
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-anion high-entropy electrolytes enabling lithium-ion batteries with enhanced voltage tolerance and wide-temperature operability†
Jia-Zhen Zhao,
Fu-Da Yu*,
Hai-Nan Wang,
Ji-Huai Wu ,
Zhang Lan,
Yi-Ming Xie and
Lan-Fang Que*
,
Zhang Lan,
Yi-Ming Xie and
Lan-Fang Que*
Engineering Research Center of Environment-Friendly Functional Materials, Ministry of Education, Institute of Materials Physical Chemistry, Huaqiao University, Xiamen 361021, China. E-mail: yufuda@hqu.edu.cn; quelanfang@hqu.edu.cn
First published on 15th July 2025
Abstract
To achieve high-voltage stability and low-temperature performance simultaneously in lithium-ion batteries, a high-entropy electrolyte with multi anions is proposed. The multi anions not only lows the solvation-free energy, but also contributes to the formation of inorganic-rich CEI layer. The optimized electrolyte enables stable cycling of NCM523 at both 25 °C and −20 °C.
Introduction
Ether-based electrolytes have garnered significant attention for lithium-ion batteries (LIBs) due to their exceptional compatibility with lithium metal anodes and high ionic conductivity at low temperatures.1–3 However, their practical application in high-voltage systems remains constrained by insufficient oxidative stability, which accelerates electrolyte decomposition and interfacial degradation at the cathode.4–8 Concurrently, achieving stable low-temperature performance necessitates addressing the trade-off between ion transport kinetics and interfacial compatibility under extreme conditions.9–14 Conventional strategies, such as single-salt formulations15–17 or localized high-concentration designs,18–20 often fail to reconcile these dual challenges, highlighting the need for innovative electrolyte engineering.21–24Emerging from high-entropy alloy paradigms, entropy-mediated stabilization has recently been transposed to electrolyte design to resolve the intrinsic voltage limitations of ether solvents. Contemporary studies frequently assert that effective entropy modulation necessitates five or more distinct constituents to maximize configurational entropy (ΔSconf). Pioneering work by Zhang et al.25 realized an ultra-broad operational thermal window (−130 °C to 25 °C) via entropy-engineered solvent coordination, delivering 0.62 mS cm−1 at −60 °C, performance attributable to entropy-driven suppression of crystallization thermodynamics. This aligns confirming that ΔSconf elevation suppresses Gibbs free energy (ΔG), thereby enhancing interfacial chemical stability and ion diffusion coefficients. Cui et al.26 elucidated that high-entropy electrolytes with reduced ionic cluster dimensions achieve superior Li+ diffusivity relative to low-entropy systems. Within the HE-DIG framework, polyanionic involvement elevates ΔSconf. Prior work confirms that lithium salts in high-entropy electrolytes operate via collective cooperation rather than dominance by single components.27 Nevertheless, the entropy-driven electrolyte mechanism and entropy-interface coupling remain poorly understood. Furthermore, research on high-entropy systems at low-temperatures and high-voltage remains insufficient, necessitating further exploration.
This study proposes a high-entropy electrolyte strategy centered on multi-component lithium salts to enhance the high-voltage resilience and low-temperature retention of ether-based systems. By leveraging synergistic interactions among diverse anions and cations, the designed electrolyte aims to stabilize both electrode–electrolyte interfaces while maintaining favorable bulk physicochemical properties. Systematic comparisons between multi-salt and conventional single-salt electrolytes are conducted, encompassing analyses of physical characteristics, theoretical simulations, and electrochemical behaviors. The stability of lithium metal anodes and high-voltage cathodes is evaluated under room-temperature and sub-zero conditions. Demonstrably, the entropy-engineered electrolyte formulation inhibits oxidative degradation pathways, endowing high-voltage cathodes with exceptional cycling resilience across both ambient and cryogenic operating conditions. This work provides a generalized approach to overcoming the intrinsic limitations of ether-based electrolytes, advancing the development of high-energy LIBs operable under extreme conditions.
Results and discussion
The high-entropy electrolyte (1 M HE-DME) reported herein is composed of multiple lithium salts (including 0.4 M LiPF6, 0.15 M LiNO3, 0.15 M LiTFSI, 0.15 M LiFSI, and 0.15 M LiDFOB) dissolved in dimethoxyethane (DME), with 1.0 M LiTFSI in DME (1 M DME) selected as a comparative electrolyte. Three HE-DME electrolytes (specific electrolyte composition provided in ESI†) with total concentrations of 0.6 M, 0.75 M, and 1.0 M were prepared by supplementing LiPF6 at 0 M, 0.15 M, and 0.4 M respectively. NCM523 half-cells with these electrolytes exhibited similar initial capacities and cycling stability at 25 °C (Fig. S1†). At 3C, the 1 M HE-DME delivered 110.2 mA h g−1, exceeding the 0.75 M (91.5 mA h g−1) and 0.6 M (86.3 mA h g−1) formulations. Crucially, 1 M HE-DME maintained 91.4 mA h g−1 at 0.5C and −20 °C with 78.2% retention after 200 cycles, while other formulations showed reduced initial capacities and accelerated decay. This systematic comparison identifies 1 M HE-DME as optimal for further study.First, molecular dynamics (MD) simulations were performed to investigate the solvation structures and Li+ coordination environments with anions/solvent molecules in both electrolytes. Structural analyses in Fig. 1a, b and S2† reveal that the multi-anion configuration in 1 M HE-DME induces elongated Li–ODME coordination bonds and reduced solvent coordination numbers relative to conventional electrolytes, confirming weakened solvation characteristics. In addition, Fig. 1c shows that 1 M HE-DME system exhibits a broader HOMO–LUMO gap (ΔE = 7.48 eV), signifying enhanced chemical stability relative to 1 M DME. Consistent with this electronic structural advantage, linear sweep voltammetry (LSV) (Fig. 1d) demonstrates significantly elevated decomposition onset potentials and suppressed oxidative currents for 1 M HE-DME under high-voltage. Furthermore, 1 M HE-DME electrolyte demonstrates superior ionic conductivity at 25 °C and −20 °C (Fig. S3†), alongside improved low-temperature adaptability, compared to 1 M DME.
 | ||
| Fig. 1 (a) Snapshot acquired through MD simulations 1 M HE-DME. (b) The coordination number N(r) and RDF g(r) of Li–ODME of 1 M DME and 1 M HE-DME. (c) The LUMO and HOMO energy of 1 M DME and 1 M HE-DME. (d) The LSV curves of 1 M DME and 1 M HE-DME. | ||
The MD simulations demonstrates an extended Li–O bond length in HE-DME compared to 1 M DME, suggesting weaker Li+-solvent binding energy in HE-DME. To verify this hypothesis, experimental validation was conducted via custom thermodynamic analysis employing an electrochemical cell (Fig. 2a).28 This apparatus eliminates liquid junction potentials using a concentrated electrolyte bridge separating reference (ELref) and test (ELtest) compartments. The solvation free energy differential (ΔGsolv) is quantified by: ΔGsolv = −zFEcell, where Ecell represents the open-circuit voltage, F is the Faraday constant, and z is the charge number (z = 1 for Li+). Electrochemical analysis reveals an inverse proportionality between solvation strength and electron affinity for solvated Li+: attenuated coordination environments enhance electron acquisition capability, manifested through progressively negative Ecell values and correspondingly elevated ΔGsolv magnitudes. As revealed by the calculated results in Fig. 2b and c, the 1 M HE-DME electrolyte possesses a weaker solvation configuration, which facilitates lithium-ion desolvation processes. And this is consistent well with the activation energy (Ea) calculated by electrochemical impedance spectroscopy (EIS), wherein Ea in 1 M HE-DME (494.6 meV) is lower than that of 1 M DME (538.8 meV), indicating improved ion transport kinetics.
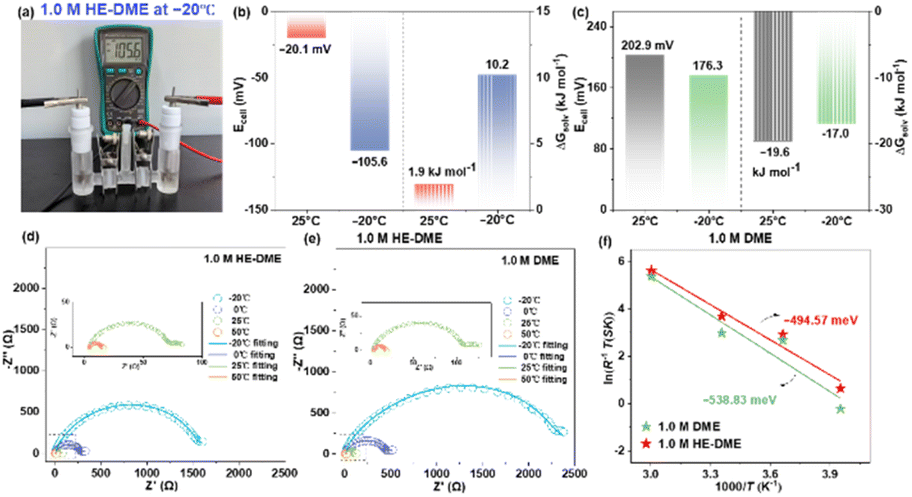 | ||
| Fig. 2 (a) The digital photo of the homemade experimental setup (1 M HE-DME as the test electrolyte). (b) The Ecell and ΔGsolv values of 1 M HE-DME at 25 °C and −20 °C. (c) The Ecell and ΔGsolv values of 1 M DME at 25 °C and −20 °C. (d and e) The separately collected EIS curves of the Li‖Li batteries based on 1 M HE-DME and 1 M DME at 50 °C, 25 °C, 0 °C and −20 °C. (f) The fitted activation energy (Ea) of 1 M HE-DME and 1 M DME. | ||
The electrochemical dynamic behaviour analysis of Li‖Li cells demonstrates the 1 M HE-DME electrolyte sustains superior cycling stability at 25 °C and −20 °C, suggesting effective anode passivation to suppress Li dendrite growth (Fig. S4†). In situ EIS analysis reveals Li‖Li cells with 1 M HE-DME maintain stable impedance during SOC cycling at 25 °C and −20 °C, contrasting with the erratic fluctuations of 1 M DME system at −20 °C due to dendrite-induced short-circuit (Fig. S5†). Electrochemical analysis identifies three characteristic relaxation processes: electrolyte resistance (Rb) at 10−5 s, SEI layer resistance (RSEI) at 10−4–10−3 s, and charge-transfer resistance (Rct) at 10−2–10−1 s as shown in Fig. S6 and S7.†29 The 1 M HE-DME system initially exhibits elevated RSEI values, with DRT deconvolution confirming this process as predominantly governing early-stage behavior. Subsequent rapid attenuation signifies stable SEI formation. Conversely, in 1 M DME, persistent RSEI dominance reflects performance degradation from dendritic propagation and parasitic reactions. The kinetic superiority of 1 M HE-DME is further manifested through comparative τ-indexed analysis, where time constants decay quantitatively corroborates accelerated interfacial charge transfer dynamics.
The electrochemical performance of LiNi0.52Co0.2Mn0.28O2 (NCM523) in the designed electrolyte is further evaluated. Fig. 3a shows that NCM523 in 1 M HE-DME electrolyte demonstrates superior rate capability at 25 °C than that of 1 M DEM. In addition, the 1 M HE-DME-based system delivers higher reversible capacity (157.3 mA h g−1) and better cycle stability (88.6% retention after 50 cycles at 0.5C) (Fig. 3b), contrasting sharply with 1 M DME counterpart (121.4 mA h g−1 and 45.5% retention). The 1 M HE-DME system maintains 150.76 mA h g−1 initial capacity and 74.8% retention after 100 cycles at 1C, outperforming the 1 M DME counterpart (Fig. S8†) (122.3 mA h g−1 and 32.9% retention). Evaluations further highlight performance disparities at −20 °C (Fig. 3c). The 1 M HE-DME system achieves 91.4 mA h g−1 initial capacity at 0.5C with 78.2% retention over 200 cycles (Fig. S9†), compared to 88.1 mA h g−1 and 35.2% retention after 50 cycles in 1 M DME system. At 1C, the 1 M HE-DME system sustains 70.5 mA h g−1 initial capacity with 80.0% retention after 350 cycles (Fig. 3d). Additionally, the 1 M HE-DME system demonstrates much higher and stable median voltage at both 25 and −20 °C (Fig. S10†).
 | ||
| Fig. 3 (a) The rate capability and (b) cycle performance at 0.5C of NCM523 in both electrolytes at 25 °C, (c) the first three charge and discharge curves at 0.1C and (d) cycle performance at 0.5C of NCM523 at both electrolytes at 0.1C and −20 °C. | ||
To elucidate the compositional distinctions of cathode electrolyte interphase (CEI) layers formed on NCM523 cathodes in both electrolytes, scanning electron microscopy (SEM) images and X-ray photoelectron spectroscopy (XPS) spectra of the cycled NCM523 after 50 cycles at 25 °C and −20 °C are collected. Elemental mapping analysis reveals reduced carbon content on the NCM523 surface based on 1 M HE-DME compared to 1 M DME (Fig. 4a). In addition, the 1 M HE-DME system reveals a more concentrated fluorine distribution on the CEI layer alongside enhanced anion diversity (Fig. S11 and S12†). Distribution analysis further indicates that fluorine species in the 1 M DME system primarily originate from parasitic degradation of lithium salts during electrolyte decomposition. While the 1 M HE-DME is concentrated on the surface of the NCM523 particles. Besides, a high content of B is observed on the surface of NCM523 cycled in HE-DME due to the reduction of LiDFOB. As shown in Fig. 4b, the DME solvent exhibits a relatively high LUMO energy level, indicating favorable reduction stability. However, its lower HOMO energy level suggests limited oxidation stability. Notably, LiDFOB and LiNO3 exhibit HOMO levels comparable to DME, potentially mitigating solvent decomposition through electron transfer regulation. LiFSI, LiTFSI, and LiPF6 display wider electrochemical stability windows, balancing decomposition resistance with sufficient ionic conduction.
 | ||
| Fig. 4 (a) The SEM mapping of NCM523 cathodes in 1 M HE-DME and 1 M DME electrolytes and the relative content of C, O, Mn elements. (b) The LUMO and HOMO energy of LiPF6, LiTFSI, LiFSI, LiNO3, LiDFOB, and DME based on MD simulations. (c) The C 1s, (d) Mn 2p, and (e) Ni 2p spectra of NCM523 in 1 M HE-DME and 1 M DME electrolytes. | ||
XPS deconvolution of the C 1s spectrum (Fig. 4c) confirms a lower proportion of organic components (C–O bonds) in the 1 M HE-DME system, while the O 1s spectrum (Fig. S13†) also demonstrates a lower proportion of organic oxygen relative to lattice oxygen. Crucially, Mn 2p and Ni 2p spectra (Fig. 4d and e) disclose elevated concentrations of high-valence Mn4+ (44.6%) and Ni3+ (31.2%) in the 1 M HE-DME system versus 39.8% Mn4+ and 23.2% Ni3+ in 1 M DME. Electrochemical stabilization of multi-valent states effectively circumvents chemo-mechanical failure by inhibiting transition metal leaching. These results conclusively establish that 1 M HE-DME promotes inorganic-dominated CEI, ensuring prolonged structural fidelity of NCM523 cathodes through entropy-optimized interfacial engineering.
The coin-type full cells incorporating NCM523 cathodes and graphite anodes with 1 M HE-DME electrolyte delivered a discharge capacity of 91.6 mA h g−1 at 25 °C with 73.9% capacity retention after 80 cycles (Fig. S14†). While at −20 °C, the system achieved 65.86 mA h g−1 initial capacity while maintaining 78.8% capacity retention through 250 cycles, demonstrating superior low-temperature endurance and cycling stability. Additionally, extended high-voltage assessments in half-cell configurations are shown detailed in Fig. S15.† The charge/discharge curves reveal an activation discharge capacity of 186.8 mA h g−1 for the NCM523 half-cell at 0.1C. Subsequent cycling at 0.5C demonstrates an initial discharge capacity of 160.0 mA h g−1. Crucially, the absence of decomposition currents or voltage anomalies between 4.3 and 4.5 V confirms exceptional oxidative stability, validating the operational integrity of the 1 M HE-DME electrolyte under elevated voltage conditions.
Conclusions
In summary, entropy-driven and multi-anion electrolyte 1 M HE-DME was developed to simultaneously enhance electrochemical stability under high-voltage conditions and boost low-temperature capacity retention and cycling durability. MD simulations reveal the 1 M HE-DME electrolyte exhibits a distinct solvation structure versus single-salt electrolyte 1 M DME, marked by enhanced oxidation stability and weakened Li–ODME interactions. Furthermore, the multi-anion coordination chemistry promotes the formation of an inorganically enriched CEI on NCM523 cathode. These synergistic effects enable NCM523 half-cells with 1 M HE-DME electrolyte to deliver 70.5 mA h g−1 initial capacity, maintaining 80.0% capacity retention after 350 cycles at −20 °C and 1C. The full-cell system achieved 65.86 mA h g−1 initial capacity while maintaining 78.8% capacity retention through 250 cycles at −20 °C and 0.5C, demonstrating superior low-temperature endurance and cycling stability.Data availability
All data supporting the findings of this study are available within the paper and its ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 52372191), and the Natural Science Foundation of Xiamen, China (Grant No. 3502Z202372036), the Natural Science Foundation of Fujian Province (2023J05047), and the Fundamental Research Funds for the Central Universities (Grant No. ZQN-1203). We acknowledge the support of the High-Performance Computing Center (HPCC) at Harbin Institute of Technology on first-principles calculations. We extend their gratitude to Ms. Xiao-Xia Xu from Shiyanjia Lab (http://https:\www.shiyanjia.com) for providing invaluable assistance with the XPS analysis.References
- S. Wang, J. Shi, Z. Liu and Y. Xia, Adv. Energy Mater., 2024, 14, 2401526 CrossRef CAS.
- T. Doi, M. Hashinokuchi and M. Inaba, Curr. Opin. Electrochem., 2018, 9, 49–55 CrossRef CAS.
- X. Ding, Q. Zhou, X. Li and X. Xiong, Chem. Commun., 2024, 60, 2472–2488 RSC.
- S. Dai, W. Fang, T. Wang, Y. Gao, T. Zhang, Z. Qin, G. Chen and X. Zhou, Chem. Eng. J., 2024, 500, 157269 CrossRef CAS.
- J. Liu, B. Li, J. Cao, X. Xing and G. Cui, J. Energy Chem., 2024, 91, 73–98 CrossRef CAS.
- D. Ouyang, K. Wang, J. Guan and Z. Wang, J. Power Sources, 2024, 615, 235095 CrossRef CAS.
- A. M. Li, O. Borodin, T. P. Pollard, W. Zhang, N. Zhang, S. Tan, F. Chen, C. Jayawardana, B. L. Lucht, E. Hu, X. Q. Yang and C. Wang, Nat. Chem., 2024, 16, 922–929 CrossRef CAS PubMed.
- Z. Wang, X. Che, D. Wang, Y. Wang, X. He, Y. Zhu and B. Zhang, Angew Chem. Int. Ed. Engl., 2024, 63, e202404109 CrossRef CAS PubMed.
- D. Hubble, D. E. Brown, Y. Zhao, C. Fang, J. Lau, B. D. McCloskey and G. Liu, Energy Environ. Sci., 2022, 15, 550–578 RSC.
- T. L. Kulova and A. M. Skundin, Int. J. Electrochem. Sci., 2020, 15, 8638–8661 CrossRef.
- Q. Li, G. Liu, H. Cheng, Q. Sun, J. Zhang and J. Ming, Chem.–Eur. J., 2021, 27, 15842–15865 CrossRef CAS PubMed.
- P. Mei, Y. Zhang and W. Zhang, Nanoscale, 2023, 15, 987–997 RSC.
- J. A. Mennel and D. Chidambaram, Frontiers in Energy, 2022, 17, 43–71 CrossRef.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- M. Mominur Rahman and E. Hu, Angew. Chem., 2023, 62, e202311051 CrossRef CAS PubMed.
- D. M. Seo, P. D. Boyle, R. D. Sommer, J. S. Daubert, O. Borodin and W. A. Henderson, J. Phys. Chem. B, 2014, 118, 13601–13608 CrossRef CAS PubMed.
- X. Wang, W. Feng, Z. Zhou and H. Zhang, Chem. Commun., 2024, 60, 11434–11449 RSC.
- K. Matsumoto, K. Inoue, K. Nakahara, R. Yuge, T. Noguchi and K. Utsugi, J. Power Sources, 2013, 231, 234–238 CrossRef CAS.
- X. Chen and D. G. Kuroda, Chem. Commun., 2023, 59, 1849–1852 RSC.
- Z. Yu, J. Zhang, T. Liu, B. Tang, X. Yang, X. Zhou and G. Cui, Acta Chim. Sin., 2020, 78, 114–124 CrossRef CAS.
- S. Mao, Q. Wu, F. Ma, Y. Zhao, T. Wu and Y. Lu, Chem. Commun., 2021, 57, 840–858 RSC.
- D. Wang, Z. Zhang, B. Hong and Y. Lai, Chem. Commun., 2019, 55, 10737–10739 RSC.
- X. Xu, X. Han, L. Lu, F. Wang, M. Yang, X. Liu, Y. Wu, S. Tang, Y. Hou, J. Hou, C. Yu and M. Ouyang, J. Power Sources, 2024, 603, 234445 CrossRef CAS.
- J.-W. Zhang, J.-L. Sun, D.-N. Zhao, Y.-J. Zhao, X.-Y. Hu, Y.-N. Wang, Y.-J. Yao, N.-S. Zhang, L.-J. Zhang, C.-L. Li, P. Wang, S.-Y. Li and X.-L. Cui, Energy Storage Mater., 2024, 72, 103698 CrossRef.
- W. Zhang, H. Xia, Z. Zhu, Z. Lv, S. Cao, J. Wei, Y. Luo, Y. Xiao, L. Liu and X. Chen, CCS Chem., 2021, 3, 1245–1255 CrossRef CAS.
- S. C. Kim, J. Wang, R. Xu, P. Zhang, Y. Chen, Z. Huang, Y. Yang, Z. Yu, S. T. Oyakhire, W. Zhang, L. C. Greenburg, M. S. Kim, D. T. Boyle, P. Sayavong, Y. Ye, J. Qin, Z. Bao and Y. Cui, Nat. Energy, 2023, 8, 814–826 CrossRef CAS.
- K. F. Ren, H. Liu, J. X. Guo, X. Sun, F. Jiang, C. Guo, W. Z. Bao, F. Yu, G. Kalimuldina, L. Kong, X. B. Cheng and J. F. Li, ACS Energy Lett., 2024, 9, 2960–2980 CrossRef CAS.
- S. C. Kim, X. Kong, R. A. Vila, W. Huang, Y. Chen, D. T. Boyle, Z. Yu, H. Wang, Z. Bao, J. Qin and Y. Cui, J. Am. Chem. Soc., 2021, 143, 10301–10308 CrossRef CAS PubMed.
- X. Chen, L. Li, M. Liu, T. Huang and A. Yu, J. Power Sources, 2021, 496, 229867 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02919h |
| This journal is © The Royal Society of Chemistry 2025 |