. Du, H. Zhang and C. Cai, J. Phys. Chem. C, 2012, 116, 20472–20479 CrossRef CAS.
- P. Wu, P. Du, H. Zhang and C. Cai, Phys. Chem. Chem. Phys., 2014, 16, 5640–5648 RSC.
- L. Hui, Y. Xue, Y. Liu and Y. Li, Small, 2021, 17, 2006136 CrossRef CAS PubMed.
- W. Wang, J. Meng, Y. Hu, J. Wang, Q. Li and J. Yang, J. Mater. Chem. A, 2022, 10, 9848–9857 RSC.
- X. Li, X. Pan, L. Yu, P. Ren, X. Wu, L. Sun, F. Jiao and X. Bao, Nat. Commun., 2014, 5, 3688 CrossRef CAS PubMed.
- A. Hashimoto, K. Suenaga, A. Gloter, K. Urita and S. Iijima, Nature, 2004, 430, 870–873 CrossRef CAS PubMed.
- V. H. Crespi, L. X. Benedict, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. phys., 1996, 53, R13303–R13305 CrossRef CAS.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Lia and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. phys., 1996, 54, 11169–11186 CrossRef.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- G. Brunetto, P. A. S. Autreto, L. D. Machado, B. I. Santos, R. P. B. Santos and D. S. Galvão, J. Phys. Chem. C, 2012, 116, 12810 CrossRef CAS.
- Hzwtech, Device Studio, Version 2021A, China, 2021, http://www.hzwtech.com Accessed on DS-PAW Search PubMed.
- Y. Ding and Y. Wang, J. Phys. Chem. C, 2013, 117, 18266–18278 CrossRef CAS.
- D. Wu, B. Yang, E. Ruckenstein and H. Chen, J. Phys. Chem. Lett., 2019, 10, 721–726 CrossRef CAS.
- D. Wu, B. Yang, H. Chen and E. Ruckenstein, Nanoscale, 2019, 11, 15472–15478 RSC.
- D. Wu, B. Yang, H. Chen and E. Ruckenstein, Energy Storage Mater., 2019, 16, 574–580 CrossRef.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- K. Toyoura, Y. Koyama, A. Kuwabara, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. phys., 2008, 78, 214303 Search PubMed.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 13921–13928 RSC.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. Shenoy, ACS Appl. Mater. Interfaces, 2013, 6, 11173–11179 CrossRef PubMed.
- Z. Wang, X. F. Zhou, X. Zhang, Q. Zhu, H. Dong, M. Zhao and A. R. Oganov, Nano Lett., 2015, 15, 6182–6186 CrossRef CAS PubMed.
- X. Cai, Q. Yang, S. Zheng and M. Wang, Energy Environ.Mater., 2021, 4, 458–464 CrossRef CAS.
- M. Winter, J. Besenhard, M. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- P. Ganesh, J. Kim, C. Park, M. Yoon, F. A. Reboredo and P. R. Kent, J. Chem. Theory Comput., 2014, 10, 5318–5323 CrossRef CAS.
- Y. Qie, J. Liu, S. Wang, Q. Sun and P. Jena, J. Mater. Chem. A, 2019, 7, 5733–5739 RSC.
- M. Mortazavi, C. Wang, J. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279–286 CrossRef CAS.
- D. Wang, Y. Liu, X. Meng, Y. Wei, Y. Zhao, Q. Pang and G. Chen, J. Mater. Chem. A, 2017, 5, 21370–21377 RSC.
- J. Zhou, L. Wang, M. Yang, J. Wu, F. Chen, W. Huang, N. Han, H. Ye, F. Zhao and Y. Li, Adv. Mater., 2017, 29, 1702061 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef.
- S. Huang, Y. Meng, Y. Cao, S. He, X. Li, S. Tong and M. Wu, Appl. Catal., B, 2019, 248, 239–248 CrossRef CAS.
- H. Wu, X. Li, R. Zhang and J. Yang, J. Mater. Chem. A, 2019, 7, 3752–3756 RSC.
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C. Sun, H. Wong, A. Guda, Y. Hao, X. Pan, K. Amine and Z. Luo, Adv. Energy Mater., 2019, 9, 1803689 CrossRef.
- Y. Ouyang, C. Ling, Q. Chen, Z. Wang, L. Shi and J. Wang, Chem. Mater., 2016, 28, 4390–4396 CrossRef CAS.
- C. Ling, L. Shi, Y. Ouyang and J. Wang, Chem. Mater., 2016, 28, 9026–9032 CrossRef CAS.
- Y. Ouyang, Q. Li, L. Shi, C. Ling and J. Wang, J. Mater. Chem. A, 2018, 6, 2289–2294 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- Z. Feng, Y. Tang, Y. Ma, Y. Li, Y. Dai, W. Chen, G. Su, Z. Song and X. Dai, Int. J. Hydrogen Energy, 2021, 46, 5378–5389 CrossRef CAS.
- R. Ku, G. Yu, J. Gao, X. Huang and W. Chen, Phys. Chem. Chem. Phys., 2020, 22, 3254–3263 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00955c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |