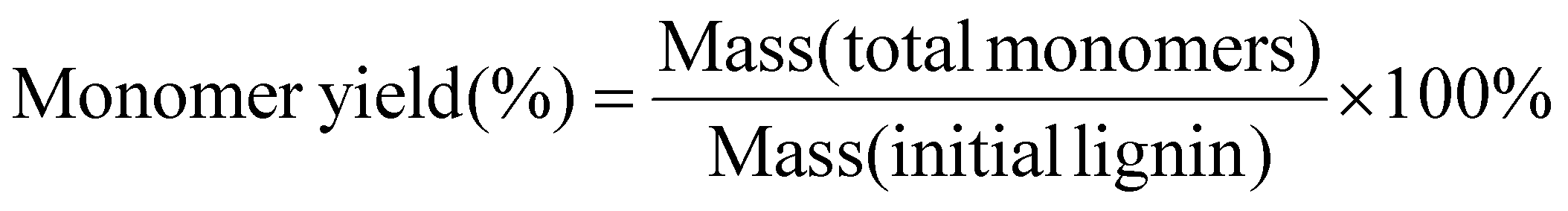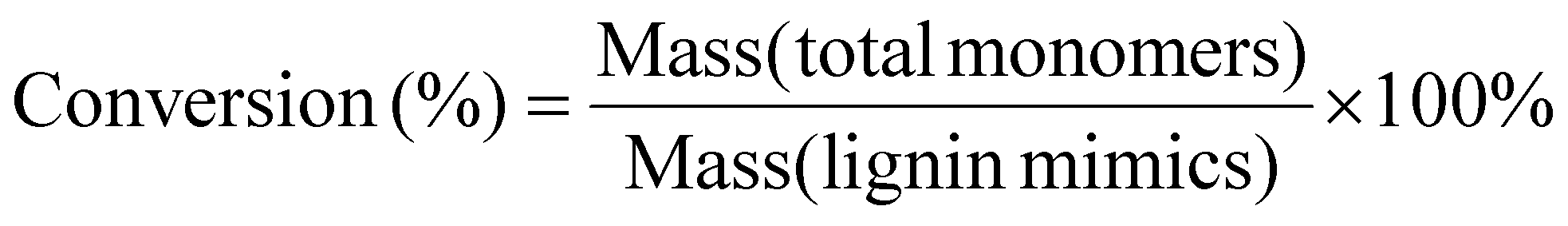DOI:
10.1039/D4GC05467A
(Paper)
Green Chem., 2025,
27, 5091-5103
Insights into the reductive catalytic deconstruction of lignin over ultralow-loading palladium–zinc catalysts derived from zinc imidazolate frameworks†
Received
30th October 2024
, Accepted 28th March 2025
First published on 1st April 2025
Abstract
The development of high-performance noble metal catalysts at the atomic scale for the selective chemical catalytic conversion of lignin into monophenolic compounds is highly desirable but remains a challenge. Herein, we report a single-atom strategy to fabricate a highly active and stable hydrogenolysis catalyst containing an ultralow Pd content (0.1 wt%) using cobalt and zinc imidazolate frameworks as precursors. The resultant Pd–Zn@NC catalyst exhibits outstanding activity in the reductive catalytic deconstruction of lignin into aromatic compounds. The catalyst affords a phenol monomer yield of up to 49.6%, which surpasses that of commercial Pd/C. Notably, it demonstrates high selectivity towards unsaturated allyl monomers, reaching a maximum of 91% under optimized conditions. Mechanistic studies using β-O-4′ mimics reveal that the high dispersion of Zn contributes to the dissociation of hydroxyl groups, while the atomically dispersed Pd significantly enhances the hydrogenation performance. The synergistic interactions between Pd and Zn active sites activate the C–O bonds, thereby enhancing reductive aryl-ether scission in lignin.
Green foundation
1. This work reports an ultralow-loading palladium–zinc catalyst derived from zinc imidazolate frameworks, which exhibits outstanding performance in the reductive catalytic deconstruction (RCD) of lignin into aromatic compounds.
2. The selective value addition of lignin to fine chemicals via catalytic hydrogenolysis plays a crucial role in sustainable green development. The resultant Pd–Zn@NC catalyst outperformed the commercial Pd/C catalyst regarding the atomic economy of Pd and selectivity. This work sheds light on the rational design of cost-effective, high-performance, and highly stable catalysts for lignin hydrogenolysis.
3. Further efforts should be devoted to scaling up the RCD process for the production of aromatics, making lignin commercially viable.
|
Introduction
Lignocellulosic biomass is a readily available and eco-friendly source of carbon for producing alternative, sustainable feedstock for energy, chemicals, and materials.1,2 One of the main components of lignocellulosic biomass is lignin, a complex oxygenated aromatic polymer that makes up 15–30% of lignocelluloses.3–5 However, the complexity of its structure has been a significant obstacle to its large-scale utilization in biorefineries.2,6,7 Reductive catalytic deconstruction (RCD) is a promising technology that can extract and decompose lignin while keeping the polysaccharides as solid residues. RCD, which is known as the “lignin-first” strategy, helps overcome biomass recalcitrance to make the biorefinery more efficient.6,8–13
The metal catalyst utilized in RCD plays a dual role in facilitating the cleavage of β-O-4′ linkages in lignin and subsequently preventing the re-polymerization of the resulting unstable monomer products through hydrogenation or deoxygenation.9,14–16 This approach results in a higher yield of phenolic monomers. To enhance the selectivity of target aromatic phenols, researchers have explored a range of catalysts.17–20 Precious metal catalysts have demonstrated superior performance in comparison with inexpensive metal catalysts, such as Mo,21,22 Ni,23,24 and Cu.25 However, the limited availability and high cost of these precious metals pose challenges for their widespread use in industrial production.26–29 As a result, there is growing interest in the development of low-loading and highly dispersed metal catalysts for lignin depolymerization as a more viable alternative.
Previous studies have reported the use of various catalysts for the depolymerization of lignin in the production of phenolic monomers.30 For example, Song et al.31 utilized a RuN/ZnO/C catalyst (0.2 wt% Ru), which achieved a yield of 46.4 wt% monophenols from birch wood. Similarly, Kim et al.26 employed a Pd0.25/CNx catalyst (0.25 wt% Pd), which resulted in a yield of 52.7 C% monophenolic products from birch. Herein, we fabricated a single-atom catalyst with an ultralow Pd content (Pd–Zn@NC) by doping trace amounts of noble metal Pd onto a metal–organic framework (MOF)32 and successfully applied it to the reductive catalytic hydrogenolysis of lignin. Under optimized reaction conditions, wood chips were converted into phenolic monomers with yields close to the theoretical maximum (pine at 16.5 wt%; eucalyptus at 49.6%). The hydrogenolysis reaction of β-O-4′ model compounds clarified the reaction pathway of lignin hydrogenolysis catalyzed by Pd–Zn@NC. Further mechanistic studies revealed that the high dispersion of Zn facilitated the dissociation of hydroxyl groups, while the atomically dispersed Pd significantly enhanced the hydrogenation performance. The synergistic effects between Pd and Zn active sites activated the scission of C–O bonds. This study developed and improved ultralow-loading and highly dispersed Pd metal catalysts toward lignin depolymerization, providing a strategy for the green and sustainable valorization of lignin (Fig. 1).
 |
| | Fig. 1 Schematic diagram of coniferous lignin and monomeric phenol structures from lignin-preferred biomass deconstruction. | |
Experimental
Materials and chemicals
All commercially available chemical reagents were used without further purification unless otherwise noted. Methanol and 1,3,5-benzenetricarboxylic acid were purchased from Energy Chemical, China. Pine (Pinus yunnanensis, 5 years old), birch (Betula alnoides, 6 years old), and eucalyptus (Eucalyptus robusta, 5 years old) were used as raw materials in this work, which were extracted, crushed, and screened into powders in the size of 40–60 meshes (0.5–1.0 mm), and dried under vacuum at 50 °C for 24 h before use. Double enzymatic lignin (DEL) was prepared from the ball-milled wood meal as previously described.26 The chemical compositions of the raw materials were analyzed according to analytical procedures (NREL/TP-510-42618).17 Dimeric lignin model compounds 1 and 4, as well as trimeric model compound 6, were prepared by following reported procedures.33
Preparation of the Pd–Zn@NC catalyst
The solubilization of 34 mg of potassium iodide (KI) and 100 mg of polyvinylpyrrolidone (PVP, Mw = 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was achieved by heating a 10 mL solution of formamide to 120 °C. Next, 59 mg of sodium palladium chloride powder was added to the mixture while stirring for 15 min. Following this, the mixture was cooled to room temperature, and 120 mL of acetone was added. The resulting dispersion was then collected through centrifugation at 10000 rpm for 10 min. The obtained palladium nanoparticles (Pd NPs) were dispersed in 21 mL of a mixed solvent consisting of a 4:1 volume ratio of N,N-dimethylformamide (DMF) to methanol. Additionally, 4276 mg of zinc nitrate hexahydrate (Zn(NO3)2·6H2O) was dissolved in 60 mL of the same mixed solvent (DMF:MeOH, 4:1) in flask A. After that, 2 mL of the above solution of palladium nanoparticles was added to flask A with a dropper. In flask B, 4664 mg of 2-methylimidazole was dissolved in 80 mL of the same mixture under sonication for 5 min, resulting in a clear solution. Then, the solution from flask B was slowly poured into flask A and set to room temperature for 12 h. The product obtained was separated through centrifugation at 10000 rpm, followed by three successive washes with methanol, and finally dried at 60 °C for 3 h. The white Pd-NPs@ZIF-8 powder, obtained after grinding, was transferred to a ceramic boat and placed in a tube furnace. The samples were then heated to 120 °C for 1 h to eliminate any moisture present and subsequently heated to 900 °C at a rate of 5 °C min−1, held at this temperature for 3 h, and cooled to room temperature at a rate of 5 °C min−1. The black powder obtained was the Pd–Zn@NC catalyst. The resulting material was ready for use without any additional treatment.
000) was achieved by heating a 10 mL solution of formamide to 120 °C. Next, 59 mg of sodium palladium chloride powder was added to the mixture while stirring for 15 min. Following this, the mixture was cooled to room temperature, and 120 mL of acetone was added. The resulting dispersion was then collected through centrifugation at 10000 rpm for 10 min. The obtained palladium nanoparticles (Pd NPs) were dispersed in 21 mL of a mixed solvent consisting of a 4:1 volume ratio of N,N-dimethylformamide (DMF) to methanol. Additionally, 4276 mg of zinc nitrate hexahydrate (Zn(NO3)2·6H2O) was dissolved in 60 mL of the same mixed solvent (DMF:MeOH, 4:1) in flask A. After that, 2 mL of the above solution of palladium nanoparticles was added to flask A with a dropper. In flask B, 4664 mg of 2-methylimidazole was dissolved in 80 mL of the same mixture under sonication for 5 min, resulting in a clear solution. Then, the solution from flask B was slowly poured into flask A and set to room temperature for 12 h. The product obtained was separated through centrifugation at 10000 rpm, followed by three successive washes with methanol, and finally dried at 60 °C for 3 h. The white Pd-NPs@ZIF-8 powder, obtained after grinding, was transferred to a ceramic boat and placed in a tube furnace. The samples were then heated to 120 °C for 1 h to eliminate any moisture present and subsequently heated to 900 °C at a rate of 5 °C min−1, held at this temperature for 3 h, and cooled to room temperature at a rate of 5 °C min−1. The black powder obtained was the Pd–Zn@NC catalyst. The resulting material was ready for use without any additional treatment.
RCD of biomass and β-O-4′ model compounds
In a typical reaction, woody sawdust (50 mg) or β-O-4′ model compounds (12.5 mg), the catalyst (6.5 mg) and MeOH (10 mL) were mixed into a 50 mL stainless steel batch reactor (Parr Instruments Co.), which was then flushed with 3 MPa H2 three times and pressurized with the desired amount of H2 at room temperature. The reaction was carried out at different temperatures for a certain time with magnetic stirring at 800 rpm. After completion, the autoclave was cooled and depressurized carefully. Then the reaction mixture was filtered, and the insoluble fraction was washed with dichloromethane (DCM). Subsequently, the solution fraction was extracted with DCM and the resulting lignin oily product was obtained after removing all volatiles under vacuum conditions. An external standard (1,3,5-trimethoxybenzene) was added to the lignin oil solution in DCM, which was subjected to GC and GC-MS for analysis (Fig. S45–S50†). The identification and quantification of lignin monomers in the oily product were assessed by comparison with authentic samples acquired through commercial purchase or independent synthesis as described previously.26
The oil yields, monomer yields and conversion of lignin were calculated using the following equations:
| |  | (1) |
| | 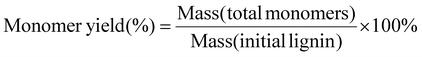 | (2) |
| | 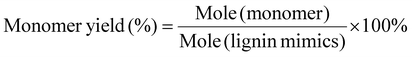 | (3) |
| |  | (4) |
| |  | (5) |
Recycling experiment of Pd–Zn@NC
The solid residue was rinsed with dichloromethane three times and drained after each reaction. To separate the fine black catalyst particles from the brown wood flour residue, the top portion of the solid residue in the funnel was separated and filtered through 100 mesh and 300 mesh sieves. Subsequently, the isolated black catalyst particles were removed under N2 and recalcined at 900 °C for 1 h in a tube furnace.
Results and discussion
Catalyst preparation and characterization
A mixture of zinc nitrate and dimethylimidazole containing Pd NPs was utilized to synthesize the catalyst. As shown in Fig. 2A, this method enabled the formation of a metal–organic framework (MOF) structure that fully encapsulated the Pd NPs. Subsequent sintering under a nitrogen atmosphere resulted in the efficient atomization of metallic Pd within the interior and surface of the MOF carrier,34 yielding a catalyst with a Pd loading of 0.1 wt% (Table S3†) as determined by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis.
 |
| | Fig. 2 Preparation and characterization of the Pd–Zn@NC catalyst. (A) Schematic diagram of catalysts. (B and C) SEM images. (D) TEM image. (E) HAADF-TEM image. (F) HAADF-TEM-EDS elemental mapping results. (G) HAADF-HRTEM image. | |
In order to characterize the catalyst, electron microscopy techniques were employed (Fig. 2). The SEM and TEM images in Fig. 2B–E showed that the structural units of the catalyst were relatively uniform in size and densely arranged, with each ZIF-8 MOF structural unit approximately 100 nm in size. This homogeneous structure resulted in a surface area of 992 cm2 g−1 (Fig. S2 and Table S2†), which was beneficial for exposing more active metal sites in multiphase catalytic reactions and improving the efficiency of the catalyst. Further energy dispersive X-ray (EDX) elemental mapping (Fig. 2F) indicated that ultralow levels of Pd and Zn in the catalyst were uniformly dispersed on the carbon and nitrogen supports. In addition, the presence of single atoms and nanoparticles with metals in the catalyst could be observed in Fig. 2G and S1.†
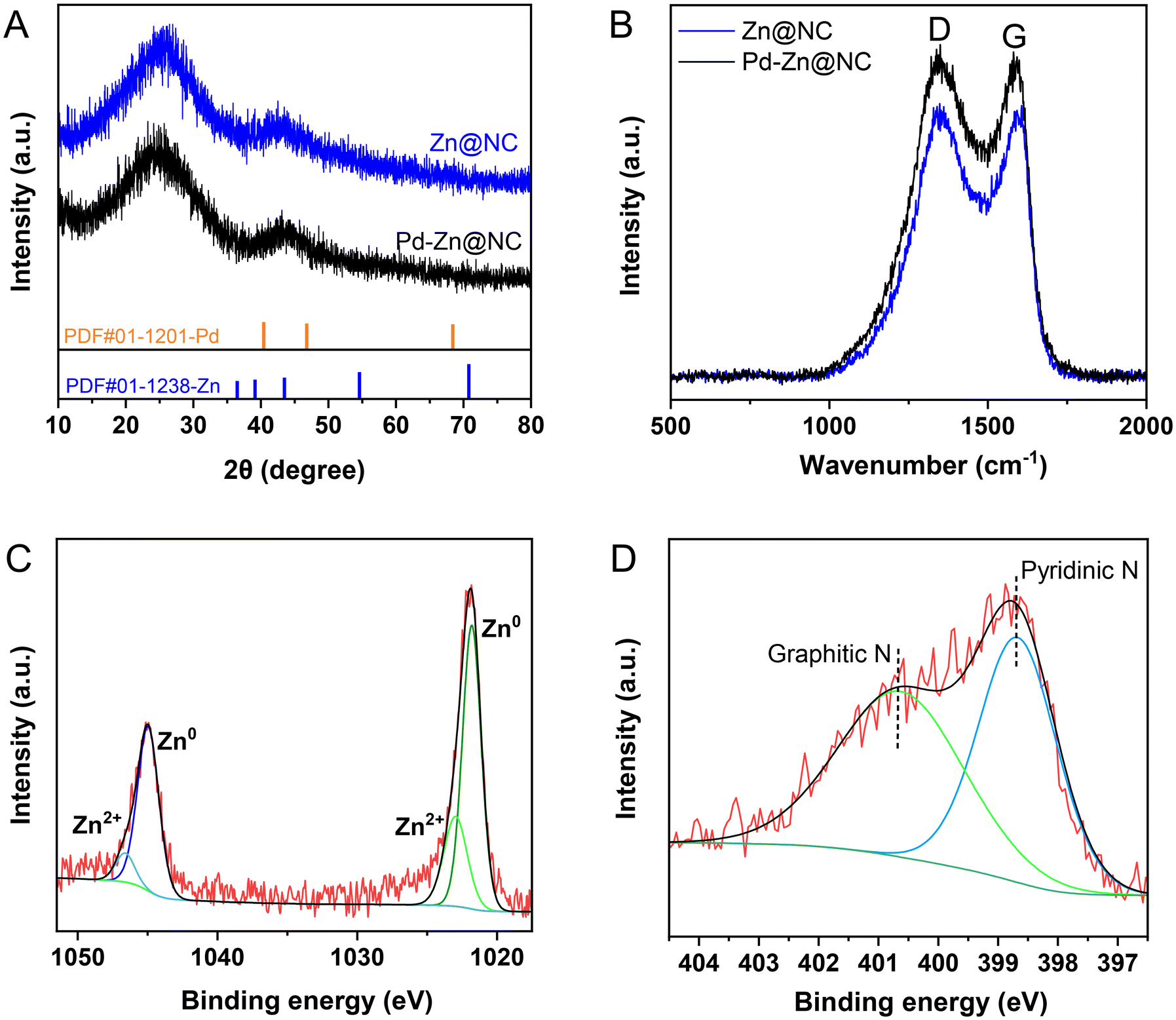
As depicted in Fig. 3A, the diffraction peak at 2θ of 23.5° was characteristic of the lattice plane of (002) and is indicative of the graphitic nature of the carbon support.35 The Pd–Zn@NC catalyst did not exhibit the characteristic diffraction peaks in the presence of metallic Pd and Zn phases. This was likely due to its good dispersion and very low content. The analysis of Fig. 3B showed that the ID/IG of sample Pd–Zn@NC (ID/IG = 1.03) was slightly larger than that of sample Zn@NC (ID/IG = 1.00), indicating that the graphitization degree of Pd–Zn@NC was lower (Fig. S3†).36 The introduction of metal NPs into the precursors of Pd–Zn@NC induced more structural defects in the carbon substrate, thus promoting the exposure of active sites.37 In the X-ray photoelectron spectroscopy (XPS) spectrum (Fig. 3C), two peaks are assigned to Zn 2p1/2 (1045.1 eV) and Zn 2p3/2 (1021.9 eV), which are attributed to the characteristic peaks of Zn. Further fitting revealed that most of them were Zn0, with a small proportion of Zn2+. The high-resolution N 1s spectrum (Fig. 3D) decomposed mainly into graphite N (400.7 eV) and pyridine N (398.7 eV).38 The C–N peak of 285.9 eV was observed in the C 1s spectrum of the catalyst (Fig. S4D†), which verified the successful doping of nitrogen and the presence of defects in the carbon structure. Electrons could be transferred from the bound nitrogen atoms to the Zn and Pd metals.39,40 In addition, there was no significant difference in the XPS results, indicating that the valence state of zinc in the catalyst was well maintained before and after the reaction (Fig. S5 and S6†). Moreover, the two peaks of Pd at 342.4 eV (3d3/2) and 337.2 eV (3d5/2) were observed with weak signals in the spectra (Fig. S4†), which was due to the very low loading of Pd and its good dispersion.
 |
| | Fig. 3 The XRD patterns (A), Raman spectra (B), and XPS spectra (C and D). | |
To further determine that most of the Pd was in the form of single atoms and its valence state, we performed extended X-ray absorption fine structure (EXAFS). The characterization of the catalyst using X-ray absorption near-edge structure (XANES) further clarified the coordination environment of Pd in the Pd–Zn@NC catalyst, with comparisons made with PdO, Pd foil and a commercial palladium–carbon (Pd/C) catalyst (Fig. 4A and S8†). The XANES analysis revealed that the valence states of Pd–Zn@NC and Pd/C were intermediate between Pd(0) and Pd(II) with the K-edge absorption position of Pd/C as similar to that of Pd-foil. The Pd–Zn@NC absorption edge was observed to be more similar to that of PdO, indicating that the Pd species had a positive charge.34 This may be ascribed to some electrons migrating from the Pd atom to the carbon and nitrogen carrier (Table S4†), thus increasing the positive charge of Pd. Moreover, the K-edge XANES showed a dominant peak at 1.50 Å and no significant peak appeared at 2.54 Å, which suggested that Pd in the Pd–Zn@NC catalyst was coordinated with N and contained only traces of Pd–Pd bonds (Fig. 4B and C). This indicated that most of the Pd was present in single atomic form. Further quantitative EXAFS analysis was carried out, and the fitting revealed that the Pd atoms were coordinated with about three adjacent N atoms (Table S4†). It could be concluded that the intensity maxima of the WT contour plots at 10 Å and 13.5 Å were attributed to Pd–N and Pd–Pd contributions, respectively (Fig. 4D), suggesting that Pd existed mainly in a single-atom form in Pd–Zn@NC, with only trace amounts of nanoparticles present. This finding was consistent with the observations from STEM and highlighted the dominance of the single atom form of Pd in the catalyst.
 |
| | Fig. 4 Characterization of the catalysts. (A) The Fourier-transformed k2-weighted EXAFS spectra in R space and the best fits of 5 wt% Pd/C, Pd foil and PdO at R space. (B) XANES spectra of the samples at the Pd L3-edge. (C) EXAFS Fourier transformed (FT) k2-weighted χ(k)-function for the Pd K-edge and its fitting. (D) Wavelet transforms for the k2-weighted Pd K-edge EXAFS signals in Pd–Zn@NC. | |
Catalytic performance of Pd–Zn@NC for the RCD of lignin
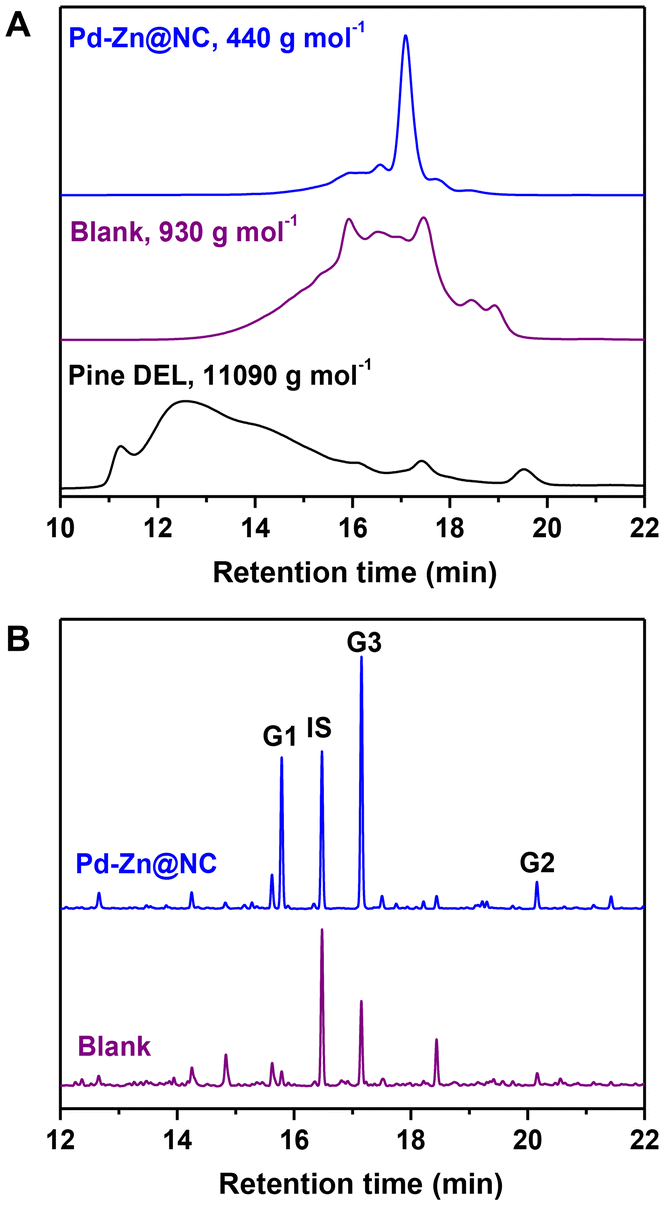
In a proof-of-concept experiment, pine wood with 27.3 wt% lignin content was used as the reactant (Table S1†). 6.5 mg of catalyst was added to MeOH with 3 MPa (25 °C) of hydrogen at 240 °C for 4 h in an autoclave. Gel permeation chromatography (GPC) analysis of the lignin oil obtained after the reaction showed a weight average molecular weight (Mw) of 440 g mol−1 (Fig. 5A) in comparison with phenolic monomers, dimers and oligomers. This was considerably less than the lignin oil derived from heating without the catalyst (Mw 930 g mol−1) and both fractions have a moderate polydispersity (PDI) index in the range of 1.55–1.79. DEL was prepared from pine wood (Mw 11090 g mol−1, PDI 5.93). The introduction of the catalyst resulted in a significant reduction in both the PDI and Mw of the lignin. Additionally, the PDI and Mw of the lignin oil after the Pd–Zn@NC reaction were found to be smaller as compared to that of Zn@NC. These findings indicated a more selective and controllable hydrogenolysis process enabled by this catalyst, resulting in a more uniform particle size distribution of the degradation products. As a consequence, these results highlighted the high catalytic reactivity of Pd–Zn@NC toward RCD of lignin. Subsequent to the reaction, the phenolic monomer products were qualitatively analyzed using GC-MS and quantified with GC. The results were surprising, with an overall yield of 16.5 wt% of phenolic monomers, including about 12 wt% of the double-bonded product (isoeugenol, G3). The selectivity of G3 was more than 70%, and the highly selective production of unsaturated monomers under hydrogenolysis conditions is relatively rare (Fig. 5B).
 |
| | Fig. 5 Characterization of pine lignin and oily products. (A) Molecular weight distribution of pine lignin and oily products obtained from the Pd–Zn@CN-catalyzed hydrogenolysis of pine sawdust via GPC analysis. (B) GC chromatogram of the monomers obtained from the Pd–Zn@CN-catalyzed hydrogenolysis of pine sawdust. | |
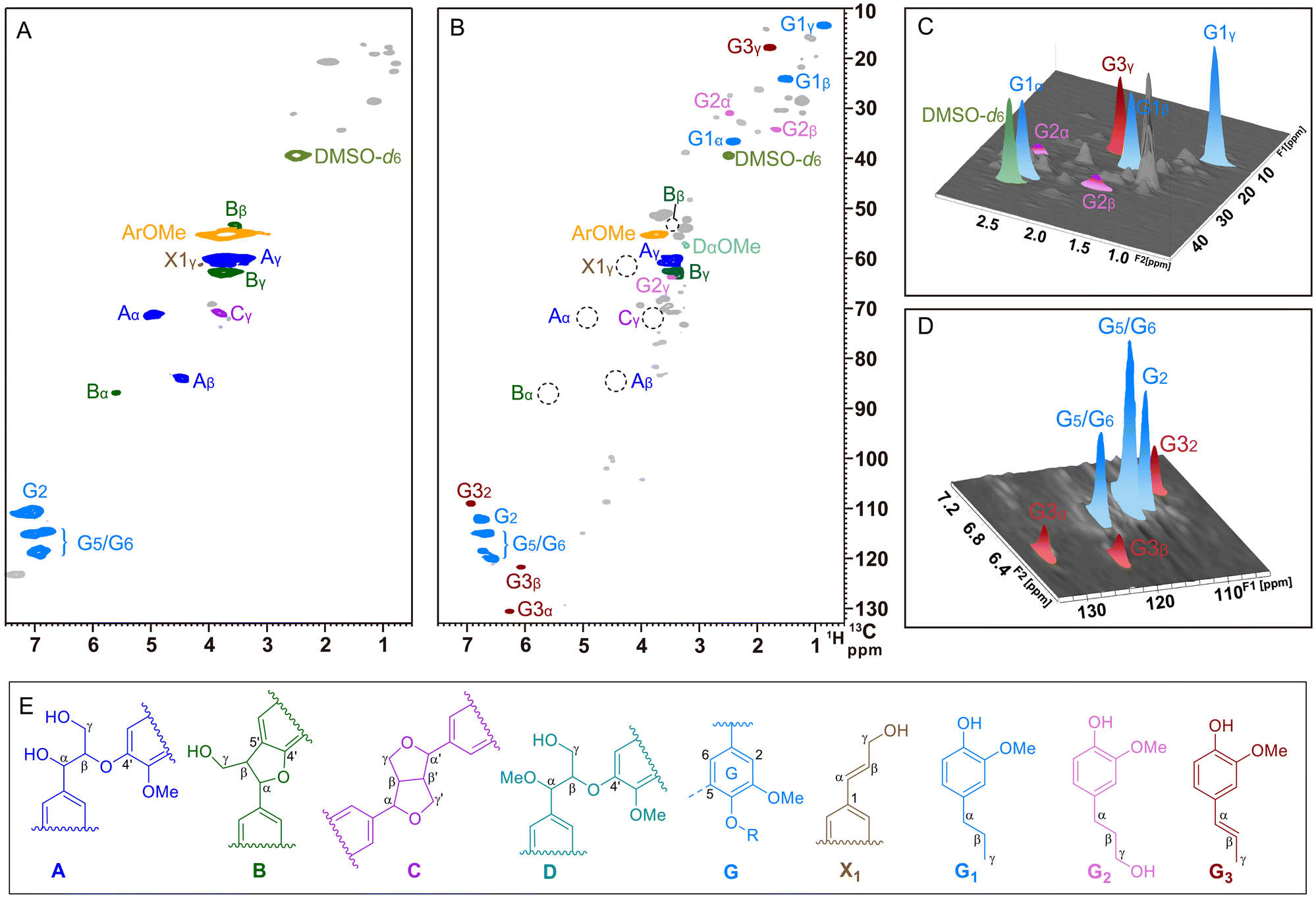
We further analyzed double enzymatic lignin (DEL) and lignin oil by 2D HSQC NMR spectroscopy (Fig. 6), which clearly revealed that the Aα and Aβ peaks attributed to β-O-4′ disappeared after the reaction (71.3/4.96 ppm, 83.9/4.45 ppm). Also, the disappearance of the 4-O-5′, α-O-γ′ and γ-OH in the spectrum indicated the occurrence of C–O bond breakage, which proved that the lignin was effectively cleaved. Notably, it was observed the appearance of the Dα-OMe molecule, associated with the etherification of Aα-OH, was in good accordance with our pervious report.25 In the side-chain region, a family of cross-peaks located at δC/δH 17.3/1.78, 121.9/6.08 and 130.8/6.26 ppm (labeled dark red) belongs to the propylene chain in G3.25,41 In addition, the cross-peaks at δC/δH 36.9/2.42, 24.1/1.51 and 14.1/0.87 ppm are assigned to the propyl chain of G1 (labeled light blue), and δC/δH 31.0/2.48, 34.1/1.67 and 63.9/3.48 ppm are the signals corresponding to the propanol side-chain of G2 (labeled pink). Notably, the signals of G1 and G3 were significantly higher than that of G2 in the 3D plot (Fig. 6C), and the characteristic peak belonging to G3 could clearly be observed in the aromatic ring region (Fig. 6D), which were consistent with the GC analysis. This distribution of products might be due to the high reactivity of γ-OH products on the highly dispersible Pd, together with the contribution of Zn to the hydrogenolysis of alcohols, as compared to that of commercial Pd/C.26,32
 |
| | Fig. 6 Characterization of pine lignin and oily products. (A) 2D HSQC NMR spectrum of double enzymatic lignin (DEL) isolated from pine wood (dissolved in DMSO-d6). (B) 2D HSQC NMR spectrum of the oily product from the Pd–Zn@CN reaction (dissolved in DMSO-d6). (C and D) 3D version of the HSQC end-chain and aromatic regions. (E) Main structures presented in the 2D HSQC NMR spectra: (A) β-O-4′ alkyl-aryl ether, (B) β-5′ phenylcoumaran, (C) β–β′ resinol, (D) α-OH with methylated β-O-4′ alkyl-aryl ether, (G) guaiacyl units, (X1) cinnamyl alcohol end-groups, (G1) dihydroeugenol, (G2) dihydroconiferyl alcohol, and (G3) isoeugenol. | |
Optimization of the reaction conditions for the RCD of lignin
To elucidate the catalytic performance of Pd–Zn@NC, we systematically investigated the influence of different parameters, including solvent, catalyst dosage, hydrogen pressure, temperature, and reaction time, on the yield of monophenols. Pine was utilized as the substrate for these investigations, and comparisons were made with different catalysts. In Fig. 7A, the reaction without a catalyst resulted in a limited amount of the G3 monomer product. This suggested that double-bonded phenolic monomeric products could be formed when lignin was depolymerised in this high-temperature system. However, upon the addition of the Pd–Zn@NC catalyst to the reaction system, the total monomer yield significantly increased to 16.5 wt%. In order to examine the impact of metallic palladium, we synthesized the Zn@NC catalyst without incorporating Pd NPs and the resulting total monomer yield was 12.3 wt%. Notably, the yield of G3 was almost identical to that achieved with Pd–Zn@NC catalysis. However, the yield of G1 decreased from 4.1 wt% to 0.6 wt%, and no G2 monomer was produced in this case (Fig. S9–11 and Table S5†). In addition, the GPC analysis of the lignin oil (Mw 480 g mol−1, Fig. S11†) showed a significant decrease in molecular weight as compared to that of DEL. This suggested that Zn and Pd may exert a synergistic effect on C–O bond cleavage.11 Subsequently, by increasing the Pd loading in the catalyst and minimising the Zn loading in the prepared Pd@NC catalyst, the yield of the G1 monomer was further increased, but the overall yield did not significantly change and could reach 16.2 wt%. Under the same reaction conditions, using a commercial Pd/C catalyst with a Pd content of approximately 5 wt%, the yield of total monomer was 14.9 wt%,42 which was lower than that of the Pd–Zn@NC, and the product was predominantly G2. In contrast, a higher amount of monomers without γ-OH was obtained in this reaction system when Pd–Zn@NC was used as a catalyst. This was probably due to the presence of Zn, which promoted the dissociation of hydroxyl groups, resulting in the production of higher amounts of G3 and G1.41 Furthermore, the Pd content in the Pd–Zn@NC catalyst is only 0.1 wt%, which is much lower than the 5 wt% in the commercial Pd/C and Ru/C catalysts.
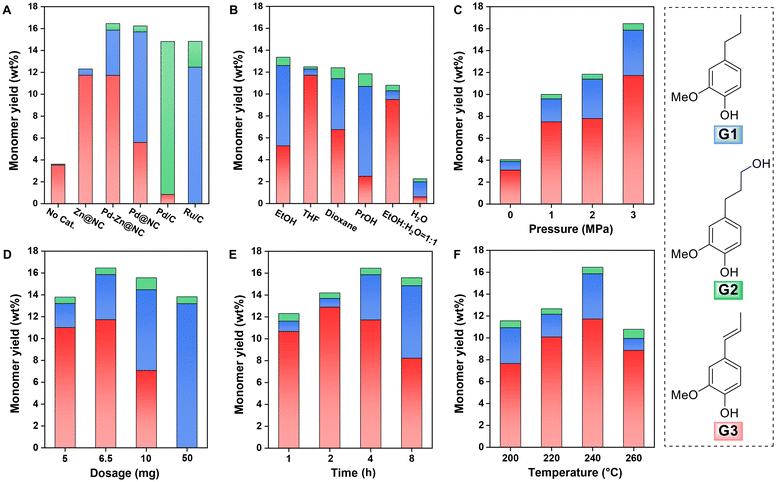 |
| | Fig. 7 Optimization of the reaction conditions for the RCD of lignin. (A) Catalytic performance of different catalysts for the RCD of lignin. Influences of solvent (B), catalyst dosage (C), hydrogen pressure (D), temperature (E) and time (F) on the catalytic hydrogenolysis of pine sawdust over a Pd–Zn@NC catalyst. Reaction conditions: (A) pine sawdust (50 mg), catalyst (6.5 mg), solvent (10 mL), H2 (3 MPa), 240 °C, 4 h. (B) Pine sawdust (50 mg), Pd–Zn@NC (5 mg), solvent (10 mL), H2 (3 MPa), 240 °C, 4 h. (C, D, E and F) under the aforementioned conditions of A, pressure, catalyst dosage, time, and temperature were varied individually. | |
The influences of solvent (B), catalyst dosage (C), hydrogen pressure (D), temperature (E) and time (F) on the catalytic hydrogenolysis of pine sawdust over a Pd–Zn@NC catalyst were investigated. Reaction conditions: (A) pine sawdust (50 mg), catalyst (6.5 mg), solvent (10 mL), H2 (3 MPa), 240 °C, 4 h. (B) Pine sawdust (50 mg), Pd–Zn@NC (5 mg), solvent (10 mL), H2 (3 MPa), 240 °C, 4 h. (C, D, E and F) under the aforementioned conditions of A, pressure, catalyst dosage, time, and temperature were varied individually.
Subsequently, we explored the impact of various solvents on monomer yields (Fig. 7B). As compared to methanol, the yields with the selected solvents were reduced to varying degrees. This could be attributed to the higher polarity of certain solvents, such as methanol, which enhances their capacity to penetrate the lignocellulosic matrix and extract lignin more effectively, thereby facilitating non-catalytic delignification. During the reductive fractionation process, alcohol solvents like MeOH may serve as hydrogen donors, thereby facilitating the reduction process (Fig. S13†).33 Consequently, lignin decomposition and subsequent fragmentation processes could potentially benefit from solvents with increased polarity. Moreover, the use of solvents such as THF and dioxane, which exhibit high solubility for lignin fragments, may further contribute to enhanced lignin decomposition to obtain higher monomer yields (Fig. S12, S13 and Table S6†). Among them, the catalyst was found to retain its high selectivity for G3 during the reaction in THF and an ethanol/water mixture. These results suggested that the catalytic hydrogenolysis reaction of pine lignin by Pd–Zn@NC could be performed in different polar solvents, demonstrating some solvent polarity adaptability. We then carried out a screening process to determine the optimum hydrogen pressure as shown in Fig. 7C, S17 and Table S9,† which was found to be 3 MPa. In this process, it was observed that the yields of all three monomers increased with hydrogen pressure, and in various organic solutions, the total yield of monomers obtained from the RCD reaction was more than 12 wt%. These results suggested that hydrogen may be the main source of hydrogen supply for the hydrogenation reactions in this system.42
The effect of varying the catalyst dosage on the catalytic reaction was then investigated (Fig. 7D). At a catalyst dosage of 6.5 mg, both the total monomer yield and G3 yield reached their maximum values (16.5 and 11.7 wt%). However, as the catalyst dosage increased, the content of G3 notably decreased, with the corresponding increase in G1 becoming the predominant product. This observation could be attributed to the elevated catalyst dosage, which augmented the exposure of metal active sites and improved the hydrogenolysis properties, leading to a pronounced reduction of carbon–carbon double bonds during the reaction. Additionally, as the reaction time increased, the proportion of G1 gradually increased, implying that G3 might serve as an intermediate product in the formation of G1 (Fig. 7E, S18, S19, and Table S10†). Upon extending the reaction time to 8 h, there was a slight decrease in yield, and the resulting lignin oil exhibited an increased Mw up to 678 g mol−1. This finding indicated a slight repolymerization phenomenon following the catalytic decomposition process (Fig. S14 and Table S7†). Nevertheless, a similar trend was observed when investigating the effect of reaction temperature. As the temperature gradually increased to 240 °C, methanol attained a supercritical state, enhancing the solubilization of lignin fragments and promoting their interaction with the catalytic active sites, resulting in the attainment of peak yields for G3 and total monomers (Fig. 7F, S15 and Table S8†).24 Remarkably, the obtained products with double bonds exhibited high selectivity (72%), thereby facilitating subsequent derivatization.5 When the temperature reached 260 °C, the yield of total monomers decreased to 10.8 wt% due to the tendency of side chains to undergo C–C condensation during the delignification phase.43 This was evident from the increased Mw (470 g mol−1) of the lignin oil with a PDI of 1.33 (Fig. S16†). In contrast, the degradation of carbohydrates with only 61 wt% retention was observed under severe reaction conditions. To sum up, the optimized conditions for Pd–Zn@NC-catalyzed hydrogenolysis of pine sawdust were as follows: pine sawdust (500 mg), Pd–Zn@NC (6.5 mg), MeOH (20 mL), H2 (3 MPa), 240 °C, and 4 h.
Stability and reusability of the Pd–Zn@NC catalyst
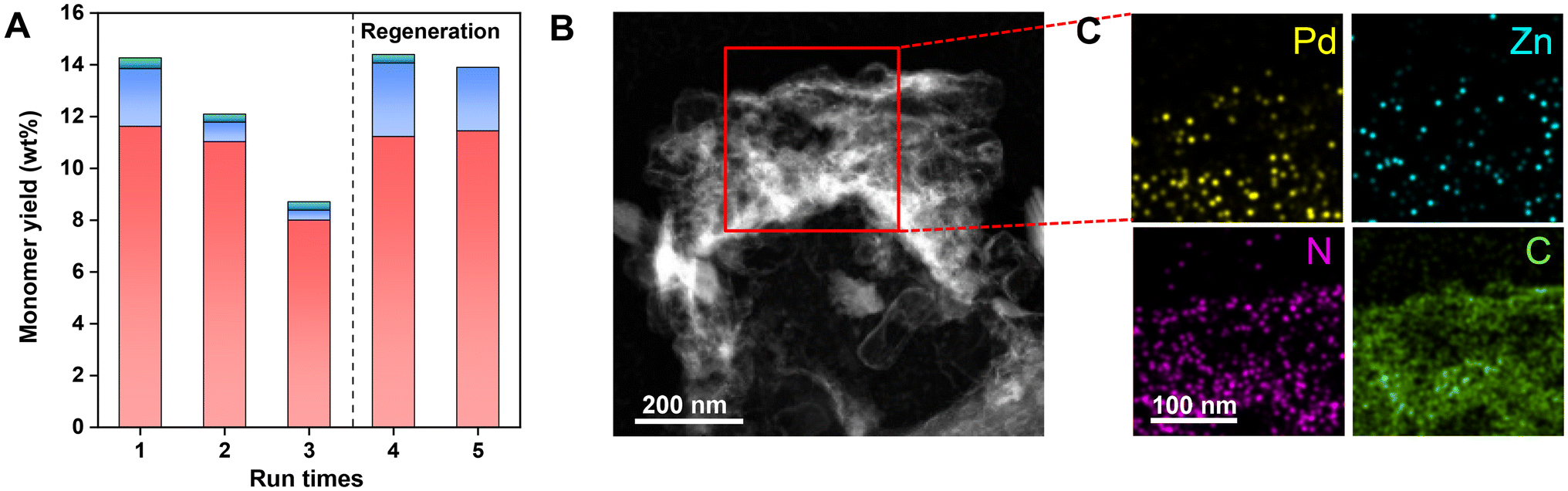
To evaluate the reproducibility of Pd–Zn@NC, we tested its cycling performance by running the reaction with a tenfold increase in substrate (0.5 g sawdust) under the optimised conditions (Fig. 8A, S21, S22 and Table S11†). A significant degree of inactivation was observed in the third cycle, with only 8.7 wt% of monophenols being produced. The ICP-OES analysis showed a Pd content of 0.05 wt% and a Zn content of 2.03 wt% (Table S3†), which was lower than the metal content in the fresh catalyst. This may be attributed to the incorporation of fine sawdust during the screening of the catalyst as well as partial loss of the catalyst during the reaction. However, the results from the second cycle reaction demonstrated that the activity of the spent catalyst could still retain 84.6% of its initial activity. This indicated that the loss of catalyst metal was not the primary cause of catalyst deactivation. Fig. 8B revealed that the morphological structure of the catalyst is disturbed. In addition, as shown in Fig. 8C and S7,† the spent catalyst showed considerable aggregates of Pd and Zn. According to XPS spectroscopy, however, the reaction did not significantly change the valence states of the metal and support (Fig. S5†). This suggested that the aggregation of Pd and Zn in the catalyst may result in its deactivation. In this case, the spent catalyst was regenerated through calcination for one hour to redisperse Pd and Zn, followed by a fourth run utilizing the regenerated Pd–Zn@NC. The total monomer yield increased to 14.5 wt%, with a selectivity of 78% for G3, suggesting that the catalytic performance was almost fully restored. This indicated that proper dispersion of the metal in the catalyst and the preservation of its pore structure were essential for the enhancement of its activity. After the fifth run, there was no significant loss of catalyst performance due to catalyst regeneration (Fig. 8A).
 |
| | Fig. 8 Stability and reusability of the Pd–Zn@NC catalyst for the RCD of lignin. (A) Reusability of the Pd–Zn@NC catalyst for the RCD of lignin. HAADF-STEM image (B) and elemental mapping (C) of Pd, Zn, N, and C of the spent Pd–Zn@NC catalyst. Reaction conditions: pine sawdust (500 mg), Pd–Zn@NC (50 mg), MeOH (20 mL), H2 (3 MPa), 240 °C, and 4 h. | |
Reactivity of β-O-4′ model compounds
To investigate how the Pd–Zn@NC catalyst could be tuned to produce double bond-containing aromatic monomers in catalytic lignin hydrogenolysis, a series of lignin model compounds were designed to test their reactivity with this catalyst (Fig. 9). First, model compounds 1 and 2 were used to separately simulate the non-phenolic and phenolic units exposed during lignin depolymerization. In the product of the hydrolysis catalyzed by the Zn@NC catalyst of model 1 (reaction (a)), 3,4-dimethoxypropene (D3, 28%) and a small amount of 3,4-dimethoxypropylbenzene (D1, 2%) were observed. However, using the Pd–Zn@NC catalyst (reaction (b)), it was found that the yield of product D3 did not change significantly (28% to 26%). The yield of product D1 increased to 34%, and 4% of 3,4-dimethoxypropanol (D2) was observed. Among the products, the propyl product has the highest yield, which differs from the distribution of RCD products from natural biomass. Because of the different chemical bonds and structures present in natural lignin, the interactions with the catalyst could affect the resulting different product yields. Next, the phenolic model compound 2 was reacted with the Zn@NC catalyst, yielding 52% guaiacol and 24% G3 (reaction (c)). The conversion of guaiacol was slightly reduced from 52% to 51% when switching to the Pd–Zn@NC catalyst, while the yield of lignin monomers was significantly increased. The yield of G3 increased to 42% and a small amount of G2 (3%) was present (reaction (d)). However, the presence of G1 was not observed. The distribution of the products differed from that of model compound 1 and natural lignin. This may be due to the fact that the temperature of reaction (d) was only 180 °C, which did not reach the required conversion temperature. Then, it was observed that G3 was converted to G1 with a high conversion rate over the Pd–Zn@NC catalyst at 240 °C (reaction (j)). When the temperature was increased to 260 °C, G3 disappeared from the product and was completely converted to G1, indicating that G3 was an intermediate of G1 in this reaction process (reaction (k), Fig. S31†). The results of the reactions of model compounds 3 and 4, which lack γ-CH2OH, showed that the C–O bond could still be cleaved in the absence of γ-OH, as compared to the direct addition of Zn2+.32,44 However, the conversion was significantly lower at 180 °C. This suggested that the absence of γ-OH affected the cleavage of the β-O-4′ structural unit on Pd–Zn@NC, making the cleavage more difficult. In the above comparison of the two types of model compounds, it was found that the non-phenolic model compounds were both converted at lower rates than the phenolic model compounds of the same type. However, the catalyst's ability to effectively cleave the β-O-4′ motif was hindered when the oxygen-containing groups on benzene were removed (model compound 5). This may be due to their ability to donate electrons, such as hydroxyl and methoxy, to the benzene ring, which was associated with a lower energy cost for the scission of β-O-4′ mimics. Finally, the product distribution from the hydrogenation of the trimer model compound 6 was similar to that of biomass, further validating the above conclusions. The results of the above two catalysts confirmed that the Zn@NC catalyst showed a certain promoting effect on the C–O bond cleavage in the lignin model compounds. Furthermore, the addition of Pd to the catalyst not only greatly improved the hydrogenation activity of the lignin model compounds but also changed the selectivity of the product.
 |
| | Fig. 9 Reactivity of β-O-4′ lignin model compounds. | |
Mechanistic analysis
In the above study of the synergistic interactions of Pd and Zn (Fig. 10 and S27–37†), we speculated that Zn actually plays a role in facilitating the preferential breaking of the C–OH bond (path 1). The Pd in the catalyst then participated in the further hydrogen supply, allowing the intermediate product to be reduced (path 2). Certainly, since the single Pd atom was mainly involved in the reaction as a hydrogenating agent, it could gradually transfer the hydrogen atom to the reactants during the reaction to promote the cleavage of the C–O bond. It is also possible that part of the G1 product of Cα–Cβ is formed directly at the same time as the C–O bond is cleaved (path 4). In order to investigate the effect of Pd particle size on the products, the reaction was carried out using a mixed catalyst of Zn@NC and Pd/C. The results were similar to the product distribution of Pd–Zn@NC. However, due to the predominance of metal particles in the commercial Pd/C, the product contained slightly more G2 (path 3, Fig. S9†), which is consistent with previous reports.26 The total yield was 14.0 wt% (Fig. S9 and Table S5†), slightly lower than that of Pd/C. This could indicate that the dispersed Zn participated in and promoted the C–O bond cleavage and that the highly dispersed Pd metal in the Pd–Zn@NC catalyst achieved a higher reactivity, which could still give a higher monomer yield with a significantly lower noble metal Pd loading. In conclusion, the mechanistic studies using β-O-4′ mimics reveal that the high dispersion of Zn significantly contributes to the dissociation of hydroxyl groups, while the atomically dispersed Pd sites significantly enhance the hydrogenation performance. Benefiting from the electron transfer effects between Pd–Zn nanoparticles, this synergistically activates the C–O bond, thus improving reductive aryl-ether scission in lignin.
 |
| | Fig. 10 Proposed reaction pathways for the catalytic hydrogenolysis of lignin in MeOH over a Pd–Zn@NC catalyst. | |
Scope of the catalyst
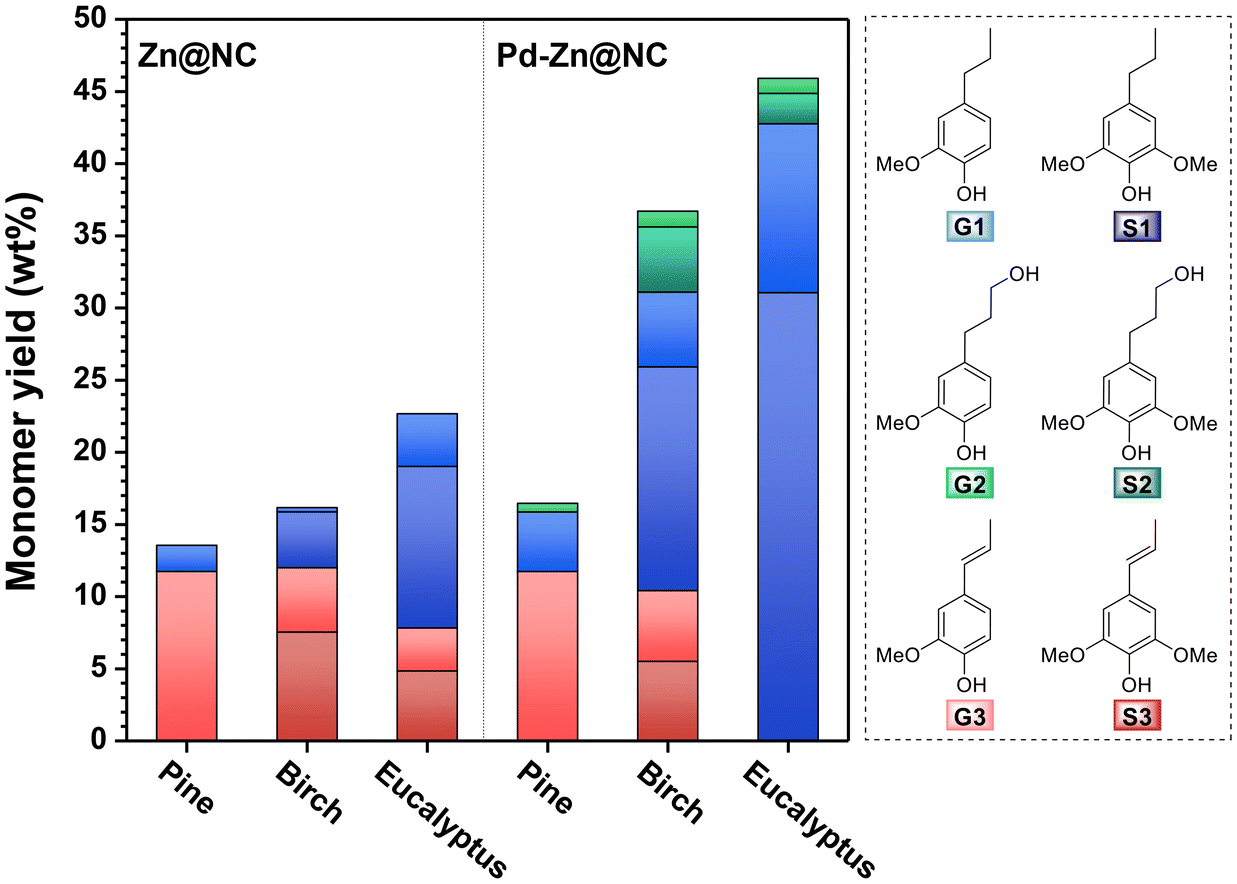
Finally, considering that the catalytic role of Pd in the hydrogenolysis of softwood lignin has not been fully clarified, we conducted RCD reactions on hardwoods with Zn@NC and Pd–Zn@NC to further verify the role of Pd in the catalyst. We first examined the hydrogenolysis of hardwood lignin under optimal reaction conditions for softwood, with a monomer yield of only 25 wt% after 4 h of reaction (Fig. S38, S39 and Table S12†). Considering that the β-O-4′ content in hardwood lignin is significantly higher than in softwood, complete hydrogenolysis requires a potentially longer reaction time. In addition to this, the higher proportion of S units in hardwood lignin and the occupation of the fifth position of its benzene ring by methoxy can effectively inhibit condensation. Extending the reaction time does not result in lower yields due to condensation side reactions as in the case of softwood. Therefore, we prolonged the reaction time and obtained the results as depicted in Fig. 11. Specifically, the Zn@NC catalyst gave a monomer yield of 16.2 wt% after 8 h of Zn@NC-catalyzed hydrogenolysis of birch, while the Pd–Zn@NC catalyst gave a higher monomer yield of 36.7 wt% (Fig. S42 and Table S14†). We further extended the reaction time for eucalyptus to 12 h over the Pd–Zn@NC catalyst, and the monomer yield increased from 22.7 wt% to 49.6 wt% as compared to the Zn@NC catalyst without added palladium metal (Table S14†). Additionally, the heavy average molecular weight, as measured by GPC, decreased from approximately 760 g mol−1 to 520 g mol−1 (Fig. S43†), supporting our initial hypothesis that the addition of Pd to the catalyst greatly enhances its catalytic activity. After extending the reaction time, the activity of the Pd–Zn@NC catalyst still outperformed that of the commercial Pd/C (Fig. S44†). The RCD enhancement by Pd was more pronounced for birch and eucalyptus in the Pd-Zn@NC catalysts compared to pine. The monomer yields were higher than previously reported data on catalyzed lignin hydrolysis using comparable ultralow-loading catalysts.29,31 We also observed a gradual decrease in the amount of products containing double bonds with increasing reaction time, which was consistent with our previous conclusion.
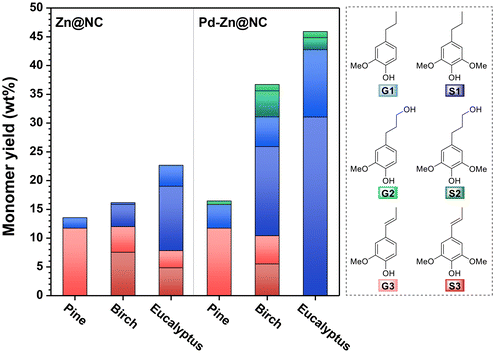 |
| | Fig. 11 Comparison of the yields of major monomers from different wood powders of the reaction over two catalysts: Zn@NC and Pd–Zn@NC. Reaction conditions: sawdust (50 mg), catalyst (6.5 mg), MeOH (10 mL), H2 (3 MPa), 240 °C, and reaction time: pine, 4 h; birch. 8 h; eucalyptus, 12 h. | |
Conclusions
In summary, we have developed a highly efficient and stable Pd–Zn@NC catalyst with an ultralow Pd amount using ZIF-8 as a precursor. The fabricated Pd–Zn@NC catalyst exhibited high catalytic performance in the RCD of lignin, affording unsaturated monomeric phenols in high yields. The resultant catalyst outperformed the commercial Pd/C catalyst regarding the atomic economy of Pd and selectivity. Notably, extensive experimental and mechanistic studies comprehensively elucidated that the synergistic interactions between Pd and Zn active sites activated the C–O bonds, thereby enhancing reductive aryl-ether scission in lignin. Specifically, the highly dispersed Zn in the catalyst played a crucial role in breaking the β-O-4′, α-OH, and γ-OH bonds, which led to the production of propenyl-substituted products. Trace amounts of Pd, in the form of atomically dispersed Pd catalysts, could greatly enhance the hydrogenation process in the reaction by promoting the C–O bond scission, allowing the saturation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. We anticipate that the insights of this work will pave a new way for further rational design of ultralow loading catalysts for lignin valorization.
C bonds. We anticipate that the insights of this work will pave a new way for further rational design of ultralow loading catalysts for lignin valorization.
Author contributions
Conceptualization, L.-P. X.; formal analysis, Y.-H. L., Q. W., and X.-J. G.; funding acquisition, L.-P. X.; investigation, Y.-H. L. and Q. W.; methodology, Q. W. and W.-Z. Y.; project administration, L.-P. X. and R.-C. S.; resources, L.-P. X.; supervision, L.-P. X. and R.-C. S.; writing – original draft, Y.-H. L. and L.-P. X.; writing – review & editing, Q. W., L.-P. X., and R.-C. S. All authors have given approval for the final version of the manuscript.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22278049 and U24A20559), the Dalian High-Level Talent Innovation Program (2024RJ017), and the Energy Revolution S&T Program of the Yulin Innovation Institute of Clean Energy (YIICEE411060316).
References
- X. Li, Y. Ding, X. Pan, Y. Xing, B. Zhang, X. Liu, Y. Tan, H. Wang and C. Li, J. Energy Chem., 2022, 67, 492–499 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- M. Carrier, A. Loppinet-Serani, D. Denux, J.-M. Lasnier, F. Ham-Pichavant, F. Cansell and C. Aymonier, Biomass Bioenergy, 2011, 35, 298–307 CrossRef CAS.
- W. Schutyser, A. T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- K. Van Aelst, E. Van Sinay, T. Vangeel, E. Cooreman, G. Van den Bossche, T. Renders, J. Van Aelst, S. Van den Bosch and B. Sels, Chem. Sci., 2020, 11, 11498–11508 RSC.
- P. J. Deuss and C. Kugge, Chem Catal., 2021, 1, 8–11 CrossRef CAS.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- S. Qiu, M. Wang, Y. Fang and T. Tan, Sustainable Energy Fuels, 2020, 4, 5588–5594 RSC.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. Huijgen, W. Dehaen and C. Courtin, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst and M. Maesen, Science, 2020, 367, 1385–1390 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- J. Chen, F. Lu, X. Si, X. Nie, J. Chen, R. Lu and J. Xu, ChemSusChem, 2016, 9, 3353–3360 CrossRef CAS PubMed.
- K. M. Torr, D. J. van de Pas, E. Cazeils and I. D. Suckling, Bioresour. Technol., 2011, 102, 7608–7611 CrossRef CAS PubMed.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- T. Renders, S. Van den Bosch, S.-F. Koelewijn, W. Schutyser and B. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS PubMed.
- L. Monsigny, E. Feghali, J.-C. Berthet and T. Cantat, Green Chem., 2018, 20, 1981–1986 RSC.
- L.-P. Xiao, S. Wang, H. Li, Z. Li, Z.-J. Shi, L. Xiao, R.-C. Sun, Y. Fang and G. Song, ACS Catal., 2017, 7, 7535–7542 CrossRef CAS.
- J. Sun, H. Li, L.-P. Xiao, X. Guo, Y. Fang, R.-C. Sun and G. Song, ACS Sustainable Chem. Eng., 2019, 7, 4666–4674 CrossRef CAS.
- X. Liu, H. Li, L.-P. Xiao, R.-C. Sun and G. Song, Green Chem., 2019, 21, 1498–1504 RSC.
- S. Van den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. Courtin and J. Thevelein, Green Chem., 2017, 19, 3313–3326 RSC.
- Q. Wang, L.-P. Xiao, Y.-H. Lv, W.-Z. Yin, C.-J. Hou and R.-C. Sun, ACS Catal., 2022, 12, 11899–11909 CrossRef CAS.
- J. Park, H. S. Cahyadi, U. Mushtaq, D. Verma, D. Han, K.-W. Nam, S. K. Kwak and J. Kim, ACS Catal., 2020, 10, 12487–12506 CrossRef CAS.
- M. V. Galkin and J. S. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- Z. Cao, M. Dierks, M. T. Clough, I. B. Daltro de Castro and R. Rinaldi, Joule, 2018, 2, 1118–1133 CrossRef CAS PubMed.
- S. Wang, K. Zhang, H. Li, L.-P. Xiao and G. Song, Nat. Commun., 2021, 12, 416 CrossRef CAS PubMed.
- T. Renders, W. Schutyser, S. Van den Bosch, S. F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- Z. Liu, H. Li, X. Gao, X. Guo, S. Wang, Y. Fang and G. Song, Nat. Commun., 2022, 13, 4716 CrossRef CAS PubMed.
- I. Klein, C. Marcum, H. Kenttämaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S.-F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis and C. Courtin, Green Chem., 2015, 17, 5035–5045 RSC.
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang and L. Zheng, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- J. K. Kim, J. K. Lee, K. H. Kang, J. C. Song and I. K. Song, Appl. Catal., A, 2015, 498, 142–149 CrossRef CAS.
- L. Yan, Y. Xu, P. Chen, S. Zhang, H. Jiang, L. Yang, Y. Wang, L. Zhang, J. Shen and X. Zhao, Adv. Mater., 2020, 32, 2003313 CrossRef CAS.
- R. Nie, H. Jiang, X. Lu, D. Zhou and Q. Xia, Catal. Sci. Technol., 2016, 6, 1913–1920 RSC.
- L. Wang, C. Yang, S. Dou, S. Wang, J. Zhang, X. Gao, J. Ma and Y. Yu, Electrochim. Acta, 2016, 219, 592–603 CrossRef CAS.
- T. Li, H. Lin, X. Ouyang, X. Qiu and Z. Wan, ACS Catal., 2019, 9, 5828–5836 CrossRef CAS.
- X. Hu, G. Luo, Q. Zhao, D. Wu, T. Yang, J. Wen, R. Wang, C. Xu and N. Hu, J. Am. Chem. Soc., 2020, 142, 16776–16786 CrossRef CAS PubMed.
- W. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han and J. Li, Nat. Nanotechnol., 2018, 13, 856–561 CrossRef PubMed.
- V. Roberts, S. Fendt, A. A. Lemonidou, X. Li and J. A. Lercher, Appl. Catal., B, 2010, 95, 71–77 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2008, 83, 78–87 CrossRef CAS.
- H. Li and G. Song, ACS Catal., 2020, 10, 12229–12238 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Run-Cang
Sun
* and
Run-Cang
Sun