
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum chemical screening of eutectic solvent components for insights into CO2 complexation mechanisms†
Stephen P.
Vicchio
 *a,
Osasumwen J.
Ikponmwosa
b and
Rachel B.
Getman
*ab
*a,
Osasumwen J.
Ikponmwosa
b and
Rachel B.
Getman
*ab
aDepartment of Chemical and Biomolecular Engineering, Clemson University, Clemson, South Carolina 29634-0909, USA. E-mail: stephen.vicchio@uky.edu
bWilliam G. Lowrie Department of Chemical and Biomolecular Engineering, Ohio State University, Columbus, OH 43210, USA. E-mail: getman.11@osu.edu
First published on 2nd May 2025
Abstract
Developing new negative emission technologies (NETs) to capture atmospheric CO2 is necessary to limit global temperature rise below 1.5 °C by 2050. The technologies, such as direct air capture (DAC), rely on sorption materials to harvest trace amounts of CO2 from ambient air. Deep eutectic solvents (DESs) and eutectic solvents (ESs), a subset of ionic liquids (ILs), are all promising new CO2 sorption materials for DAC. However, the experimental design space for different DESs/ESs/ILs is vast, with the exact CO2 complexation pathways difficult to elucidate; this creates significant limitations in rationally designing new materials with targeted CO2 sorption energetics. Herein, the CO2 complexation pathways for a structural library of different DES/ES components are computed using quantum chemical calculations (i.e., density functional theory). For the entire structure library, we report the energies of elementary CO2 binding and proton transfer reactions as these reactions are fundamental in DAC within DESs and ESs. These elementary reactions are combined to generate CO2 complexation pathways and calculate their free energies. The different elementary steps and reaction pathways demonstrate the range of CO2 complexation free energies and the significance between CO2 binding and proton transfer reactions. We also report the CO2 complexation free energies with different functional groups around the CO2 sorption site, supporting the concept of functionalization for tuning CO2 complexation thermodynamics. Additionally, our findings suggest potential descriptors, such as proton affinity or pKa, could be useful when identifying candidate species for ESs and predicting/rationalizing product distributions. Our work has implications for experimental synthesis, characterization, and performance evaluation of new DAC sorption materials.
Design, System, ApplicationThe development of negative emission technologies (NETs) is crucial for mitigating global climate change by capturing atmospheric CO2. Among these technologies, direct air capture (DAC) relies on advanced sorbent materials to efficiently harvest CO2 from ambient air. Deep eutectic solvents (DESs) and eutectic solvents (ESs), as subsets of ionic liquids (ILs), show significant promise as CO2 sorbents due to their unique properties, including negligible vapor pressures, high thermal stabilities, and chemical tunability. However, the vast experimental design space and complex CO2 complexation pathways present challenges in rationally designing new materials with targeted CO2 sorption energetics. In this work, we employ quantum chemical calculations, specifically density functional theory (DFT), to investigate the CO2 complexation mechanisms within a structural library of DES/ES components. By computing the energies of elementary CO2 binding and proton transfer reactions, we generate comprehensive CO2 complexation pathways and calculate their free energies. This approach allows us to explore the impact of various functional groups on CO2 binding thermodynamics, providing insights into the tunability of CO2 sorption properties. Our findings highlight the potential of functionalization to enhance CO2 complexation by modifying the chemical environment around the sorption site. Additionally, we identify potential descriptors, such as proton affinity or pKa, that could aid in predicting and rationalizing CO2 sorption performance. This work lays the groundwork for the experimental synthesis, characterization, and optimization of new DAC sorbent materials, ultimately contributing to the advancement of NETs and the global effort to combat climate change. |
Introduction
Reducing CO2 emissions from burning non-renewable fossil fuels is insufficient to remediate global climate change.1 Therefore, the development of new negative emission technologies (NETs)2–5 to remove previously emitted CO2 is essential in limiting global temperature rise.6 Direct air capture (DAC), one such NET, is a carbon capture process that relies on novel sorbent materials to efficiently remove atmospheric CO2 under ambient conditions.6–9 Promising DAC materials include ionic liquids (ILs),10–12 deep eutectic solvents (DESs),10,13–15 metal–organic frameworks (MOFs),16–20 and covalent organic frameworks (COFs).21–23 These solid- and liquid-sorbent materials exhibit promising physical and chemical properties for DAC and are tunable. For state-of-the-art DAC materials,24 higher gravimetric CO2 uptake is achieved when CO2 binds chemically (e.g., ILs25) instead of physically (e.g., MOFs26). This is particularly important for DAC, where the amount of CO2 is very small. For DAC, there is a need to develop new materials that exhibit high CO2 capacities, which requires a deeper understanding of the structure–function relationships that govern the CO2 sorption process.Both ILs and DESs are promising liquid sorbents for DAC due to their negligible vapor pressures, high thermal stabilities, and chemical tunabilities.27–30 ILs are liquid salts comprising of both cationic and anionic components (charges of ±1) that melt below 100 °C.31 Both these cationic and anionic components are hydrogen bond acceptors (HBAs). Examples of cationic components (i.e., [HBAc]+) are 1-ethyl-3-alkylimidazolium ([EMIM]+) and choline ([Ch]+), and examples of anionic components (i.e., [HBAa]−) are imidazolide ([Im]−) and pyrrolide ([Pyr]−).32 DESs are similar to ILs in that they contain the cationic and anionic components, but DESs also contain a hydrogen bond donor (HBD), such as ethylene glycol (EG) that contributes to further melting point suppression33,34 and, generally, lowers the viscosity of the solvent.14,35 However, even eutectic solvents (ESs), where the exact ‘deep’ eutectic composition is not necessarily known, still exhibit promising properties for DAC.
The CO2 uptake performance for ILs and DESs/ESs has been previously studied using experimental and computational approaches.11,36–46 For example, Lee et al. elucidated the different CO2 binding pathways for an imidazolium [HBAc]+ and pyrrolide [HBAa]− in the presence of ethylene glycol (EG), finding that CO2 prefers to bind to the [HBAc]+.47 Further advancements by Klemm et al. demonstrate how the CO2 uptake capacity can be tuned by converting amino acid-based ILs to ESs through the addition of EG.14 Supplemental modeling insights demonstrate how the HBD alters the interactions within the solvent leading to a difference in uptake for the IL vs. the ES.
These studies motivate the need for research aimed at identifying the CO2 complexation mechanisms associated with CO2 uptake. Developing strategies to predict CO2 uptake performance a priori is of critical importance due to the large experimental design space for these materials. There are multiple mechanisms for CO2 complexation within an ES,37,48 as defined by the CO2 binding site (i.e., to the [HBAc]+, the [HBAa]−, and/or the HBD species). The simplest mechanism is complexation to the [HBAa]−, where CO2 can bind directly to the species without the need for proton transfer to create the binding site. However, the [HBAc]+ and the HBD species can also accept CO2 following proton transfer to create a site for complexation.14,38,49,50 The thermodynamics and kinetics behind these different complexation pathways are influenced by several properties, including the chemical nature of the various components of the sorbent material. Ultimately, the goal is to understand how to tune CO2 complexation in ILs/DESs/ESs via chemical functionality.
Along these lines, in this work we combine high-throughput screening and density functional theory (DFT) calculations to probe CO2 complexation by identifying the CO2 binding locations and thermodynamics for CO2 binding and proton transfer reaction steps. Quantifying the complexation thermodynamics of the various species within ILs and DESs/ESs is fundamental for rationally designing tunable CO2 sorbent materials. We specifically focus on ESs by computing the CO2 complexation thermodynamics for ∼420 combinations of ESs (i.e., unique cations, anions, and HBDs species). A library of hypothetical ESs provides information into the CO2 complexation thermodynamics associated with different complexation pathways and the intermolecular proton transfers. Additionally, [HBAa]− functionalization is also systematically explored by varying the functional groups around the CO2 binding site with electron-donating and electron-withdrawing groups. Overall, our work provides fundamental insights into the CO2 sorption for ESs while laying the groundwork for rationally constructing solvents for CO2 capture.
Methodology
The following sections discuss CO2 complexation to the HBA and HBD components of ESs. For the HBA species, the cationic component is denoted as HBAc and the anionic component is denoted as HBAa. Charged species are denoted with brackets (i.e., [ ]), and positive and negative charges are indicated using [ ]+ for cationic and [ ]− and anionic species, respectively. Species without brackets are neutral. This delineation is essential when defining the different fundamental reactions since these reactions can involve proton transfers, which alter the charges of the species. For example, a proton transfer from the [HBAc]+ or the HBD is needed before CO2 complexation to those species. These proton transfers can neutralize or charge these species (i.e., [HBAc]+ → HBAcf + [H]+ or HBD → [HBD]− + [H]+). Given the more favorable kinetics for intermolecular proton transfers over intramolecular proton transfers,14,47,51 only intermolecular proton transfers are considered in this work. Protons that are transferred are hence transferred to the [HBAa]− (i.e., [HBAa]− + [H]+ → HBAa).Structure library
A library of structures consisting of 5 [HBAc]+, 12 [HBAa]−, and 7 HBD species is investigated. Fig. 1 provides the full and abbreviated naming conventions and the structural representations for each species investigated, with the ball-and-stick representations for all species provided in ESI† Section S1. Hypothetical ESs are constructed by combining a single [HBAc]+, [HBAa]−, and HBD species, producing a total of 420 different hypothetical mixtures. However, the total number of structures considered is even larger due to the multiple binding sites for CO2 sorption and proton binding. For example, on a [HBAa]− comprising multiple N binding sites, CO2 can bind to each N atom. All possible binding sites are considered to ensure the lowest energy conformer is found; the lowest energy conformers are reported in this manuscript. All structures are stored in an open, online repository.52 | ||
| Fig. 1 The (a) hydrogen bond acceptor cations ([HBAc]+), (b) hydrogen bond acceptor anions ([HBAa]−), and (c) hydrogen bond donors (HBD) considered in this work. Both the full- and abbreviated-naming conventions are provided; the cationic and anionic species are identified with brackets and charges, respectively. | ||
A subset of the [HBAa]− library (i.e., [Im]− and [Pyr]−) comprises similar species with different functional groups to investigate how different functional groups influence complexation. The functional groups are selected based on their electron-donating (e.g., R–OH) and electron-withdrawing (e.g., R–CHO) capabilities to understand the influence on CO2 binding. The different functional groups are attached adjacent to the N binding site on the [Im]− and [Pyr]− species (Fig. 2a) at ortho positions relative to the CO2 binding site. Specifically for [Im]−, the functional group attachment occurs between the ring N atoms. These functionalized [HBAa]− species are compared to the unfunctionalized [Im]− and [Pyr]− species (which have a hydrogen atom in the ortho position). The left column in Fig. 2b shows the electron-donating groups (i.e., R–Me, R–Et, R–OH, R–MeOH, and R–EtOH), and the right column in Fig. 2b shows the electron-withdrawing groups (R–F, R–CN, R–CHO, R–COCH3, and R–COOCH3) considered in this work.
 | ||
| Fig. 2 [Pyr–X]− and [Im–X]− (a) were investigated for the influence of functionalization with the ten different functional groups (X) shown in (b). In part (b), electron-donating groups are shown in the left column while electron-withdrawing groups are shown in the right column. | ||
Fundamental reactions
For a given [HBAc]+[HBAa]−:HBD ES, the hypothetical reaction mechanisms for CO2 complexation, consisting of CO2 binding and proton transfer steps, are presented in Table 1. The proton transfer reactions are included to account for cases where a species must donate a proton for CO2 complexation. The generic, hypothetical reaction mechanisms determine the individual thermodynamics of each step and, when combined, the subsequent CO2 complexation pathway free energies (Fig. 3).| Reaction mechanism | |
|---|---|
| All proton transfer reactions are written as affinity reactions for consistency. | |

|
R1 |
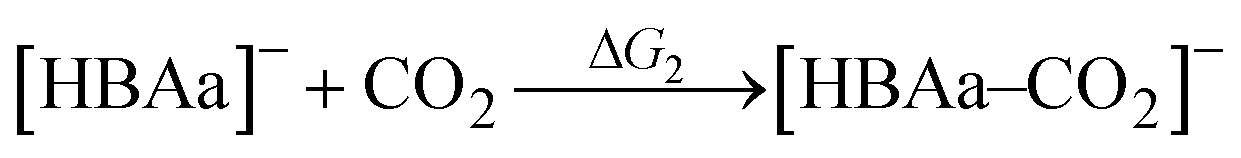
|
R2 |

|
R3 |

|
R4 |

|
R5 |

|
R6 |

|
R7 |

|
R8 |
 | ||
| Fig. 3 The different CO2 complexation pathways considered for a [HBAc]+[HBAa]−:HBD ES. Complexation occurs via the [HBAa]− (pink), HBAc (mauve), [HBD]− (purple), and HBD (blue) pathways. Reaction numbers are the same as in Table 1. Only the [HBAa]− pathway does not require a proton transfer for CO2 complexation. The other three pathways all involve an intermolecular proton transfer, which is modeled here as occurring to the [HBAa]− species. | ||
Hypothetical reaction pathways
There are four distinct CO2 complexation pathways considered in this work, which are outlined in Fig. 3. We label these pathways as the ‘[HBAa]−’, ‘HBAc’, ‘[HBD]−’, and ‘HBD’ pathways. The pathways are defined by the species that CO2 binds to (i.e., to the [HBAa]−, HBAc, [HBD]−, or HBD species). In the [HBAa]− pathway, CO2 complexes directly to the [HBAa]−via reaction R2. CO2 binding to the [HBAc]+ requires proton transfer via the reverse of reaction R3 (all proton transfer reactions in Table 1 are written as affinity reactions for consistency) prior to complexation via reaction R4. CO2 can complex to the HBD via two pathways. In the [HBD]− pathway, the HBD species first transfers a proton via the reverse of reaction R5. CO2 then complexes to the deprotonated HBD species (i.e., [HBD]−) via reaction R6. An additional HBD pathway is considered where CO2 first forms a weak complex with the neutral HBD molecule (reaction R7) prior to proton transfer via reaction R8. Theoretically, protons could be transferred to any negatively charged species in the solvent. We consider the case where they are transferred to the [HBAa]−, converting it to the HBAa via reaction R1. The total free energy of complexation is denoted ΔGc and is the sum of all necessary reactions en route to CO2 complexation. For example, considering the [HBAc]+ pathway (mauve color in Fig. 3), ΔGc = −ΔG3 + ΔG1 + ΔG4.Quantum chemical (QM) calculations
The entire hypothetical reaction library is screened using the Gaussian16 Revision C.02 software,53 with the structures, energies, and vibrational frequencies calculated at the B3LYP/6-311+G(d,p) level of theory. Test calculations performed at the ωB97XD/6-311+G(d,p) level of theory indicate a maximum 3.3 kJ mol−1 difference in free energy. Dispersion corrections formulated by Grimme et al. with Becke–Johnson (BJ) damping are included.54,55 Solvation is needed to stabilize the electronic and geometric structures of the ionic species. We use implicit solvation for simplicity. However, explicit intermolecular interactions could contribute to the reported free energies. To learn how this could influence the results, we investigated the influence of hydrogen bonded HBD species on the [HBAa]− complexation pathway free energies. This analysis is summarized in ESI† Section S3. We find that, generally, explicit stabilization leads to a decrease in the CO2 complexation free energies to the [HBAa]− species (i.e., CO2 complexation is more favorable). Exploring this further would be an excellent topic for future work. Our simulations utilize ethanol (ε = 24.8520) as the implicit solvent, since it is representative of the range of dielectric constants of the hypothetical ESs. Further rationale for this choice is provided in ESI† Section S2.The computational workflow is fully automated, thus enabling the quick generation of Gaussian16 input files, the collection and analysis of the energies, and the creation of the pathway energies. The workflow, which heavily utilizes the atomic simulation environment (ASE),56 ensures that all potential binding sites are explored for each structure. For every structure presented in Fig. 1, the necessary reactions for each component presented in Table 1 are calculated. Free energies are estimated by adding the vibrational free energy contributions from the harmonic oscillator approximation to the electronic energies. The vibrational contributions are calculated using the pMuTT57 python package. All vibrational frequencies below 50 cm−1 are set equal to 50 cm−1 to avoid overestimating the entropic contribution of low-frequency modes, which is a limitation of the harmonic oscillator approximation.58 The energy of [H]+ is calculated relative to H3O+/H2O, i.e., G[H]+ = G[H3O]+ − GH2O, since it is not possible to compute the energy of an isolated proton (G[H]+) in most electronic structure software. This is a modeling choice that does not impact the results, but enables the calculation of relative proton transfer reaction free energies.
Results
CO2 complexation
 | ||
| Fig. 4 The [HBAa]− CO2 complexation free energies (in kJ mol−1), where ΔGc = ΔG2. The figure is organized by structural similarity to compare how functionalization impacts CO2 binding. Chemical structures are included to illustrate structural similarities and differences. Atom color key: gray = carbon, blue = nitrogen, red = oxygen, white = hydrogen. Different families of [HBAa]− species are denoted with the purple, blue, teal, and orange brackets. | ||
The influence of chemical functionality on CO2 complexation is further investigated by systematically adding different functional groups around the CO2 binding site on both the [Pyr]− and [Im]− species. The influence of different functional groups on CO2 complexation free energies to [Pyr–X]− and [Im–X]− species, where X represents the different functional groups, is presented in Fig. 5. We find that alkyl functional groups (i.e., R–Et and R–Me), which are considered weakly electron-donating groups, exhibit similar free energies as the parent [Pyr]− and [Im]− structures. The main electron donating groups considered are R–OH, R–MeOH, and R–EtOH. For [Pyr–X]−, only R–OH results in stronger CO2 complexation (by ∼15 kJ mol−1). In contrast, R–MeOH leads to a similar complexation energy, and R–EtOH has a CO2 complexation energy that is less favorable (more positive) by ∼11 kJ mol−1. For [Im–X]−, the presence of all electron donating groups leads to more negative (stronger) CO2 complexation energies, ranging from ∼12 to 20 kJ mol−1 compared to [Im]−.
 | ||
| Fig. 5 The [HBAa]− CO2 complexation free energies (in kJ mol−1), where ΔGc = ΔG2 for the (a) [Pyr–X]− and (b) [Im–X]− families with different functional groups (as shown in Fig. 2). The single hatch indicates electron-donating groups and the double hatch indicates electron-withdrawing groups. The naming conventions for structures appearing in Fig. 4 are modified according to the naming convention in Fig. 2 (i.e., [Pyr–CN]− = [2–CNpyr]−, [Im–Me]− = [MeIm]−, and [Im–Et]− = [EtIm]−). Atom color key: gray = carbon, blue = nitrogen, red = oxygen, white = hydrogen. | ||
Conversely, the presence of electron withdrawing groups leads to more positive (i.e., less thermodynamically favorable) CO2 complexation energies compared to the parent species. Electron withdrawing groups remove electron density from the ring structure, resulting in a less nucleophilic N binding sites for CO2 complexation. These results suggest that CO2 complexation free energies to [HBAa]− species may be tuned with careful selection of the functional groups adjacent to the N binding site. In general, the CO2 complexation results for the [HBAa]− species highlight the potential tunability of CO2 complexation to the [HBAa]− species.
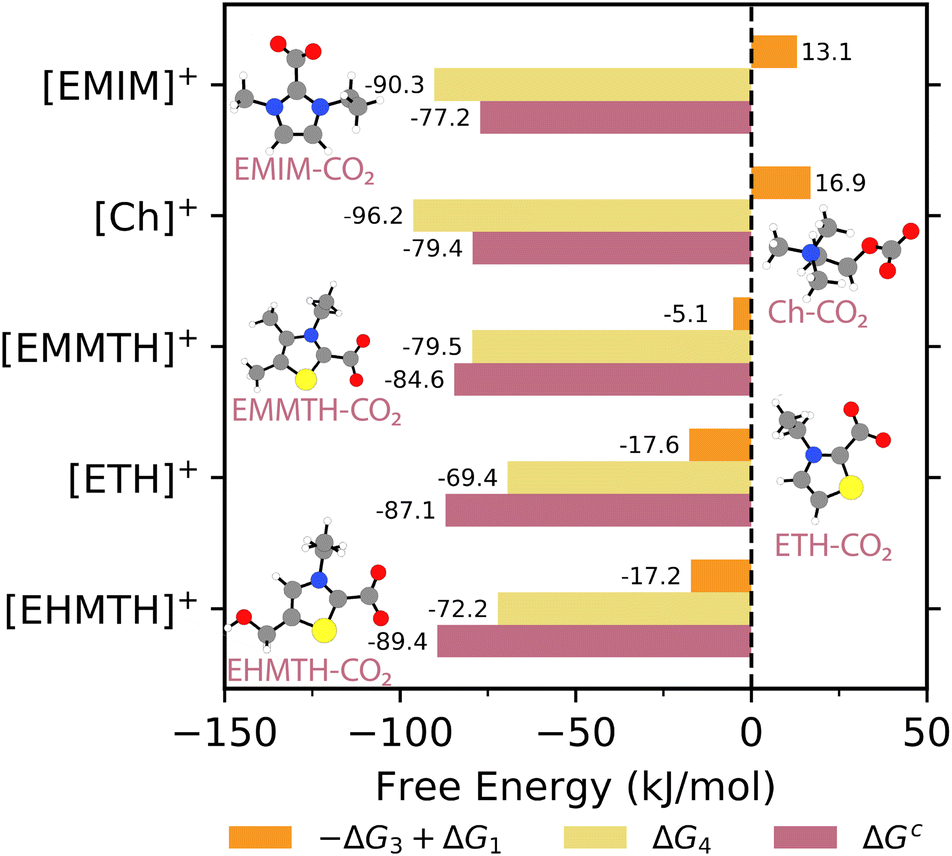 | ||
| Fig. 6 The HBAc pathway free energies (ΔGc = −ΔG3 + ΔG1 + ΔG4; mauve), the proton transfer free energies (−ΔG3 + ΔG1; orange), and the CO2 binding free energies (ΔG4; yellow). ΔG1 is calculated using [Im]− as the proton acceptor. The structure insets illustrate the CO2 complexation final products. Atom color key: gray = carbon, blue = nitrogen, red = oxygen, white = hydrogen, yellow = sulfur. | ||
The overall CO2 complexation pathway free energies (ΔGc) for the [HBAc]+ species are presented by the mauve bars in Fig. 6. The pathway free energies are obtained by summing the proton transfer free energies with the CO2 binding energy, i.e., −ΔG3 + ΔG1 + ΔG4. Overall, the CO2 complexation pathway free energies range from −77.2 kJ mol−1 to −89.4 kJ mol−1. They are strongly negative due to the favorable/negative CO2 binding free energies (i.e., ΔG4), which span from −69.4 kJ mol−1 to −96.2 kJ mol−1 (yellow bars in Fig. 6). As previously mentioned, the sulfur-containing [HBAc]+ species exhibit deviations in proton transfer energies. However, these deviations are not observed in the ΔGc free energies (−84.6 kJ mol−1 to −89.4 kJ mol−1). The CO2 binding free energies (ΔG4) compensate for the differences in proton transfer free energies for the [EMMTH]+, [EHMTH]+, and [ETH]+ species, such that the overall complexation free energies for all [HBAc]+ species are similar.
Free energies involved in CO2 complexation to the different HBD species are reported in Fig. 7. Overall, the CO2 complexation pathways for the different HBD species (green bars in Fig. 7) are thermodynamically similar, with ΔGc of ∼−65 kJ mol−1, despite variations in the chain length of the diamine species (increasing in length from EDA to TDA to PDA). The exception is GOL, which forms an intramolecular hydrogen bond between the complexed CO2 and the adjacent –OH group and exhibits ΔGc of −87.5 kJ mol−1, but even this difference is minor. All HBDs considered in this work prefer the pathway where weak CO2 complexation occurs initially. This is due to the proton transfer free energies being more favorable from the weakly complexed species than for the uncomplexed species (i.e., −ΔG5 + ΔG1 ≫ ΔG7; more positive purple bars than blue bars in Fig. 7). This is particularly true for the HBDs with the –N site, where proton transfer from the HBD is significantly uphill (∼160 kJ mol−1) compared to the free energy to form HBD–CO2 species (∼−20 kJ mol−1), and because the free energies of proton transfer from the weakly complexed HBD–CO2 species (i.e., −ΔG8 + ΔG1) are negative for all HBD species.
 | ||
| Fig. 7 The HBD/[HBD]− CO2 complexation pathway free energies (ΔGc = −ΔG5 + ΔG1 + ΔG6 = ΔG7 − ΔG8 + ΔG1; green), the free energies for transferring a proton from the HBD to [Im]− (−ΔG5 + ΔG1; purple), the free energy of weak CO2 complexation (ΔG7; blue), and the free energies of proton transfer from HBD–CO2 to [Im]− (−ΔG8 + ΔG1; light blue). ΔG1 is calculated using [Im]− as the proton acceptor. The structure inserts demonstrate the different intermediates for the HBD and [HBD]− complexation pathways. Atom color key: gray = carbon, blue = nitrogen, red = oxygen, white = hydrogen. | ||
 | ||
Fig. 8 Heat maps comparing the free energies between the HBD and HBAc pathways: (a) difference between the weakly complexed CO2 step (ΔG7) and the proton transfer step (−ΔG3 + ΔG1) free energies and (b) difference between HBD (ΔGcHBD) and HBAc  complexation pathway free energies. For the proton transfer steps, the energy (ΔG1) is calculated using [Im]− as the proton acceptor. The color bar indicates the more favorable pathway. For (a), purple indicates that weak complex is more favorable (ΔG7 < −ΔG3 + ΔG1) and mauve indicates the proton transfer is more favorable (ΔG7 > −ΔG3 + ΔG1). For (b), purple indicates that the HBD complexation is preferred (ΔGcHBD < ΔGcHBAc) and mauve indicates that the HBAc complexation pathway is preferred (ΔGcHBD > ΔGcHBAc). complexation pathway free energies. For the proton transfer steps, the energy (ΔG1) is calculated using [Im]− as the proton acceptor. The color bar indicates the more favorable pathway. For (a), purple indicates that weak complex is more favorable (ΔG7 < −ΔG3 + ΔG1) and mauve indicates the proton transfer is more favorable (ΔG7 > −ΔG3 + ΔG1). For (b), purple indicates that the HBD complexation is preferred (ΔGcHBD < ΔGcHBAc) and mauve indicates that the HBAc complexation pathway is preferred (ΔGcHBD > ΔGcHBAc). | ||
The HBD vs. [HBAc]+ pathway complexation free energies  are presented in Fig. 8b. Specifically, Fig. 8b plots the difference between the HBD complexation pathway (ΔGcHBD) and the HBAc complexation pathway (ΔGcHBAc) free energies. For all species except GOL, the reported free energies differences are positive (ΔGcHBD − ΔGcHBAc > 0), with free energies ranging from ∼11 kJ mol−1 to ∼26 kJ mol−1. These results indicate that for all HBD species except GOL, CO2 complexation to the HBAc is favored (i.e., ΔGcHBAc < ΔGcHBD).
are presented in Fig. 8b. Specifically, Fig. 8b plots the difference between the HBD complexation pathway (ΔGcHBD) and the HBAc complexation pathway (ΔGcHBAc) free energies. For all species except GOL, the reported free energies differences are positive (ΔGcHBD − ΔGcHBAc > 0), with free energies ranging from ∼11 kJ mol−1 to ∼26 kJ mol−1. These results indicate that for all HBD species except GOL, CO2 complexation to the HBAc is favored (i.e., ΔGcHBAc < ΔGcHBD).
The free energies for the [HBAa]− and HBD complexation pathways are compared in Fig. 9. Since the [HBAa]− does not require proton transfer, the free energy of CO2 binding to the [HBAa]− species (ΔG2) is compared to the free energy of weak CO2 complexation to the HBD (ΔG7) in Fig. 9a. The difference in free energies ranges from −18 kJ mol−1 to +76 kJ mol−1. These differences are strongly influenced by the [HBAa]− species, which exhibits large variations in CO2 complexation free energies (ΔG2) as reported in Fig. 4. However, the overall complexation pathway free energies are presented in Fig. 9b and show a more neutral thermodynamic landscape  for most [HBAa]−/HBD pairs with only GOL breaking this trend. Overall, this suggests that thermodynamically, these species are approximately equally likely to complex CO2.
for most [HBAa]−/HBD pairs with only GOL breaking this trend. Overall, this suggests that thermodynamically, these species are approximately equally likely to complex CO2.
 | ||
Fig. 9 Heat maps comparing the free energies between the HBD and [HBAa]− pathways: (a) difference between the weak CO2 complexation free energy (ΔG7) and the [HBAa]− CO2 complexation free energy (ΔG2) and (b) difference between HBD (ΔGcHBD) and [HBAa]− complexation pathway free energies. For the proton transfer steps, the energy (ΔG1) is calculated using the corresponding [HBAa]− species as the proton acceptor. The color bar indicates the more favorable pathway. For (a), purple indicates that weak complexation to the HBD is more favorable (ΔG7 < ΔG2) and pink indicates that complexation to the [HBAa]− is more favorable (ΔG7 > ΔG2). For (b), purple indicates that the HBD complexation pathway is preferred complexation pathway free energies. For the proton transfer steps, the energy (ΔG1) is calculated using the corresponding [HBAa]− species as the proton acceptor. The color bar indicates the more favorable pathway. For (a), purple indicates that weak complexation to the HBD is more favorable (ΔG7 < ΔG2) and pink indicates that complexation to the [HBAa]− is more favorable (ΔG7 > ΔG2). For (b), purple indicates that the HBD complexation pathway is preferred  and pink indicates that the [HBAa]− complexation pathway is preferred and pink indicates that the [HBAa]− complexation pathway is preferred  . . | ||
Discussion
The values of ΔGc calculated in this work support that, with the exception of GOL, CO2 complexation to the [HBAc]+ is favored, but that multiple complexation pathways are present within eutectic solvents, with CO2 complexation occurring to the [HBAc]+, [HBAa]−, and HBD species. This suggests that multiple CO2 sorption products could be present in IL/ES/DES solvents. This poses significant challenges when designing different sorbents with specific CO2 uptake properties since identifying the mechanisms and thermodynamics for complexation is challenging. Although not explored here, understanding CO2 complexation is also further complicated by a single species potentially complexing multiple CO2. For example, while our structure library only considers a single CO2 complexation for each HBD, experimentally, certain HBD species have been observed to complex multiple CO2 molecules.10,59 When rationalizing experimental uptake data, understanding the locations and potential for a single species that complexes multiple CO2 molecules is important to identify the different mechanisms.Furthermore, our findings demonstrate that functional groups around the complexation site result in large deviations (∼70 kJ mol−1) of the CO2 complexation energies. The electron-donating groups display more favorable (i.e., more negative values of ΔGc); electron-withdrawing groups display less favorable (i.e., more positive values of ΔGc). Generally, the species with more negative complexation free energies display intramolecular hydrogen bonds between the complexed CO2 and adjacent ligands. As reported in Fig. S7,† these intramolecular hydrogen bonds lower the CO2 complexation free energies by ∼30 kJ mol−1 for the [IM–EtOH]−, [IM–MeOH]−, and [Pyr–OH]− species. This observation supports the observation made by Klemm et al. that intramolecular hydrogen bond formation stabilizes complexed CO2 reaction intermediates.14 Overall, these findings suggest that rationally selecting the functional groups around the CO2 binding site provides significant opportunities for tuning CO2 complexation.
Another potential mechanism for tuning CO2 complexation is considering the nucleophilicity of a given molecule, which is well established for CO2 complexation.60 Most CO2 sorption materials rely on nucleophiles (i.e., containing electron lone pairs) to capture CO2. However, the same strong, electron-rich character also promotes proton transfer reactions. The relationship between nucleophilicity to both CO2 binding and proton transfer reactions presents a challenge in designing solvents (Fig. S8 and S9†). For the [HBAa]− species (Fig. S9a†), a more negative (i.e., stronger) proton transfer free energy indicates a more negative (i.e., stronger) CO2 binding energy, and this is attributed to the increased nucleophilicity of the [HBAa]−. A similar observation is established for both [HBAc]+ (Fig. S9b†) and [HBD]− species (Fig. S8†). However, since the HBAc and [HBD]− rely on intermolecular proton transfers for CO2 complexation, a stronger (more negative) proton transfer energy leads to unfavorable CO2 binding thermodynamics as these species must donate a proton. These findings support the idea that there is a competitive process between CO2 binding and proton affinity for an [HBAa]− species, which can be attributed to the nucleophilicity of the species. The interplay between nucleophilicity and the CO2 binding and proton transfer steps is fundamental to the CO2 complexation process. A strong correlation between the CO2 binding and proton transfer reactions for all [HBAc]+, [HBAa]− and HBD species suggests that experimental parameters (i.e., proton affinity or pKa)61 might be a reasonable descriptor for CO2 binding.
Additionally, the hydrogen bond network62–64 within ILs and DESs/ESs poses further challenges in understanding CO2 complexation. In ILs, the hydrogen bond network is relatively simple, with hydrogen bonds forming between the [HBAc]+ and [HBAa]−.65,66 The inclusion of the HBD complicates the hydrogen bond network by adding additional hydrogen bond interactions ([HBAc]+/[HBAa]−, [HBAa]−/HBD, and [HBAa]−/HBD).67,68 The hydrogen bond network can have a significant influence on how and where CO2 complexation occurs.14,39,69,70 Tuning CO2 complexation via the rational selection of HBDs is another potential ‘design’ variable outside the scope of this particular work.
Overall, our findings demonstrate a large range of complexation free energies to the different ILs/DESs/ESs components. Prior literature suggests reaction enthalpies ranging from ∼−50 kJ mol−1 to −65 kJ mol−1 for energy-efficient DAC.71,72 Using this range of values as a guide, systems consisting of imidazolide (i.e., [Im]−, [MeIm]−, [EtIm]−, and [BIm]−) and 2-cyanopyrrolide ([2-CNpyr]−) as [HBAa]− species and all HBD species considered exhibit complexation free energies within the target range. These findings are supported by experimental observations in reaction enthalpies for an [EMIM]+[2-CNpyr]− IL73 and potential production distribution for an [MEAH]+[Im]−:EG DES.74 Elucidating the thermodynamic landscape by investigating the CO2 complexation is a necessary first step towards designing DAC materials but cannot fully explain CO2 sorption. Additional information about mass transfer75 and reaction kinetics76 and mechanisms would be needed to fully clarify CO2 uptake. Such investigations would be good topics for future work.
Conclusion
DFT calculations provide insights into the thermodynamics for CO2 complexation to hypothetical ESs ([HBAc]+[HBAa]−:HBD). A structure library, comprised of different [HBAc]+, [HBAa]−, and HBD species is used to model CO2 binding and proton transfer reaction steps, given that these fundamental reactions are involved in CO2 complexation. The [HBAa]− complexation pathway is the simplest since no intermolecular proton transfer is required for CO2 binding. However, both the [HBAc]+ and the HBD species require an intermolecular proton transfer for CO2 complexation. The complexation thermodynamics behind these pathways are explored, with the HBAc pathway being slightly more thermodynamically favorable compared to the [HBAa]− and HBD pathways. However, differences in the experimentally, observed product distributions could be influenced by free energy effects related to solvent structure that are beyond the scope of this work.Furthermore, the functional groups surrounding the CO2 binding site influence on complexation is also studied for the [HBAa]− species and [HBAc]+ species. For [HBAa]−, the N binding site is ‘tuned’ by electron-withdrawing and electron-donating groups near the binding site. The findings demonstrate that electron-withdrawing groups lead to weaker (less favorable) complexation free energies, whereas electron-donating groups lead to stronger (more favorable) complexation free energies. For [HBAc]+, deviations for the [ETH]+-based species (i.e., different functional groups R–H, R–CH2OH, and R–CH3) is observed. Interestingly, these findings suggest that there might be differences in the functional groups that impact complexation to the [HBAc]+ and [HBAa]− species. Our findings suggest that tuning CO2 complexation via functionalization is possible.
Data availability
The data supporting this article have been included as part of the ESI.† Additionally, all structures are located on a GitHub repository.Author contributions
SPV designed the study, performed the calculations, analyzed the data, wrote the initial manuscript draft, and reviewed and edited the manuscript. OI performed calculations and reviewed and edited the manuscript. RBG designed the study, supervised the work, reviewed and edited the manuscript, and provided financial support.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work is supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award number DESC0022214. SPV was also supported by a GAANN Fellowship (Award Number: P200A180076) from the United States Department of Education. SPV and RBG would like to acknowledge the Clemson Computing and Information Technology (CITI) Advanced Computing and Data Science (ACDS) group for the generous allotment of compute time on the Palmetto cluster. OI and RBG would also like to thank the Ohio Supercomputer Center for its high-performance computing resources and expertise.Notes and references
- T. Stocker, Q. Dahe, G. Plattner, M. Tignor, S. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. Midgley, The Intergovernmental Panel on Climate Change (IPCC) 2013: Summary for Policymakers, in Climate Change 2013: The Physical Science Basis, Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, 2013 Search PubMed.
- D. McLaren, Process Saf. Environ. Prot., 2012, 90, 489–500 CrossRef CAS.
- N. A. of Sciences Engineering and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, National Academies Press, 2019, pp. 1–510 Search PubMed.
- J. Rosen, Science, 2018, 359, 733–737 CrossRef CAS PubMed.
- G. Iyer, L. Clarke, J. Edmonds, A. Fawcett, J. Fuhrman, H. McJeon and S. Waldhoff, Energy Clim. Change, 2021, 2, 1–5 Search PubMed.
- I. P. on Climate Change (IPCC), in Summary for Policymakers, Cambridge University Press, 2022, pp. 1–24 Search PubMed.
- Y. Qiu, P. Lamers, V. Daioglou, N. McQueen, H. S. de Boer, M. Harmsen, J. Wilcox, A. Bardow and S. Suh, Nat. Commun., 2022, 13, 1–13 Search PubMed.
- J. F. Wiegner, A. Grimm, L. Weimann and M. Gazzani, Ind. Eng. Chem. Res., 2022, 12649–12667 CrossRef CAS.
- G. Li and J. Yao, Engineering, 2024, 5, 1298–1336 CrossRef.
- R. Dikki, E. Cagli, D. Penley, M. Karayilan and B. Gurkan, Chem. Commun., 2023, 12027–12030 RSC.
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS PubMed.
- B. Peric, J. Sierra, E. Martí, R. Cruañas, M. A. Garau, J. Arning, U. Bottin-Weber and S. Stolte, J. Hazard. Mater., 2013, 261, 99–105 CrossRef CAS PubMed.
- M. Chen and J. Xu, Molecules, 2023, 28, 1–10 Search PubMed.
- A. Klemm, S. P. Vicchio, S. Bhattacharjee, E. Cagli, Y. Park, M. Zeeshan, R. Dikki, H. Liu, M. K. Kidder, R. B. Getman and B. Gurkan, ACS Sustainable Chem. Eng., 2023, 11, 3740–3749 CrossRef CAS.
- V. Migliorati, F. Sessa and P. D'Angelo, Chem. Phys. Lett., 2019, 2, 1–7 Search PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- S. Bose, D. Sengupta, T. M. Rayder, X. Wang, K. O. Kirlikovali, A. K. Sekizkardes, T. Islamoglu and O. K. Farha, Adv. Funct. Mater., 2023, 1–19 Search PubMed.
- I. B. Orhan, T. C. Le, R. Babarao and A. W. Thornton, Commun. Chem., 2023, 6, 1–12 CrossRef PubMed.
- A. Sinha, L. A. Darunte, C. W. Jones, M. J. Realff and Y. Kawajiri, Ind. Eng. Chem. Res., 2017, 56, 750–764 CrossRef CAS.
- L. Li, Z. Xiao, C. Xu, Y. Zhou and Z. Li, Environ. Res., 2024, 262, 1–10 Search PubMed.
- H. Lyu, H. Li, N. Hanikel, K. Wang and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 12989–12995 CrossRef CAS PubMed.
- H. Li, A. Dilipkumar, S. Abubakar and D. Zhao, Chem. Soc. Rev., 2023, 52, 6294–6329 RSC.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- M. Zanatta, ACS Mater. Au, 2023, 3, 576–583 CrossRef CAS PubMed.
- L. Lombardo, H. Yang, K. Zhao, P. J. Dyson and A. Züttel, ChemSusChem, 2020, 13, 2025–2031 CrossRef CAS PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 RSC.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed.
- J. Zhang, J. Sun, X. Zhang, Y. Zhao and S. Zhang, Greenhouse Gases:Sci. Technol., 2011, 1, 142–159 CrossRef CAS.
- X. Zhang, X. Zhang, H. Dong, Z. Zhao, S. Zhang and Y. Huang, Energy Environ. Sci., 2012, 5, 6668–6681 RSC.
- Y. Gu, Y. Hou, S. Ren, Y. Sun and W. Wu, ACS Omega, 2020, 5, 6809–6816 CrossRef CAS PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- L. J. Kollau, M. Vis, A. V. D. Bruinhorst, A. C. C. Esteves and R. Tuinier, Chem. Commun., 2018, 54, 13351–13354 RSC.
- N. F. Gajardo-Parra, V. P. Cotroneo-Figueroa, P. Aravena, V. Vesovic and R. I. Canales, J. Chem. Eng. Data, 2020, 65, 5581–5592 CrossRef CAS.
- B. E. Gurkan, J. C. D. L. Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- G. Latini, M. Signorile, V. Crocellà, S. Bocchini, C. F. Pirri and S. Bordiga, Catal. Today, 2019, 336, 148–160 CrossRef CAS.
- M. Pan, Y. Zhao, X. Zeng and J. Zou, Energy Fuels, 2018, 32, 6130–6135 CrossRef CAS.
- M. Chen, S. Zhang, Q. Mei, H. Zhou and D. Yang, Chem. Commun., 2025, 1–4 Search PubMed.
- S. Oh, O. Morales-Collazo and J. F. Brennecke, J. Phys. Chem. B, 2019, 123, 8274–8284 CrossRef CAS PubMed.
- S. Seo, M. A. Desilva and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 14870–14879 CrossRef CAS PubMed.
- B. Yoon, S. Chen and G. A. Voth, J. Am. Chem. Soc., 2024, 146, 1612–1618 CrossRef CAS PubMed.
- W. Kong, X. Zhong, K. Yang, Z. Dong, T. Song, M. Fang, T. Wang, S. Zhang, W. Li and S. Li, Chem. Eng. J., 2025, 504, 1–15 CrossRef.
- J. Ma, Y. Du, M. Liu, Y. Zhou, X. Wang, B. Wang, J. Zhu and M. Zhu, J. Phys. Chem. B, 2024, 372–384 Search PubMed.
- A. Al-Bodour, N. Alomari, S. Aparicio and M. Atilhan, J. Mol. Liq., 2023, 390, 1–13 CrossRef.
- B. Yoon and G. A. Voth, J. Am. Chem. Soc., 2023, 145, 15663–15667 CrossRef CAS PubMed.
- Y. Y. Lee, D. Penley, A. Klemm, W. Dean and B. Gurkan, ACS Sustainable Chem. Eng., 2021, 9, 1090–1098 CrossRef CAS.
- X. Li, M. Hou, Z. Zhang, B. Han, G. Yang, X. Wang and L. Zou, Green Chem., 2008, 10, 879–888 RSC.
- S. Onofri and E. Bodo, J. Phys. Chem. B, 2021, 125, 5611–5619 CrossRef CAS PubMed.
- W. T. Zheng, K. Huang, Y. T. Wu and X. B. Hu, AIChE J., 2018, 64, 209–219 CrossRef CAS.
- B. Li, Y. Fu, Z. Yang, S. Dai and D. E. Jiang, J. Phys. Chem. B, 2024, 10207–10213 CrossRef CAS PubMed.
- DAC-Screening-Data-Repository, 2025, https://github.com/getman-research-group/DAC_Screening_Data_Repository, [Online; accessed 2025-02-22].
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr , J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision C.02, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, W. Hujo and B. Kirchner, Phys. Chem. Chem. Phys., 2012, 14, 4875–4883 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 1–31 Search PubMed.
- J. Lym, G. R. Wittreich and D. G. Vlachos, Comput. Phys. Commun., 2019, 106864 Search PubMed.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- X. Wu, J. Ruan, K. Wang, X. Zhang, M. Ma, L. Chen and Z. Qi, Green Chem. Eng., 2024, 1–13 Search PubMed.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- I. M. Bernhardsen, I. R. Krokvik, C. Perinu, D. D. Pinto, K. J. Jens and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2018, 68, 68–76 CrossRef CAS.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- U. L. Abbas, Q. Qiao, M. T. Nguyen, J. Shi and Q. Shao, AIChE J., 2022, 68, 1–15 CrossRef.
- D. K. Panda and B. L. Bhargava, J. Mol. Graphics Modell., 2022, 113, 1–9 CrossRef PubMed.
- K. Dong and S. Zhang, Chem. – Eur. J., 2012, 18, 2748–2761 CrossRef CAS PubMed.
- S. Bhattacharjee, R. Dikki, B. Gurkan and R. B. Getman, J. Mol. Liq., 2024, 410, 125569 CrossRef CAS.
- R. Stefanovic, M. Ludwig, G. B. Webber, R. Atkin and A. J. Page, Phys. Chem. Chem. Phys., 2017, 19, 3297–3306 RSC.
- Y. Zhang, D. Poe, L. Heroux, H. Squire, B. W. Doherty, Z. Long, M. Dadmun, B. Gurkan, M. E. Tuckerman and E. J. Maginn, J. Phys. Chem. B, 2020, 124, 5251–5264 CrossRef CAS PubMed.
- C. Zhu, H. Wood, P. Carbone, C. D'Agostino and S. P. de Visser, Phys. Chem. Chem. Phys., 2025, 2381–2394 RSC.
- Z. Wang, M. Chen, B. Lu, S. Zhang and D. Yang, ACS Sustainable Chem. Eng., 2023, 11, 6272–6279 CrossRef CAS.
- Z. Yang and S. Dai, Green Chem. Eng., 2021, 2, 342–345 CrossRef.
- R. P. Lively and M. J. Realff, AIChE J., 2016, 62, 3699–3705 CrossRef CAS.
- Y. Y. Lee, K. Edgehouse, A. Klemm, H. Mao, E. Pentzer and B. Gurkan, ACS Appl. Mater. Interfaces, 2020, 12, 19184–19193 CrossRef CAS PubMed.
- J. Cheng, C. Wu, W. Gao, H. Li, Y. Ma, S. Liu and D. Yang, Int. J. Mol. Sci., 2022, 23, 1–7 Search PubMed.
- M. H. Abdellah, A. Kiani, W. Conway, G. Puxty and P. Feron, Chem. Eng. J., 2024, 481, 1–13 CrossRef.
- M. Zanatta, N. M. Simon, F. P. dos Santos, M. C. Corvo, E. J. Cabrita and J. Dupont, Angew. Chem., Int. Ed., 2019, 58, 382–385 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5me00034c |
| This journal is © The Royal Society of Chemistry 2025 |