
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient chemical synthesis of mirror-image DNA polymerase Dpo4 assisted by one-pot multi-segment condensation†
Miao
Wang‡
a,
Tingting
Cui‡
c,
Shuqing
Huang
a,
Dongyang
Han
b,
Xiangyu
Deng
b,
Yanbo
Liu
 b and
Chaowei
Shi
*a
b and
Chaowei
Shi
*a
aHefei National Research Center for Interdisciplinary Sciences at the Microscale, University of Science and Technology of China, 230027 Hefei, China. E-mail: scwei@ustc.edu.cn
bDepartment of Chemistry, Tsinghua University, Beijing 100084, China
cSchool of Food and Biological Engineering, Engineering Research Center of Bio-process, Ministry of Education, Hefei University of Technology, 230009 Hefei, China
First published on 7th March 2025
Abstract
The mirror-image DNA polymerase Dpo4 is a valuable enzymatic tool in biomedical research; however, its chemical synthesis has been hindered by low efficiency. Here, we describe an efficient chemical synthesis of D-Dpo4 using a one-pot multi-segment condensation strategy, which was achieved using Fmoc-masked peptide thioester. By minimizing the need for isolating and handling intermediates through one-pot three- and four-segment ligations, we were able to obtain D-Dpo4 with higher efficiency, achieving an improved overall yield of 9.8%, representing a twofold improvement from previous work. Our work provides a practical and streamlined approach for the synthesis of mirror-image proteins with larger sizes and underscores the utility of one-pot multi-segment condensation in chemical protein synthesis.
Introduction
Mirror-image proteins (D-proteins), composed entirely of D-amino acids and achiral glycine, have emerged as valuable tools in biomedical research due to their unique biochemical properties.1–3 These unique biochemical properties make mirror-image proteins useful biological tools with applications in drug discovery, protein crystallography, and the development of mirror-image biochemical systems.4–7 The original studies on mirror-image proteins began with Kent et al.8 who pioneered the chemical synthesis of mirror image enzymes to develop and explore biochemical systems based on biomolecules of opposite chirality. Unlike natural L-proteins, these chirally inverted D-enantiomers can only be obtained by chemical synthesis.3,8–21 While recent advances in chemical protein synthesis have enabled the routine production of moderately sized D-proteins (typically 100–200 residues),4,10,21–39 the preparation of larger D-proteins (e.g., those containing more than 300 amino acids) still often suffers from low efficiency.40,41 A representative example is the 352-residue D-enantiomer of Sulfolobus solfataricus P2 DNA polymerase IV (D-Dpo4),42 which can be used as a tool to develop therapeutically promising nuclease-resistant L-nucleic acid aptamers through mirror-image systematic evolution of ligand by exponential enrichment (miSELEX) technology.43 In a recent study, D-Dpo4 was assembled from nine peptide segments, requiring more than 15 steps of intermediate purification during its preparation.44 These tedious and time-consuming purification procedures heavily complicated the synthetic workflows and compromised the overall efficiency (with a low yield of less than 5%), limiting the practical accessibility of this molecular tool.In this context, here we report the study on the use of a one-pot multi-segment condensation strategy to facilitate the chemical synthesis of D-Dpo4. Through the strategic implementation of one-pot three-segment and one-pot four-segment ligations to minimize the need for isolation and handling of intermediates, D-Dpo4 was obtained with an improved overall yield of 9.8%, which is 2-fold higher than that of the previous study.44 Our work provides a practical synthetic route for obtaining a useful mirror-image protein tool for biomedical research and underlines the utility of one-pot multi-segment condensation in streamlining the chemical synthesis of D proteins with larger sizes.
Results and discussion
The D-Dpo4 protein (352 amino acids) with an N-terminal His6-tag was strategically divided into nine peptide segments (segments 1–9), each spanning 20 to 50 residues (Fig. 1A). To enable native chemical ligation (NCL), seven cysteine residues (42, 123, 155, 207, 229, 268, and 313) were introduced as temporary ligation sites (which were later reverted to alanine after ligation), with a subsequent plan to revert these positions to alanine post ligation.45–48 A His6 tag was incorporated at the N-terminus of D-Dpo4 to facilitate final protein purification (Fig. 1A).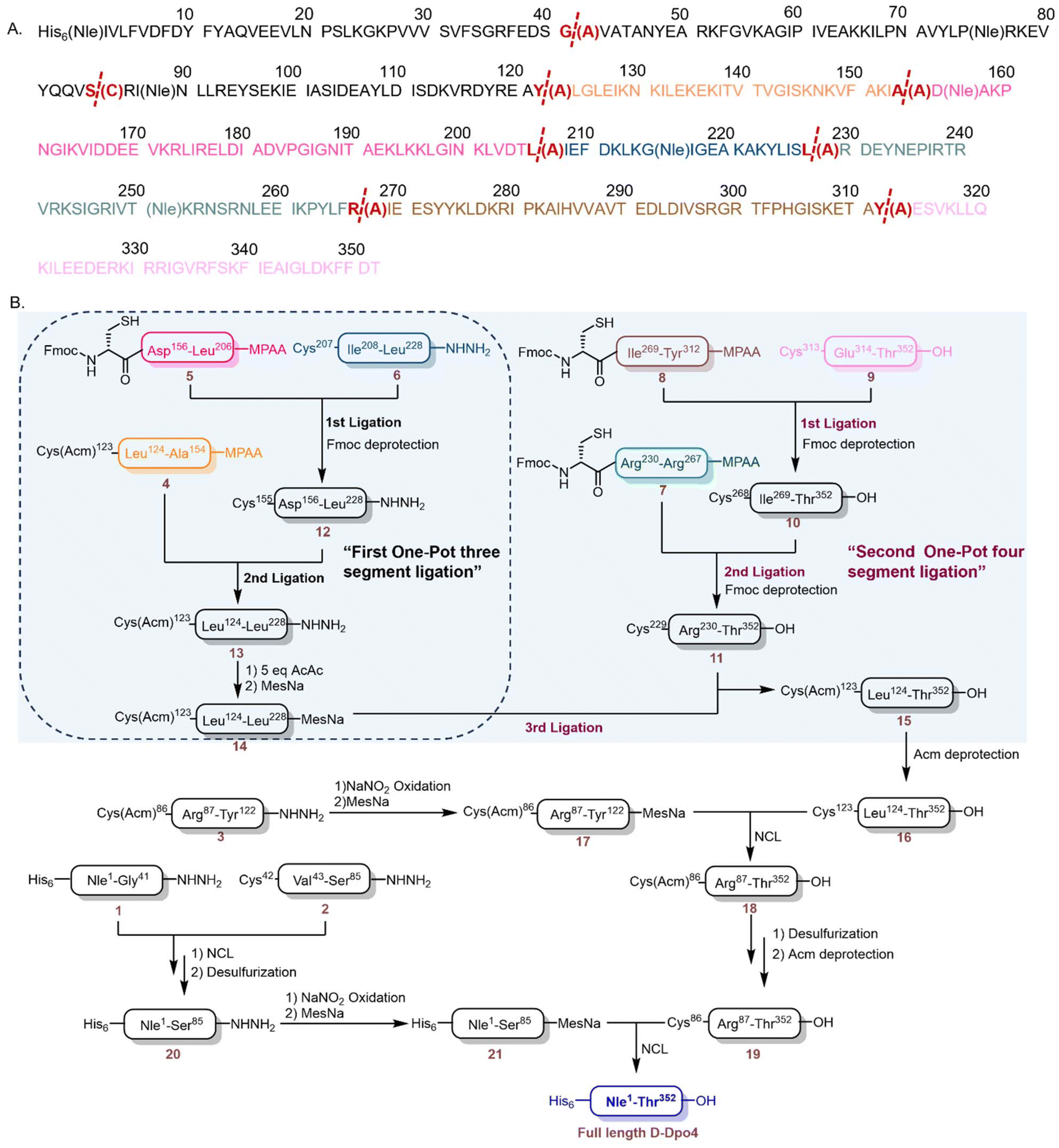 | ||
| Fig. 1 (A) The amino acid sequence of His6-D-Dpo4. The full-length protein is divided into nine segments and ligation sites are highlighted in bold. (B) Synthetic route of His6-D-Dpo4. | ||
We synthesized all segments (1–9) using standard Fmoc (9-fluorenylmethyloxycarbonyl)-based solid-phase peptide synthesis (SPPS), purified by reversed-phase high-performance liquid chromatography (RP-HPLC) and verified by electrospray ionization mass spectrometry (ESI-MS) analysis (Fig. 2). To facilitate the one-pot C-to-N sequential ligation strategy, segments 4, 5, 7, and 8 were designed with C-terminal aryl thioester precursors to facilitate one pot ligation.49,50 The N-terminal cysteine of segment 5 was protected with the Fmoc group for preventing cyclization,51 while segments 7 and 8 similarly carried Fmoc-protected N-terminal cysteines to enable sequential ligation with segment 9. Additionally, orthogonal protection was implemented for Cys86 and Cys123 using acetamidomethyl (Acm) groups,52 ensuring compatibility with the Fmoc-based ligation strategy (Fig. 1B).
 | ||
| Fig. 2 Analytical RP-HPLC chromatogram (λ = 214 nm) and ESI-MS spectra of segments 1–9. (A) Segment 1. (B) Segment 2. (C) Segment 3. (D) Segment 4. (E) Segment 5. (F) Segment 6. (G) Segment 7, “*” corresponds to the trifluoroacetate of 7. (H) Segment 8. (I) Segment 9. | ||
We have successfully achieved the efficient synthesis of the 105-amino-acid fragment 14 by adopting a one-pot ligation–Fmoc deprotection–thioester transfer strategy (Fig. 3A). Initially, three functional modules were meticulously prepared through Fmoc-SPPS: a 32-amino-acid (AA) fragment 4 (Cys (Acm)123-Ala154-COSC6H4CH2COOH) with a C-terminal thioester precursor, a 51-AA thioesterified fragment 5 (Fmoc-Cys155-Leu206-COSC6H4CH2COOH) with the N-terminal Fmoc protection intact, and a 22-AA fragment 6. Considering the relatively hydrophobic nature of its sequence, Acm was selected as the cysteine protecting group in fragment 4 to minimize its impact on solubility. Notably, the N-terminal Fmoc group on Cys155 of fragment 5 was retained as a removable protecting group for the terminal cysteine, which effectively suppressed the thioester-induced intramolecular cyclization of fragment 5. Subsequently, an acetylacetone-mediated activation method was employed to directly transform the crude peptides of fragments 4 and 5 into MPAA thioesters (COSC6H4CH2COOH).49 This approach remarkably reduced the separation and purification steps in precursor preparation, thus facilitating a more streamlined one-pot ligation process.
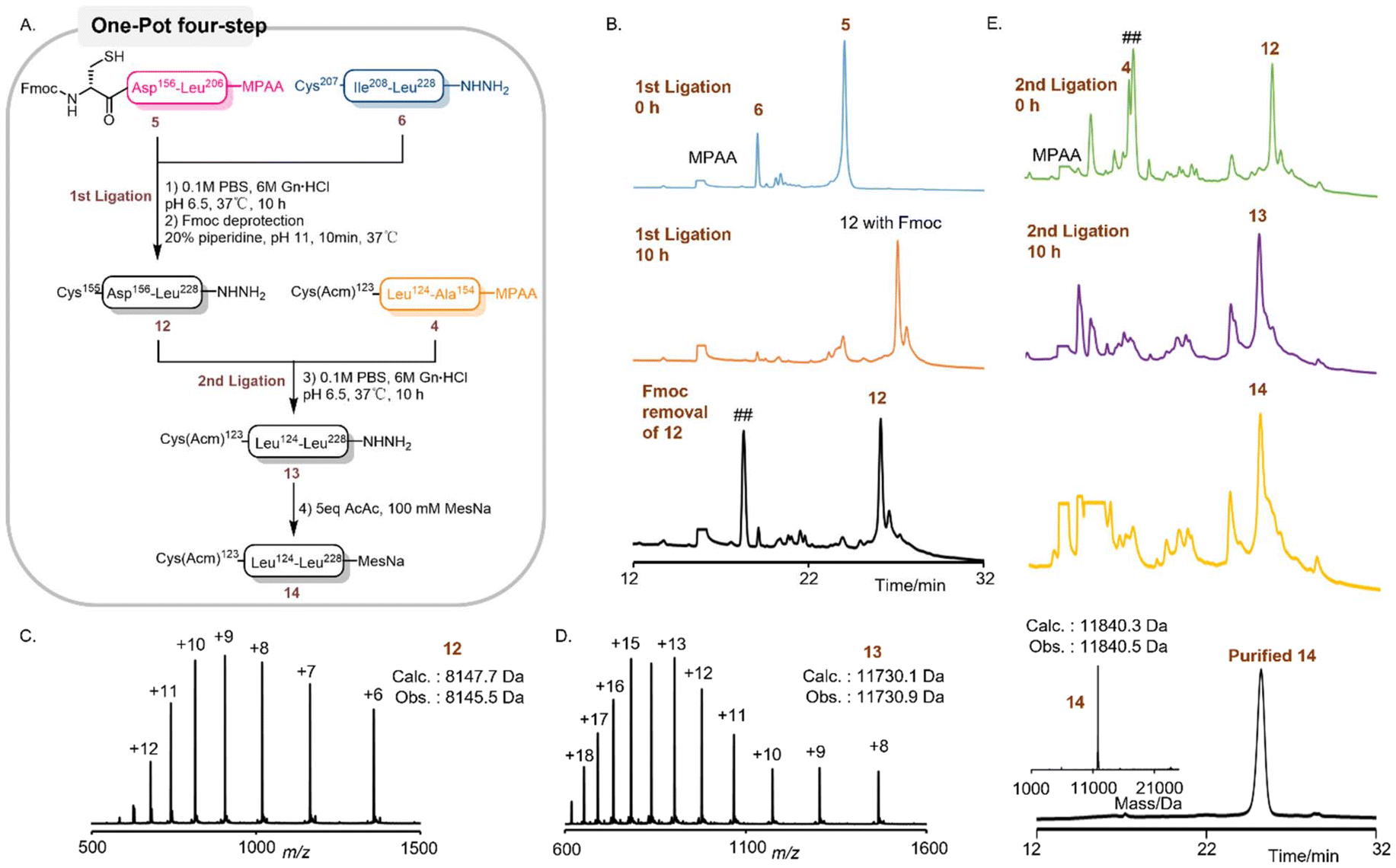 | ||
| Fig. 3 One-pot three segment ligation of segment 14. (A) Synthetic route of segment 14. (B) Analytical RP-HPLC trace (λ = 214 nm) of segment 12. (C) ESI-MS spectrum of segment 12. (D) ESI-MS spectrum of segment 13. (E) Analytical RP-HPLC trace (λ = 214 nm) of segment 14 and the ESI-MS deconvoluted spectrum of segment 14. “##” corresponds to the dibenzofulvene–piperidine adduct. | ||
With all three peptides in hand, we commenced the process by conducting native chemical ligation (NCL) between fragment 5 (1.0 equiv.) and fragment 6 (1.0 equiv.) (Fig. 3B). The ligation reaction was carried out in a ligation buffer solution with a pH of 6.9, containing 100 mM MPAA. The reaction progress was monitored by RP-HPLC. After 10 hours, the ligation reaction reached completion. Immediately following the NCL reaction, without any intermediate separation steps, 20% (by volume) of piperidine was directly added to the ligation system. This addition enabled the efficient removal of the Fmoc group at the N-terminus of the ligation product by adjusting the pH of the system to approximately 11. The reaction progress was continuously monitored by RP-HPLC, and within 10 minutes, all Fmoc groups were completely removed, yielding fragment 12 (Fig. 3B and C). Subsequently, the pH of the system was precisely adjusted back to 6.9. This step protonated the piperidine in the system, ensuring that it would not interfere with the subsequent second NCL reaction. Then, fragment 4 was added to the reaction system, and the reaction progress was continuously monitored using RP-HPLC. After another 10 hours, the reaction concluded, resulting in the formation of the ligation product 13 (Fig. 3D). After this reaction, without any purification procedures, the pH of the system was adjusted to the range of 2–3, and 5 equivalents of acetylacetone(acac) were added. The reaction mixture was then incubated at 37 °C for 1 hour. Subsequently, 100 mM MesNa was introduced, and the pH was adjusted to 5.0. The reaction was maintained at 37 °C for an additional hour. Under these conditions, the majority of the peptide hydrazide 13 was successfully converted into product 14 (Fig. 3E).
The overall isolated yield of this four-step one-pot process, involving three-fragment ligation, Fmoc-deprotection, a second ligation, and thioesterification, was 18.1%. This innovative scheme effectively avoided the use of custom-made, non-commercial TFA–Thz protecting groups, which were utilized in previous methodologies. Moreover, it streamlined the previously cumbersome synthetic route. The original route, which involved 8 reaction steps, 3 HPLC purification steps, and 3 lyophilization treatments, was simplified to just four reaction steps, along with only 1 HPLC purification and 1 lyophilization.44 This substantial simplification of the synthetic process led to a remarkable reduction in the total time required for fragment acquisition. It took merely two days to obtain fragment 14, which was three to four times faster than the original synthetic route, vividly demonstrating the distinct superiority of the one-pot method.
Inspired by the outcome achieved with segment 14, we further explored the one-pot strategy for the synthesis of compound 15 (Fig. 4A). The sequence of 15 was strategically partitioned into 4 segments, as depicted in Fig. 1: 7 (Cys229-Arg267, 38AA), 8 (Cys268-Tyr312, 44AA), 9 (Cys313-Thr352, 39AA), and 14 (Cys123-Leu228, 105AA). These segments were successively ligated to generate the target polypeptide. Both segments 7 and 8, prepared as MPAA thioesters, were synthesized through the acetylacetone-mediated activation of peptidyl hydrazides,49 which were derived from the crude peptides following Fmoc SPPS and cleavage. Segment 14, a MesNa thioester, was obtained via a previously described one-pot ligation process (Fig. 3). The synthesis was initiated by the ligation of peptides 8 (1.0 equiv.) and 9 (1.0 equiv.) to yield peptide 10 (Fig. 4B). The reaction was conducted in pH 6.9 buffer containing 100 mM MPAA, and its progress was monitored by RP-HPLC. After 10 hours, the ligation reaction reached completion, and then the in situ removal of the Fmoc group at the N-terminus was carried out in a one-pot manner. The Fmoc group was smoothly removed within 10 minutes by adding 20% (by volume) piperidine by adjusting the final pH to approximately 11, and the reaction progress was monitored by RP-HPLC. Subsequently, the pH of the reaction mixture was adjusted to 6.9 for the next ligation step. Without any purification, peptide 7 (1.0 equiv.) was directly added for ligation with 10 to obtain 11 in a one-pot reaction (Fig. 4C). The subsequent Fmoc deprotection and pH adjustment were carried out following the same procedure as that for fragment 10. Finally, 14 (1.3 equiv.) was added for the final ligation under a pH of 6.9, affording polypeptide 15 with an isolated yield of 41.7% (Fig. 4D).
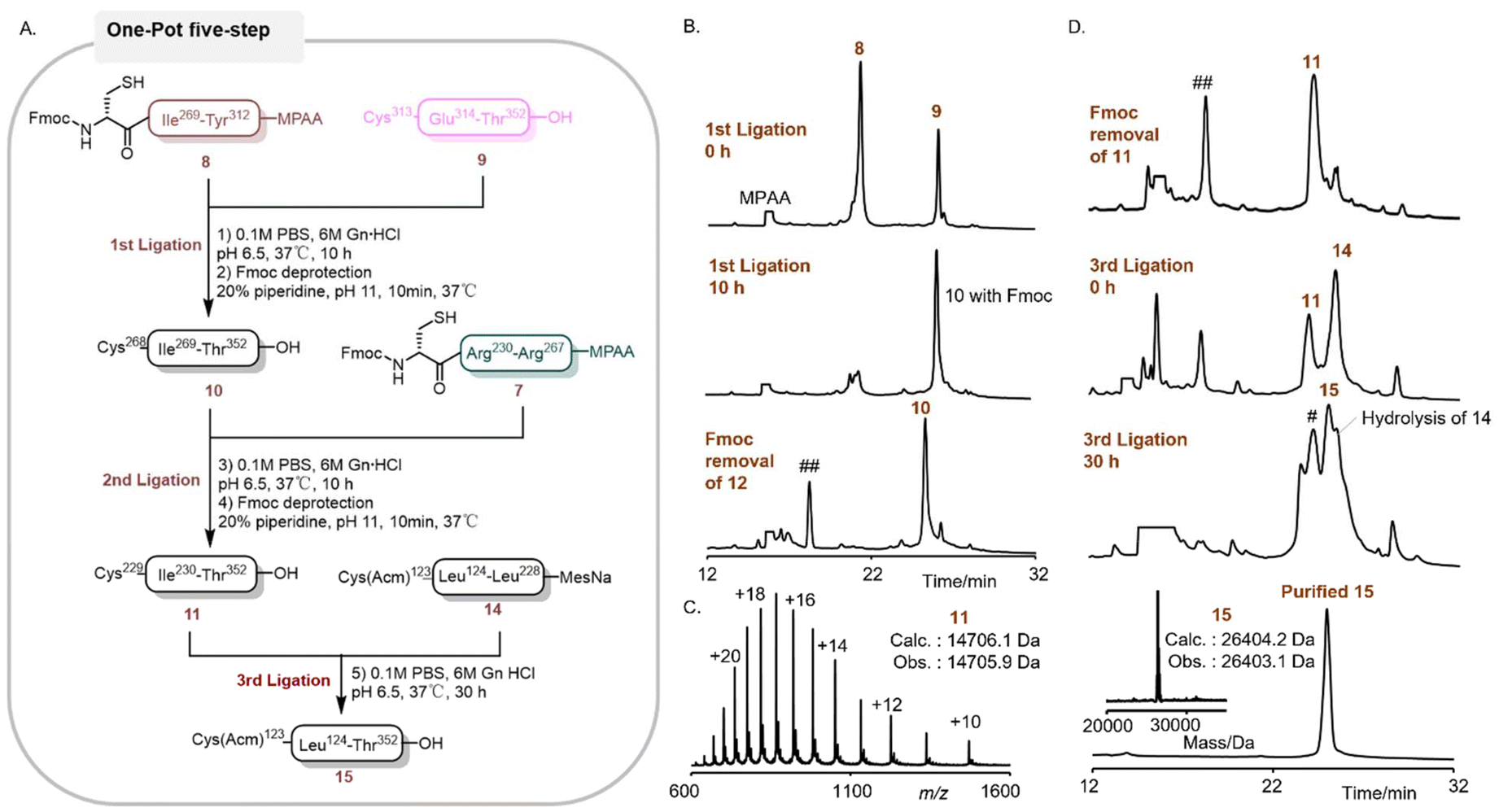 | ||
| Fig. 4 One-pot three segment ligation of segment 15. (A) Synthetic route of segment 15. (B) Analytical RP-HPLC trace (λ = 214 nm) of segment 10. (C) ESI-MS spectrum of segment 11. (D) Analytical RP-HPLC trace (λ = 214 nm) of segment 15 and the ESI-MS deconvoluted spectrum of segment 15. “#” corresponds to the excess of 14 ligated with 9. “##” corresponds to the dibenzofulvene–piperidine adduct. | ||
During this one-pot process, the formation of the main by-product was ascribed to the reaction of 9 with an excess of 14. Notably, the overall yield (41.7% in one-pot) was significantly enhanced compared to the reported multi-step synthetic route (18.9%). Furthermore, the purification and lyophilization steps has been substantially reduced from six times to two times, thereby greatly improving the overall synthetic efficiency of D-Dpo4.
Hydrazide-based native chemical ligation (NCL) between 1 (1.1 equiv.) and 2 (1 equiv.) was carried out at pH 6.6 for 12 hours. Subsequently, desulfurization was performed to yield 20 (Fig. S2 and S3†). After activation with NaNO2 at pH 3, a clean conversion of the hydrazide 20 to the MesNa thioester 21 was detected (Fig. S4†). Similarly, 3 was transformed into 17 through the same activation and thiolysis procedure (Fig. S1†). Then, 17 was ligated with 16 at pH 6.5 for 16 hours, smoothly and cleanly forming 18 with an isolated yield of 78% (Fig. S6†). Subsequently, free-radical-based desulfurization of 18 was conducted using the VA-044-based method for 14 hours. This process successfully converted all six ligation-site Cys residues back to Ala with an isolated yield of 79% (Fig. S7†). Thereafter, the Acm protecting group on Cys86 was removed by PdCl2 with an isolated yield of 75% (Fig. S8†).
Finally, the purified 19 (1.0 equiv.) was ligated with 21 (2.0 equiv.) at pH 6.5 for 18 hours, affording the full-length product D-Dpo4 with an isolated yield of 70% (Fig. S9†). The final product was characterized by analytical RP-HPLC and ESI-MS (Fig. 5A). Then the D-protein was folded by successive dialysis against a series of renaturation buffer solutions containing 4 M, 2 M, 1 M, 0.5 M, 0.25 M, and 0 M Gn·HCl, respectively. After dialysis, the folded D-Dpo4 was heated and purified using Ni-NTA. Additionally, the synthetic D-Dpo4 was characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), which showed a single band at the expected molecular weight of approximately 40 kDa, identical to that of recombinant L-Dpo4 (Fig. 5B).
 | ||
| Fig. 5 Chemical synthesis and characterization of D-Dpo4. (A) Analytical RP-HPLC chromatogram (λ = 214 nm) and ESI-MS deconvoluted spectra of D-Dpo4. (B) SDS-PAGE analysis of synthetic D-Dpo4 and recombinant L-Dpo4, stained with Coomassie Brilliant Blue. M, protein maker. (C) PCR and miPCR amplification of a 128 bp D/L-DNA sequence using L/D-Dpo4, analyzed by 3% agarose gel electrophoresis and stained with 4S Gelred. M, DNA maker. (D) The products of miPCR and PCR were digested with DNase I and analysed by 3% agarose gel electrophoresis. M, DNA maker. | ||
With the synthetic mirror-image Dpo4 obtained, we utilized the miPCR system to test its enzyme activity. The miPCR reactions were carried out in 25 μL reaction systems containing 25 mM HEPES (pH 7.5), 5 mM MgCl2, 50 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 10% glycerol, 3% DMSO, 0.1 mg/mL BSA, 160 μM (each) L-dNTPs, 0.2 μM (each) L-primers, 60 nM L-ssDNA template, and ∼1 μM D-Dpo4 polymerase. Considering that the mirror-image DNA polymerization was less efficient than the corresponding natural system, the PCR reaction was carried out in two phases of 15 cycles each, with the addition of fresh enzyme between the phases. The PCR program settings were as follows: 86 °C for 30 s (initial denaturation); 86 °C for 15 s (denaturation), 54 °C for 15 s (annealing), and 65 °C for 2 min (extension) for 30 cycles; 65 °C for 5 min (final extension). The miPCR products exhibited a clear band in the agarose gel with the expected length of 128 bp, and the intensity of the band increased with the number of cycles (Fig. 5C). To test the chirality of the miPCR product, we used an endonuclease (DNase I) to digest the products of the miPCR and PCR. Both the products of miPCR and PCR were digested with 5 U DNase I at 37 °C for 30 min and then analyzed by 3% agarose gel electrophoresis. In this case, the products of miPCR were completely resistant to digestion by the endonuclease of natural chirality (Fig. 5D). It shows promising potential for generating L-nucleic acid aptamers for research and therapeutic purposes.
Conclusions
In summary, we reported the efficient total chemical synthesis of the 352-residue D-Dpo4 with 6 His tag using a one-pot multi-segment condensation strategy. To simplify the intermediate purification, peptides 4, 5 and 6 were assembled into 14 by one-pot three-segment ligation, and peptides 7, 8, 9 and 14 were then assembled into 15 by one-pot four-segment ligation. In this way, D-Dpo4 can be obtained more efficiently with an overall yield of 9.8%. Our work provides a practical means to chemically synthesize mirror-image proteins with larger sizes.Author contributions
Miao Wang: most part of synthesis, original draft, data analysis, investigation. Tingting Cui: data analysis, manuscript review, investigation. Shuqing Huang: writing – review & editing. Dongyang Han: writing – review & editing. Xiangyu Deng: writing – review & editing. Yanbo Liu: writing – review & editing. Chaowei Shi: resources, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFA0909400) and the National Natural Science Foundation of China (22422411).References
- L. Zhao and W. Lu, Curr. Opin. Chem. Biol., 2014, 22, 56–61 CrossRef CAS.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS.
- K. Harrison, A. S. Mackay, L. Kambanis, J. W. C. Maxwell and R. J. Payne, Nat. Rev. Chem., 2023, 7, 383–404 CrossRef CAS PubMed.
- X. Zhou, C. Zuo, W. Li, W. Shi, X. Zhou, H. Wang, S. Chen, J. Du, G. Chen, W. Zhai, W. Zhao, Y. Wu, Y. Qi, L. Liu and Y. Gao, Angew. Chem., Int. Ed., 2020, 59, 15114–15118 CrossRef CAS PubMed.
- A. J. Callahan, S. Gandhesiri, T. L. Travaline, R. M. Reja, L. L. Salazar, S. Hanna, Y. C. Lee, K. Li, O. S. Tokareva, J. M. Swiecicki, A. Loas, G. L. Verdine, J. H. McGee and B. L. Pentelute, Nat. Commun., 2024, 15, 1813 CrossRef CAS PubMed.
- C. Zuo, W. W. Shi, X. X. Chen, M. Glatz, B. Riedl, I. Flamme, E. Pook, J. W. Wang, G. M. Fang, D. Bierer and L. Liu, Sci. China: Chem., 2019, 62, 1371–1378 CrossRef CAS.
- Y.-K. Qi, J.-S. Zheng and L. Liu, Chem, 2024, 10, 2390–2407 CAS.
- R. C. Milton, S. C. Milton and S. B. Kent, Science, 1992, 256, 1445–1448 CrossRef CAS PubMed.
- M. T. Weinstock, M. T. Jacobsen and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11679–11684 CrossRef CAS.
- A. M. Levinson, J. H. McGee, A. G. Roberts, G. S. Creech, T. Wang, M. T. Peterson, R. C. Hendrickson, G. L. Verdine and S. J. Danishefsky, J. Am. Chem. Soc., 2017, 139, 7632–7639 CrossRef CAS PubMed.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- J. S. Zheng, S. Tang, Y. K. Qi, Z. P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS.
- M. Pan, S. Gao, Y. Zheng, X. Tan, H. Lan, X. Tan, D. Sun, L. Lu, T. Wang, Q. Zheng, Y. Huang, J. Wang and L. Liu, J. Am. Chem. Soc., 2016, 138, 7429–7435 CrossRef CAS PubMed.
- Z. Wang, W. Xu, L. Liu and T. F. Zhu, Nat. Chem., 2016, 8, 698–704 CrossRef CAS PubMed.
- A. Pech, J. Achenbach, M. Jahnz, S. Schulzchen, F. Jarosch, F. Bordusa and S. Klussmann, Nucleic Acids Res., 2017, 45, 3997–4005 CrossRef CAS.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS PubMed.
- G. M. Fang, J. X. Wang and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 10347–10350 CrossRef CAS.
- Y. C. Huang, G. M. Fang and L. Liu, Natl. Sci. Rev., 2016, 3, 107–116 CrossRef CAS.
- H. Ai, M. Sun, A. Liu, Z. Sun, T. Liu, L. Cao, L. Liang, Q. Qu, Z. Li, Z. Deng, Z. Tong, G. Chu, X. Tian, H. Deng, S. Zhao, J. B. Li, Z. Lou and L. Liu, Nat. Chem. Biol., 2022, 18, 972–980 CrossRef CAS PubMed.
- S. Tang, Y. Y. Si, Z. P. Wang, K. R. Mei, X. Chen, J. Y. Cheng, J. S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 5713–5717 CrossRef CAS PubMed.
- R. Ding, W. W. Shi and J. S. Zheng, Org. Lett., 2023, 25, 4857–4861 CrossRef CAS PubMed.
- H. N. Chang, B. Y. Liu, Y. K. Qi, Y. Zhou, Y. P. Chen, K. M. Pan, W. W. Li, X. M. Zhou, W. W. Ma, C. Y. Fu, Y. M. Qi, L. Liu and Y. F. Gao, Angew. Chem., Int. Ed., 2015, 54, 11760–11764 CrossRef CAS.
- P. S. Marinec, K. E. Landgraf, M. Uppalapati, G. Chen, D. Xie, Q. Jiang, Y. Zhao, A. Petriello, K. Deshayes, S. B. H. Kent, D. Ault-Riche and S. S. Sidhu, ACS Chem. Biol., 2021, 16, 548–556 CrossRef CAS PubMed.
- M. Uppalapati, D. J. Lee, K. Mandal, H. Li, L. P. Miranda, J. Lowitz, J. Kenney, J. J. Adams, D. Ault-Riche, S. B. Kent and S. S. Sidhu, ACS Chem. Biol., 2016, 11, 1058–1065 CrossRef CAS PubMed.
- S. W. Dong, J. S. Zheng, Y. M. Li, H. Wang, G. Chen, Y. X. Chen, G. M. Fang, J. Guo, C. M. He, H. G. Hu, X. C. Li, Y. M. Li, Z. G. Li, M. Pan, S. Tang, C. L. Tian, P. Wang, B. Wu, C. L. Wu, J. F. Zhao and L. Liu, Sci. China: Chem., 2024, 67, 1060–1096 CrossRef CAS.
- Y. Zheng, B. Zhang, W. W. Shi, X. Deng, T. Y. Wang, D. Han, Y. Ren, Z. Yang, Y. K. Zhou, J. Kuang, Z. W. Wang, S. Tang and J. S. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202318897 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS.
- J. S. Zheng, M. Yu, Y. K. Qi, S. Tang, F. Shen, Z. P. Wang, L. Xiao, L. Zhang, C. L. Tian and L. Liu, J. Am. Chem. Soc., 2014, 136, 3695–3704 CrossRef CAS.
- J. X. Wang, G. M. Fang, Y. He, D. L. Qu, M. Yu, Z. Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 2194–2198 CrossRef CAS PubMed.
- C. Zuo, R. Ding, X. Wu, Y. Wang, G. C. Chu, L. J. Liang, H. Ai, Z. B. Tong, J. Mao, Q. Zheng, T. Wang, Z. Li, L. Liu and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202201887 CrossRef CAS PubMed.
- W. W. Shi, C. Shi, T. Y. Wang, Y. L. Li, Y. K. Zhou, X. H. Zhang, D. Bierer, J. S. Zheng and L. Liu, J. Am. Chem. Soc., 2022, 144, 349–357 CrossRef CAS.
- Y. M. Li, Y. T. Li, M. Pan, X. Q. Kong, Y. C. Huang, Z. Y. Hong and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 2198–2202 CrossRef CAS PubMed.
- W. Shi, T. Wang, Z. Yang, Y. Ren, D. Han, Y. Zheng, X. Deng, S. Tang and J. S. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202313640 CrossRef CAS PubMed.
- J. Zhao, X. Liu, J. Liu, F. Ye, B. Wei, M. Deng, T. Li, P. Huang and P. Wang, J. Am. Chem. Soc., 2024, 146, 2615–2623 CrossRef CAS PubMed.
- F. Ye, J. Zhao, P. Xu, X. Liu, J. Yu, W. Shangguan, J. Liu, X. Luo, C. Li, T. Ying, J. Wang, B. Yu and P. Wang, Angew. Chem., Int. Ed., 2021, 60, 12904–12910 CrossRef CAS PubMed.
- T. Li, H. Liu and X. Li, Org. Lett., 2016, 18, 5944–5947 CrossRef CAS PubMed.
- Z. Sun and X. Li, Chin. J. Chem., 2021, 39, 2795–2800 CrossRef CAS.
- J. S. Zheng, J. Liang, W. W. Shi, Y. Li, H. G. Hu, C. L. Tian and L. Liu, Sci. Bull., 2021, 66, 1542–1549 CrossRef CAS.
- Y. Xu and T. F. Zhu, Science, 2022, 378, 405–412 CrossRef CAS PubMed.
- J. Cramer and T. Restle, J. Biol. Chem., 2005, 280, 40552–40558 CrossRef CAS PubMed.
- J. Chen, M. Chen and T. F. Zhu, Nat. Biotechnol., 2022, 40, 1601–1609 CrossRef CAS PubMed.
- W. Jiang, B. Zhang, C. Fan, M. Wang, J. Wang, Q. Deng, X. Liu, J. Chen, J. Zheng, L. Liu and T. F. Zhu, Cell Discovery, 2017, 3, 17037 CrossRef CAS.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef CAS PubMed.
- Z. Q. Sun, W. J. Ma, Y. H. Cao, T. Y. Wei, X. Y. Mo, H. Y. Chow, Y. Tan, C. H. P. Cheung, J. M. Liu, H. K. Lee, E. C. M. Tse, H. Liu and X. C. Li, Chem, 2022, 8, 2542–2557 CAS.
- D. Han, Y. Cui, X. Deng, C. Li, X. Zhu, B. Wang, G. C. Chu, Z. A. Wang, S. Tang, J. S. Zheng, L. J. Liang and L. Liu, J. Am. Chem. Soc., 2025, 147, 4135–4146 CrossRef CAS PubMed.
- K. Jin, T. Li, H. Y. Chow, H. Liu and X. Li, Angew. Chem., Int. Ed., 2017, 56, 14607–14611 CrossRef CAS PubMed.
- D. T. Flood, J. C. J. Hintzen, M. J. Bird, P. A. Cistrone, J. S. Chen and P. E. Dawson, Angew. Chem., Int. Ed., 2018, 57, 11634–11639 CrossRef CAS.
- B. Zhang, Y. Zheng, G. Chu, X. Deng, T. Wang, W. Shi, Y. Zhou, S. Tang, J. S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306270 CrossRef CAS PubMed.
- A. Kar, J. Mannuthodikayil, S. Singh, A. Biswas, P. Dubey, A. Das and K. Mandal, Angew. Chem., Int. Ed., 2020, 59, 14796–14801 CrossRef CAS PubMed.
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00243e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |