
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriphilic organochalcogen compounds for high-capacity and stable solid-state lithium–sulfur batteries†
Daiwei
Wang
 b,
Mitsutoshi
Otaki
b,
Atif S.
Alzahrani
bc,
Yue
Gao
d,
Jennifer L.
Gray
e,
Qian
Lu
f,
Meng
Liao
b,
Timothy S.
Arthur
g and
Donghai
Wang
*ab
b,
Mitsutoshi
Otaki
b,
Atif S.
Alzahrani
bc,
Yue
Gao
d,
Jennifer L.
Gray
e,
Qian
Lu
f,
Meng
Liao
b,
Timothy S.
Arthur
g and
Donghai
Wang
*ab
aDepartment of Mechanical Engineering, Southern Methodist University, Dallas, TX 75205, USA. E-mail: donghaiwang@smu.edu
bDepartment of Mechanical Engineering, The Pennsylvania State University, University Park, PA 16801, USA
cInterdisciplinary Research Center for Sustainable Energy Systems (IRC-SES), King Fahd University of Petroleum & Minerals (KFUPM), Dhahran 31261, Saudi Arabia
dDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16801, USA
eMaterials Research Institute, The Pennsylvania State University, University Park, PA 16801, USA
fDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16801, USA
gToyota Research Institute of North America, Ann Arbor, Michigan 48105, USA
First published on 15th April 2025
Abstract
The triple-phase interface among the active material, conductive host, and solid-state electrolyte is critical for achieving high-performance solid-state sulfur cathodes. However, solid–solid contact often creates unfavorable interfaces with low ion/electron transport efficiency and limited durability, leading to reduced discharge capacity and compromised cycling stability. To overcome this challenge, we introduce a novel hybrid inorganic–organic cathode design that uses sulfur and organochalcogen compounds (i.e., phenyl disulfide/diselenide) as active materials. These organochalcogen compounds, with a low melting point of 60 °C, exhibit a good affinity for sulfur, carbon, and solid-state electrolytes in their molten state. This triphilic feature enables uniform integration among cathode components with efficient and robust interfaces. Consequently, introducing a small amount of organochalcogen compounds (2 wt% of cathode weight) enhances discharge capacity (>1000 mA h g−1), rate capability, and cycling stability (>400 cycles) in full cells.
Broader contextLithium–sulfur all-solid-state batteries have emerged as a promising candidate for next-generation energy storage technology, offering higher energy density, lower cost, and enhanced safety to meet the demands of electric vehicles and aviation. However, the intrinsically poor solid–solid interfacial contacts among cathode components often lead to reduced specific capacity and limited cycle life. To address this challenge, we present a novel hybrid inorganic–organic cathode design utilizing inorganic elemental sulfur and organic organochalcogen compounds as active materials. We discovered the triphilic nature of the organochalcogen compounds, which helps establish intimate and durable interfacial contacts among cathode components, thereby enhancing ion and electron transport, increasing discharge capacity, and improving cycling stability. These findings could potentially inspire the development of hybrid inorganic–organic sulfur cathodes with superior interfacial properties and enhanced reaction kinetics, advancing lithium–sulfur all-solid-state battery technology. |
Introduction
Lithium–sulfur (Li–S) all-solid-state batteries (ASSBs) are gaining increasing attention as a low-cost, energy-dense, and safer energy storage solution for electric vehicles and aviation.1–5 However, unlocking the full potential of the sulfur conversion reaction (S + 2Li ↔ Li2S, 1675 mA h g−1, theoretical) remains a significant challenge.6,7 Sulfur and its lithiation products possess inherently low ionic and electronic conductivity,8 making the conversion reaction heavily dependent on the ion/electron transport efficiency at the three-phase interfaces among sulfur, the conductive host, and solid-state electrolyte (SSE). Unfortunately, the transport efficiency is naturally poor due to the rigid nature of solids, resulting in sluggish reaction kinetics and limited sulfur utilization (Fig. 1a).9–11 Additionally, sulfur's repeated large volume change during cycling further exacerbates the issue, leading to the failure of solid–solid interfaces and continuous capacity decay.1,12,13 | ||
| Fig. 1 Triphilic nature of organochalcogen compounds. (a) Schematic illustration of the mechanism of triphilic organochalcogen molecules (i.e., PDS and PSE) in promoting uniformity and facilitating favorable interfacial contact among cathode components. (b) DSC curves of PDS and PSE. (c) Photographs of the sulfur–PDS mixture before and after melting at 80 °C, demonstrating that sulfur can be dissolved into PDS melts. (d) Photographs of the carbon–PDS mixture before and after melt infiltration (heat treatment at 80 °C), along with a schematic illustration depicting the infiltration of PSE into the pores of porous carbon. (e) Contact angle measurements of PDS, PSE, and sulfur melts on the surface of the LPS pellet. The results show that PDS and PSE can wet the surface with smaller contact angles at 80 °C than molten sulfur at 140 °C. (f) SEM images of the PDS–sulfur mixture and LPS pellets before (left) and after heat treatment at 80 °C (right). The pellets were prepared by pressing the powders together under 400 MPa. The results demonstrate that the melt-precipitation process of PDS–sulfur enhances intimate interfacial contact between the PDS–sulfur mixture and LPS powders during cathode preparation. | ||
Various strategies have been developed to address this interfacial challenge, including tailoring the sulfur structure,1,14,15 optimizing cathode preparation,12,16–18 incorporating metal sulfides,19,20 engineering SSEs,2,9,21–24 and adjusting the carbon architecture.25–28 Significant advancements have been made in enhancing interfacial ion/electron transport and sulfur utilization, but they still fall short of meeting the technical requirements for practical applications at high sulfur content and mass loading.29
While a comprehensive solution to solve all the challenges remains elusive, approaching the problem from a different perspective could be a promising alternative. Compared to inorganic compounds, organic materials offer distinct advantages, such as superior mechanical properties. For example, Lu et al. demonstrated that integrating flexible polymer binders effectively accommodates volume changes, thereby improving the resilience of the triple-phase interface and achieving improved cycling stability.30 Nevertheless, strategies leveraging organic materials to solve the interfacial problems in solid-state sulfur cathodes remain scarce, leaving this area largely unexplored.
Here, we present the design of hybrid inorganic–organic cathodes using elemental sulfur and organochalcogen compounds, diphenyl disulfide (PDS) or diphenyl diselenide (PSE), as active materials (AMs). PDS and PSE possess a low melting point of around 60 °C, and their melts display a good affinity for sulfur, SSEs, and carbon. Sulfur readily dissolves in molten PDS and PSE, and the melts can effectively wet the surface of SSEs and infiltrate porous carbon. This distinctive triphilic characteristic enhances the uniformity of cathode materials and facilitates the formation of seamless, durable triple-phase interfaces with improved ion transport efficiency (Fig. 1a), as confirmed by microscopic and electrochemical characterization. Further evaluation of ASSBs demonstrates that incorporating only 2 wt% of PDS or PSE into sulfur cathodes (50 wt% AM) boosts discharge capacity beyond 1000 mA h g−1AM at room temperature and enables stable cycling for over 400 cycles. Notably, this improved performance is maintained in ASSBs with high AM loadings exceeding 4 mg cm−2.
Experimental
Synthesis of solid-state electrolytes
Li2S (99.98%), P2S5 (99%), and LiI (99.99%) were obtained from Sigma-Aldrich, and LiBr (99.995%) was sourced from Alfa Aesar. Solid-state electrolytes (SSEs), including 75Li2S·25P2S5 (LPS) glass and Li7P2S8Br0.5I0.5 (LPSBI) glass ceramic,31 were synthesized by mechanically milling stoichiometric amounts of precursors in ZrO2 jars (FRITSCH PULVERISETTE 7 premium line) with anhydrous heptane (99%, Sigma-Aldrich) as the medium. The powders were milled at 500 rpm for 20 hours (for LPS) or 40 hours (for LPSBI) and then collected and vacuum-dried at 120 °C to remove heptane. LPSBI powders were further annealed at 160 °C for 60 hours. All experiments were performed under an argon atmosphere (H2O < 1 ppm, O2 < 5 ppm) within a glovebox. Li6PS5Cl (LPSC, D50–1 μm) was purchased from MSE Supplies.Preparation of cathode composites
Active material–carbon (AM–carbon) composites were prepared by mixing carbon (C, Ketjenblack EC-600J), sulfur (S, ≥99.0%, Sigma-Aldrich), phenyl disulfide (PDS, 99%, Sigma-Aldrich), and diphenyl diselenide (PSE, 98%, Sigma-Aldrich) with the desired weight ratios (C/S/PDS or PSE = 10/50–x/x, w/w/w, x = 0, 2, and 5) using mechanical ball milling at 300 rpm for 10 hours. Subsequently, the resulting powders were mixed with LPS at a weight ratio of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 and subjected to additional ball milling at 350 rpm for 10 hours in ZrO2 jars under an argon atmosphere, yielding the final cathode composite powders (denoted as Sulfur, S-PDS, and S-PSE cathodes).
:40 and subjected to additional ball milling at 350 rpm for 10 hours in ZrO2 jars under an argon atmosphere, yielding the final cathode composite powders (denoted as Sulfur, S-PDS, and S-PSE cathodes).
Materials characterization
Thermogravimetric analysis (TGA) of AM–carbon composites was conducted using a Discovery TGA 550 (TA Instruments) at a heating rate of 10 °C min−1 up to 500 °C under a nitrogen flow. Differential scanning calorimetry (DSC) measurements were performed using a Discovery 2500 (TA Instruments) at a heating rate of 10 °C min−1 under a nitrogen flow in Tzero Hermetic pans. An Apreo scanning electron microscope (SEM) was used to collect SEM and energy-dispersive spectroscopy (EDS) mapping images. Transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and EDS mapping images were obtained using an FEI Talos F200X (S)TEM instrument with a cryogenic (Cryo) holder. X-ray diffraction (XRD) patterns of the materials and electrodes were recorded using a Malvern Panalytical Empyrean III instrument. An XRD holder with a polycarbonate dome was utilized to load air-sensitive samples inside an argon-filled glovebox. A LabRAM HR Evolution Confocal Raman Microscope using a 532 nm laser was employed for Raman characterization. For X-ray photoelectron spectroscopy (XPS) characterization, the samples were loaded onto a stage inside an argon-filled glovebox and transferred to the instrument (PHI VersaProbe II Scanning XPS Microprobe) through a vacuum transfer vessel to prevent air exposure. 31P magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectra of the cathode powders were collected on an Avance-III-HD ss500 instrument with a 4 mm Bruker CPMAS probe, operating at a spinning speed of 10 kHz and using CaHPO4·2H2O as the reference.Solid-state battery fabrication
Four types of cells, steel|cathode|steel, steel|Li|LPSC|cathode |LPSC|Li|steel, Li–In|LPSBI|cathodes, and steel|Li|LPSC|Li|steel, were fabricated using Swagelok-type cells in an argon-filled glovebox.Steel|cathode|steel cells were prepared by pressing 100 mg of cathode powders at 294 MPa for 3 minutes in the cell. Subsequently, the cells were compressed using three screws under 55 MPa for further testing.
To prepare steel|Li|LPSC|cathode|LPSC|Li|steel cells, 70 mg of LPSC, 60 mg of cathode, and 70 mg of LPSC powders were pressed at 294 MPa for 3 minutes in the cell successively to form a pellet. 1 cm2, 100 μm-thick Li foils were then attached to each side of the pellet, and the cells were compressed at approximately 8 MPa using screws for subsequent evaluation. Steel|Li|LPSC|Li|steel cells were prepared similarly with 140 mg of LPSC powders.
For ASSBs, i.e., Li–In|LPSBI|cathodes cells, the preparation involved several steps: (1) 80 mg of LPSBI powders was weighed and pressed in the cell at 294 MPa for 1 minute; (2) cathode powders with the desired weight were evenly spread and pressed onto one side of the SSE pellet at 294 MPa for 3 minutes; (3) a 4 mg piece of lithium and an indium chip (127 μm thick, 10 mm diameter, 99.99%, Sigma-Aldrich) were successively attached to a stainless steel rod and pressed onto the opposite side of the SSE pellet at 100 MPa. Finally, the assembled cells were compressed at ∼55 MPa for further evaluation.
Electrochemical evaluation
All the electrochemical cycling tests of the cells were performed using a Landt cycler between 0.5 and 2.5 V vs. Li–In/Li+ at room temperature or 60 °C. The electronic conductivity of the cathodes was determined using the d.c. polarization method by applying ±0.1, 0.2, and 0.3 V voltage biases on the steel|cathode|steel symmetric cells and recording the current evolution for 1 hour. Similarly, the ionic conductivity of the cathodes was measured via the DC polarization method by applying ±0.1, 0.2, and 0.4 V voltage biases to steel|Li|LPSC|cathode|LPSC|Li|steel symmetric cells or ±10 and 20 mV to steel|Li|LPSC|Li|steel cells. Li–In|LPSBI| cathode cells were discharged/charged at 0.2 A g−1AM to various potentials, followed by a 20-minute rest prior to the electrochemical impedance spectroscopy (EIS) measurement. The EIS data were collected using a Solartron Modulab from 0.1 Hz to 1 MHz, with a voltage bias of 10 mV. The distribution of relaxation times (DRT) analysis was carried out using DRTtools (https://github.com/ciuccislab/pyDRTtools).32 For galvanostatic intermittent titration technique (GITT) measurements, the cells were cycled at 0.2 A g−1AM for 2 cycles first and then discharged/charged with current pulses of 0.1 A g−1AM for 30 minutes, followed by a 4-hour rest.Results and discussion
Triphilic nature of organochalcogen compounds
PDS and PSE are redox-active materials, with S–S or Se–Se bonds reversibly cleaving and reforming upon cycling, corresponding to theoretical capacities of 245 and 171 mA h g−1, respectively (Fig. S1†).33,34 They exhibit a melting temperature of ∼60 °C, lower than that of sulfur (>100 °C), as confirmed by the differential scanning calorimetry (DSC) measurement (Fig. 1b and Fig. S2†). This characteristic of PDS and PSE allows them to melt easily during cathode preparation, facilitating their interaction with other cathode components to form favorable interfaces.The interactions of molten PDS and PSE with sulfur, carbon, and SSEs were investigated to understand their behavior during cathode preparation. Sulfur could readily dissolve in PDS/PSE melts, forming a yellowish solution (Fig. 1c and Fig. S3a†). This melt-dissolution behavior allows sulfur and PDS/PSE melts to infiltrate the pores of porous carbon during ball milling, achieving a uniform distribution within the structure (Fig. 1d and Fig. S3b†). Notably, no chemical reaction between sulfur and PDS/PSE occurs in this process, as suggested by Raman spectroscopy (Fig. S4†). Furthermore, the wettability of molten PDS and PSE on the LPSLPS glass SSE pellet surface was evaluated, revealing their stronger affinity for LPS than molten sulfur (Fig. 1e). This enhanced affinity facilitates sulfur–PDS/PSE to form intimate, void-free interfacial contact with the SSE during cathode preparation (Fig. 1f and Fig. S5†). Collectively, these results highlight the triphilic nature of PDS and PSE. The induced melt-dissolution–precipitation behavior promotes the formation of seamless interfaces among cathode components, enhancing overall cathode integration.
Characterization of cathode composites
Next, we prepared carbon–AM composites (carbon/sulfur/PDS or PSE = 10/50–x/x, w/w/w, x = 0 or 2) and cathode materials (carbon–AM/LPS = 60/40, w/w, denoted as sulfur cathode, S-PDS cathode, and S-PSE cathode, respectively) using mechanical ball milling. Their properties were examined using thermal, spectroscopic, and microscopic characterization techniques. Thermal gravimetric analysis (TGA) results indicate a high AM content of ∼83 wt% in all carbon–AM composites (Fig. 2a). Incorporating PDS and PSE with low melting points (Fig. S6†) resulted in weight loss at a slightly lower temperature than the carbon–sulfur composite. Because of the high AM content, sulfur cannot be fully confined within the porous carbon, and crystalline sulfur peaks were observed in X-ray diffraction (XRD) patterns of the carbon–AM composite and cathode powders (Fig. 2b and Fig. S7†). No peaks associated with PDS or PSE were seen owing to their low content. Furthermore, in the prepared cathode powders, no chemical bonding was observed between PDS/PSE and other cathode components, as confirmed by XPS and 31P MAS NMR characterization (Fig. S8†).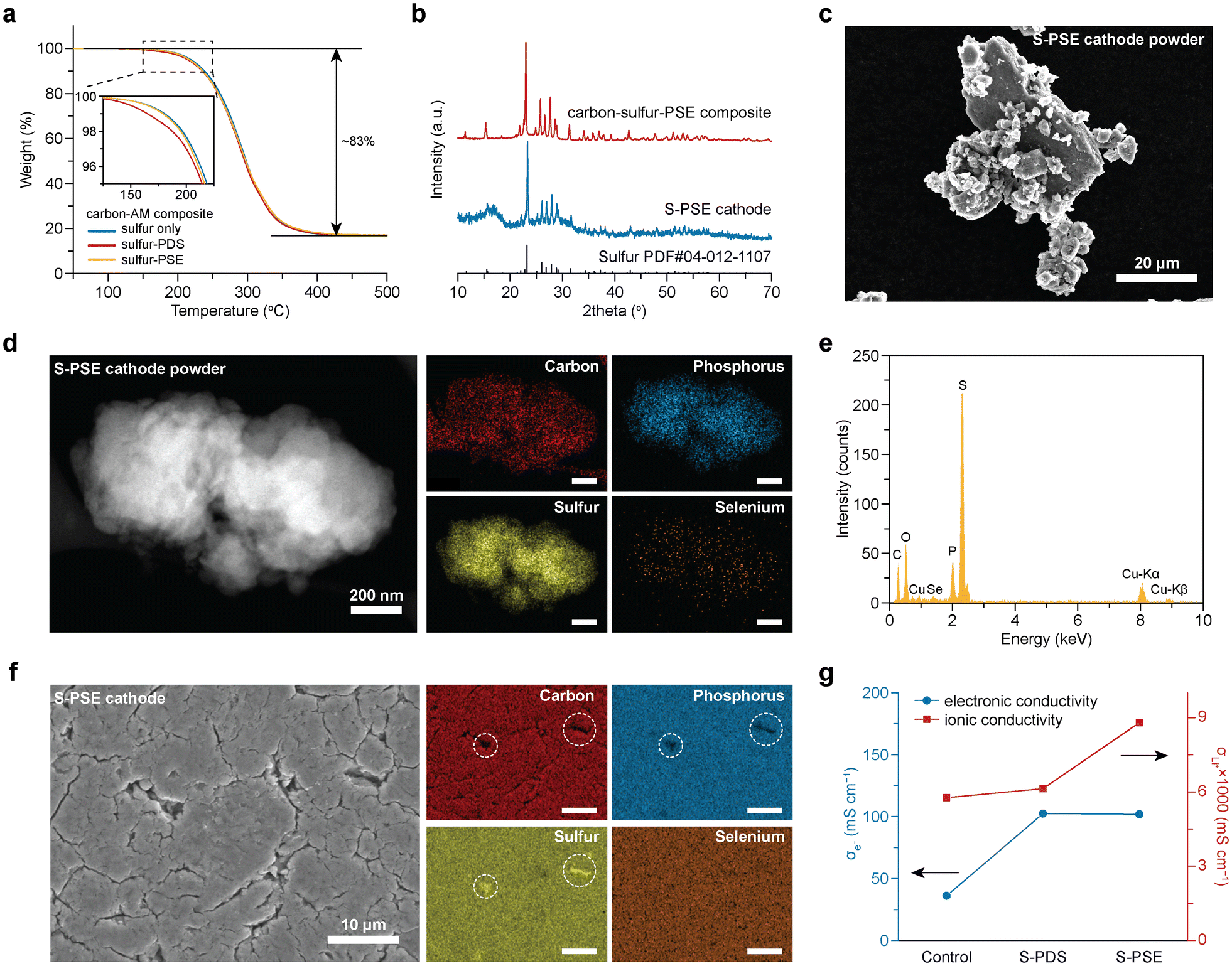 | ||
| Fig. 2 Characterization of cathode composites. (a) TGA curves of different sulfur–AM composites. (b) XRD patterns of the sulfur-PSE composite and the S-PSE cathode. (c) SEM image of S-PSE powders. (d) STEM and EDS mapping images of an S-PSE particle obtained under cryogenic conditions and (e) corresponding EDS spectrum. (f) STEM and EDS mapping images of the top surface of the S-PSE cathode. (g) Electronic and ionic conductivity of different cathodes at 30 °C. | ||
All the prepared composite cathodes are micrometer-sized particles with uniform elemental distribution across each particle, as observed by scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS) mapping images (Fig. 2c and Fig. S9†). To further elucidate elemental distribution at the submicrometer scale, we employed cryogenic transmission electron microscopy (cryo-TEM) with scanning transmission electron microscopy (STEM) and EDS. The results show that all elements evenly distribute across the S-PSE cathode particles without noticeable phase separation (Fig. 2d, e and Fig. S10†), verifying the uniform integration among cathode components.
Upon pressing the powders into the S-PSE electrode, micrometer-sized sulfur particles were observed on the electrode surface, whereas PSE remained uniformly distributed (Fig. 2f). The lithiation of these unconfined bulk sulfur particles is challenging, which will result in reduced sulfur utilization.21 Encouragingly, compared to sulfur cathodes, there are relatively fewer bulk sulfur particles on the surface of cathodes with PDS and PSE (Fig. S11†). Additionally, we evaluated the ionic and electronic conductivity of different cathodes using d.c. polarization methods (Fig. S12†).35,36 The measured electronic and ionic conductivities of S-PDS (0.102 S cm−1 and 6.13 × 10−6 S cm−1) and S-PSE (0.102 S cm−1 and 8.79 × 10−6 S cm−1) cathodes are all higher than those of the sulfur cathode (0.036 S cm−1 and 5.77 × 10−6 S cm−1) at 30 °C (Fig. 2g). It shows that incorporating PSD and PSE can promote ion and electron transport through the electrode, which might result from the improved interfacial contact and optimized electrode microstructure.16,17
Electrochemical evaluation of ASSBs
Next, ASSBs were assembled and tested to evaluate the electrochemical performance of different cathodes with an areal AM loading of ∼2 mg cm−2. S-PDS and S-PSE cathodes delivered higher discharge capacities (957.2 and 982.4 mA h g−1AM) and better reversibility (98.3% and 94.7%) than the sulfur cathode (922.8 mA h g−1AM and 89.1%) during the first cycle at 0.2 A g−1AM (Fig. 3a). Note that all applied current rates and reported specific capacities were calculated based on the total weight of sulfur and PDS/PSE in the cathode. | ||
| Fig. 3 Electrochemical evaluation of ASSBs at room temperature. (a) Galvanostatic discharge–charge curves of different cathodes at the initial cycle at 0.2 A g−1AM and (b) corresponding differential capacity (dQ/dV) curves. (c) Rate capability of different cathodes. (d) Cycling performance of different cathodes at 0.6 A g−1AM in CCCV mode with a cutoff current of 0.2 A g−1AM at 2.5 V. The areal AM loading is 1.6–1.8 mg cm−2. (e and f) Corresponding dQ/dV curves of (e) the sulfur cathode and (f) the S-PDS cathode at different cycles. (g) Cycling performance of a cell with the S-PDS cathode at an areal AM loading of 4.565 mg cm−2. The cells were cycled at 0.1 A g−1AM for the first 10 cycles and then at 0.2 A g−1AM for subsequent cycles. (h) Comparison of S-PDS and S-PSE cathode performance with that of state-of-the-art Li–S ASSBs under similar conditions (cell condition details in Table S2†).14,17,19,20,25 | ||
The corresponding differential capacity (dQ/dV) curves were analyzed to elucidate the reaction kinetics (Fig. 3b). Two reduction peaks (I and II, S8 → Li2S) were observed during the discharge, along with one oxidation peak (III, Li2S → S8) during charge in all cells, suggesting similar redox reactions. The sharper peaks with higher intensity, along with reduced voltage polarization (detailed peak positions in Table S1†), observed in cells with S-PDS and S-PSE cathodes suggest enhanced solid-state reaction kinetics. Notably, the redox peaks associated with PDS and PSE cannot be clearly distinguished due to their similar redox potentials to sulfur, as well as their low content and limited discharge capacity.
The rate performance of the cells was also evaluated. S-PDS and S-PSE cathodes exhibited higher discharge capacity at all current rates compared to sulfur cathodes (Fig. 3c and Fig. S13†). Specifically, the S-PDS cathode delivered discharge capacities of 1149.2, 990.4, 823.0, and 602.6 mA h g−1AM at 0.2, 0.4, 1.0, and 2.0 A g−1AM, respectively. Similarly, under the same conditions, the S-PSE cathode achieved 1202.6, 1005.6, 759.8, and 537.4 mA h g−1AM discharge capacities. In contrast, at these current rates, the sulfur cathode showed discharge capacities of only 994.0, 821.6, 607.4, and 413.8 mA h g−1AM. Moreover, S-PDS and S-PSE cathodes retained good rate capability at 60 °C (Fig. S14†).
Furthermore, the cycling stability of these cathodes with an areal AM loading of 1.6–1.8 mg cm−2 was examined at 0.6 A g−1AM using the constant current/constant voltage (CC/CV) charging method with a cutoff current of 0.2 A g−1AM at 2.5 V (Fig. 3d). The discharge capacities of all cells increased in the first few cycles before declining, primarily due to the degradation of SSEs.13 Compared to sulfur cathodes, S-PDS and S-PSE cathodes exhibited a higher discharge capacity and enhanced cycling stability. The maximum discharge capacity of S-PDS and S-PSE cathodes reached 1063.2 and 999.8 mA h g−1AM, respectively, surpassing that of the sulfur cathode (869.4 mA h g−1AM). After 400 cycles, the S-PDS and S-PSE cathodes retained 84.3% and 89.4% of their maximum discharge capacity, respectively, both outperforming sulfur cathodes (75.9%). This result demonstrates the beneficial impact of PDS and PSE in improving cycling stability. Moreover, the capacity decay in sulfur cathodes is accompanied by a significant increase in voltage polarization with cycling (Fig. 3e and Fig. S15†). In contrast, S-PDS and S-PSE cathodes exhibited a less pronounced voltage polarization buildup. Instead, their capacity decay was primarily attributed to active material loss, as indicated by the decline in the redox reaction peak intensity (Fig. 3f and Fig. S15b–d†). These distinct behaviors possibly suggest different capacity fading mechanisms, which will be discussed later.
We also evaluated the cycling performance of S-PDS and S-PSE cathodes with areal AM loading above 4 mg cm−2. The cells were cycled at 0.1 A g−1AM for the first 10 cycles and 0.2 A g−1AM thereafter, demonstrating high discharge capacity and good cycling stability for over 200 cycles (Fig. 3g and Fig. S16†). Specifically, the cell with the S-PDS cathode delivered the highest specific discharge capacity of 1184 mA h g−1AM at 0.2 A g−1AM, which remained at 941.8 mA h g−1AM after 200 cycles. Overall, the room-temperature performance of S-PDS and S-PSE cathodes is among the best in the state-of-the-art Li–S ASSBs (Fig. 3h and Table S2†). Although substituting sulfur with PDS or PSE reduces the overall theoretical capacity of the AM, it favorably improves AM utilization (Table S3†).
Nevertheless, it should be noted that increasing the PDS and PSE content does not always guarantee higher discharge capacity. Due to their lower density (PDS, 1.35 g cm−3; PSE, 1.84 g cm−3) compared to sulfur (2 g cm−3), a higher weight percentage of PDS or PSE increases the volumetric content of the AM in the cathode. If excessive, this can negatively impact battery performance by limiting ion transport pathways.22 For instance, when the PDS/PSE content increases to 5 wt%, the S-PDS cathode exhibited a lower discharge capacity and AM utilization than sulfur cathodes, whereas the S-PSE cathode maintained good performance with enhanced discharge capacity and AM utilization (Fig. S17 and Table S3†).
Characterization of ASSBs
To assess ion transport efficiency within cathodes during (de)lithiation, impedance spectra of ASSBs with different cathodes were collected during the initial cycle and interpreted using the distribution of relaxation times (DRT) model (Fig. 4a–f and Fig. S18†).37,38 Three prominent peaks (A, B, and C) were observed in the DRT curves, corresponding to ion transport across the anode interface (peak A), ion transport across the cathode interface (peak B), and solid-state ion diffusion within the cathode (peak C).39 During discharge, the intensities of peaks B and C initially decrease and then keep increasing in cells with sulfur cathodes (Fig. 4b). In contrast, in cells with S-PDS and S-PSE cathodes, these peaks decrease and then quickly stabilize (Fig. 4e and Fig. S18b†). The distinct evolution behaviors might result from the interfacial changes at the first cycle and suggest improved interfacial durability and ion transport efficiency in S-PDS and S-PSE cathodes compared to sulfur cathodes. Different from the discharging process, the intensity of all peaks keeps increasing following a similar trend during charging, regardless of the cathode type (Fig. 4c, f and Fig. S18c†). Notably, the peak intensities are relatively lower in cells with S-PDS/PSE cathodes at different states of charge compared to those with sulfur cathodes, indicating reduced resistance and improved ionic transport.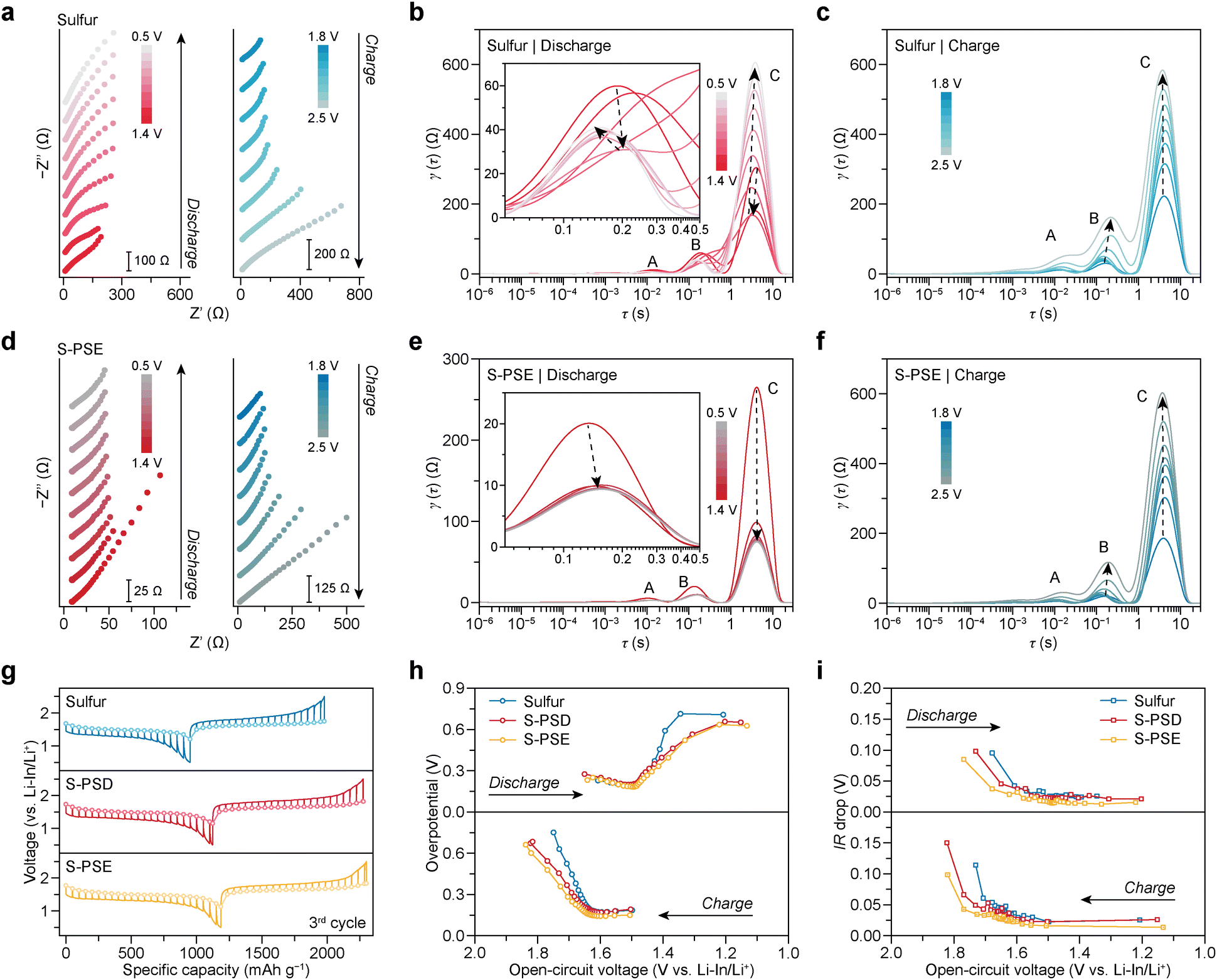 | ||
| Fig. 4 Electrochemical characterization of Li–S ASSBs. (a) Impedance spectra of the ASSB with the sulfur cathode at different potentials during the first discharge/charge cycle (cycled at 0.2 A g−1AM and rested for 20 min before each measurement). (b and c) Corresponding DRT analysis of the impedance spectra during the discharge (b) and charge process (c). (d) Impedance spectra of the ASSB with the S-PSE cathode at different potentials during the first discharge/charge cycle. (e and f) Corresponding DRT analysis of the impedance spectra during the discharge (e) and charge process (f). (g) GITT and OCV curves of cells with different cathodes at the 3rd cycle. The cells were cycled at 0.1 A g−1AM for 30 min during each pulse and rested for 4 hours. (h and i) Overpotential (h) and ohmic resistance (i) evolution curves obtained from the GITT test. | ||
The galvanostatic intermittent titration technique (GITT) was employed to study the electrochemical redox behavior of ASSBs with different cathodes. Cells with S-PDS and S-PSE cathodes showed higher discharge capacity (Fig. 4g) as well as smaller overpotential (Fig. 4h) and reduced ohmic resistance (IR drop, Fig. 4i) throughout the discharge/charge process compared to cells with sulfur cathodes. These findings corroborate enhanced sulfur utilization, improved reaction kinetics, and better ion transport in S-PDS and S-PSE cathodes. Additionally, the open-circuit voltage (OCV) curve indicates only a single plateau at 1.48 and 1.65 V (vs. Li–In/Li+; 2.10 and 2.27 V vs. Li/Li+) during discharge and charge, respectively, for all cells (Fig. 4g and Fig. S19†). This behavior corresponds to the solid-phase sulfur conversion reaction.7 Notably, diphenyl selenopolysulfides (C6H5SeSxSeC6H5) formed during cycling, if any, are relatively minimal, as evidenced by the Raman spectra (Fig. S20†). It is possibly caused by the sluggish solid-state reaction kinetics. Therefore, unlike in liquid electrolyte systems,40 the role of PSE and PDS as redox mediators in ASSBs, in terms of altering the reaction pathway, appears to be limited.
Spectroscopic, microscopic, and electrochemical characterization studies of the cycled electrodes and cells were also conducted to elucidate the capacity fading mechanism. XRD patterns of cycled S-PSE electrodes in the charged state revealed crystalline Li2S peaks, which were absent in the cycled sulfur cathode (Fig. S21†). This suggests that capacity decay in the S-PSE cathode is primarily induced by the accumulation of irreversible Li2S.10 In contrast, in the sulfur cathode, it may be attributed to deteriorated interfacial contact. SEM images of the cycled electrodes’ surface (Fig. S22a and b†) further unveiled fewer cracks on the cycled S-PDS/PSE electrodes than on the pristine ones (Fig. 2f). The disappearance of cracking in S-PDS/PSE electrodes after cycling is likely due to the accumulation of inactive Li2S, which induces cathode expansion through volume change, thereby eliminating cracks and promoting stable interfacial contact. In contrast, numerous cracks were observed on the cycled sulfur cathode surface (Fig. S22c†), indicating deteriorated interfacial contact. This degradation is likely to contribute to the increased voltage polarization and capacity decay observed after cycling. These observations are further supported by the DRT analysis results of the impedance spectra (Fig. S23†), which uncovered more efficient interfacial ion transport and solid-state ion diffusion within cycled S-PDS/PSE cathodes than cycled sulfur cathodes.
Conclusions
In summary, our findings highlight the combination of inorganic elemental sulfur and organochalcogen compounds as a promising strategy for developing high-performance cathodes for ASSBs. The triphilic nature of PDS and PSE promotes favorable interfacial contact among cathode components, enhancing AM utilization and improving cycling stability. Given the abundance of organochalcogen compounds, various hybrid inorganic–organic sulfur cathodes can be explored in the future to optimize interfacial properties and enhance reaction kinetics.Author contributions
Conceptualization: Da.W. and Do.W.; investigation: Da.W., M.O., A.S.A., Y.G., J.L.G., and L.Q.; formal analysis: Da.W., M.O., A.S.A., Y.G., J.L.G., L.Q., and M.L.; writing – original draft: Da.W. and Do.W.; writing – review and editing: Da.W., M.O., T.S.A. and Do.W.; supervision: Do.W.Data availability
All relevant data are included in this article and the ESI.†Conflicts of interest
Do.W., Da.W., M.S., and A.S.A. are inventors on US patent application no. 17/806,351 filed by Toyota Motor Corp and Penn State Research Foundation regarding the formula of the cathode mixtures described in this article. The other authors declare no competing interests.Acknowledgements
This work was supported by the Toyota Motor Corporation and the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the US Department of Energy, through US–Germany Collaboration Projects and Argonne National Laboratory subcontracting award no. 0F-60008. We would like to thank Dr Christy George from the Department of Chemistry at the Pennsylvania State University for her help with the NMR measurement.References
- J. Zhou, M. L. Holekevi Chandrappa, S. Tan, S. Wang, C. Wu, H. Nguyen, C. Wang, H. Liu, S. Yu, Q. R. S. Miller, G. Hyun, J. Holoubek, J. Hong, Y. Xiao, C. Soulen, Z. Fan, E. E. Fullerton, C. J. Brooks, C. Wang, R. J. Clément, Y. Yao, E. Hu, S. P. Ong and P. Liu, Nature, 2024, 627, 301–305 CrossRef CAS PubMed.
- H. Song, K. Münch, X. Liu, K. Shen, R. Zhang, T. Weintraut, Y. Yusim, D. Jiang, X. Hong, J. Meng, Y. Liu, M. He, Y. Li, P. Henkel, T. Brezesinski, J. Janek and Q. Pang, Nature, 2025, 637, 846–853 CrossRef CAS PubMed.
- J. Lee, C. Zhao, C. Wang, A. Chen, X. Sun, K. Amine and G.-L. Xu, Chem. Soc. Rev., 2024, 53, 5264–5290 RSC.
- F. Liang, S. Wang, Q. Liang, A. Zhong, C. Yang, J. Qian, H. Song and R. Chen, Adv. Energy Mater., 2024, 14, 2401959 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- D. Cao, X. Sun, F. Li, S.-M. Bak, T. Ji, M. Geiwitz, K. S. Burch, Y. Du, G. Yang and H. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202302363 CrossRef CAS PubMed.
- J. Gu, W. Hu, Y. Wu, F. Ren, Z. Liang, H. Zhong, X. Zheng, R. Ma, Y. Luo, X. Chen, J. Shi and Y. Yang, Chem. Mater., 2024, 36, 4403–4416 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- D. Wang, B. Gwalani, D. Wierzbicki, V. Singh, L.-J. Jhang, T. Rojas, R. Kou, M. Liao, L. Ye, H. Jiang, S. Shan, A. Silver, A. T. Ngo, Y. Du, X. Li and D. Wang, Nat. Mater., 2025, 24, 243–251 CrossRef CAS PubMed.
- R. Bradbury, G. F. Dewald, M. A. Kraft, T. Arlt, N. Kardjilov, J. Janek, I. Manke, W. G. Zeier and S. Ohno, Adv. Energy Mater., 2023, 13, 2203426 CrossRef CAS.
- S. Ohno and W. G. Zeier, Acc. Mater. Res., 2021, 2, 869–880 CrossRef CAS.
- A. S. Alzahrani, M. Otaki, D. Wang, Y. Gao, T. S. Arthur, S. Liu and D. Wang, ACS Energy Lett., 2021, 6, 413–418 CrossRef CAS.
- S. Ohno, R. Koerver, G. Dewald, C. Rosenbach, P. Titscher, D. Steckermeier, A. Kwade, J. Janek and W. G. Zeier, Chem. Mater., 2019, 31, 2930–2940 CrossRef CAS.
- X. Li, J. Liang, J. Luo, C. Wang, X. Li, Q. Sun, R. Li, L. Zhang, R. Yang, S. Lu, H. Huang and X. Sun, Adv. Mater., 2019, 31, 1808100 CrossRef PubMed.
- W. Zhao, Y. Zhang, Q. Liu, Y. Song, X. Li, L. Ren, G. Yin, S. Lou and J. Wang, Angew. Chem., Int. Ed., 2025, 64, e202413670 CrossRef CAS PubMed.
- H. Kim, H.-N. Choi, J.-Y. Hwang, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2023, 8, 3971–3979 CrossRef CAS.
- L.-P. Hou, H. Yuan, C.-Z. Zhao, L. Xu, G.-L. Zhu, H.-X. Nan, X.-B. Cheng, Q.-B. Liu, C.-X. He, J.-Q. Huang and Q. Zhang, Energy Storage Mater., 2020, 25, 436–442 CrossRef.
- X. Yao, N. Huang, F. Han, Q. Zhang, H. Wan, J. P. Mwizerwa, C. Wang and X. Xu, Adv. Energy Mater., 2017, 7, 1602923 CrossRef.
- S. Xu, C. Y. Kwok, L. Zhou, Z. Zhang, I. Kochetkov and L. F. Nazar, Adv. Funct. Mater., 2021, 31, 2004239 CrossRef CAS.
- U. Ulissi, S. Ito, S. M. Hosseini, A. Varzi, Y. Aihara and S. Passerini, Adv. Energy Mater., 2018, 8, 1801462 CrossRef.
- H. Guo, J. Li, M. Burton, J. Cattermull, Y. Liang, Y. Chart, G. J. Rees, J. Aspinall and M. Pasta, Cell Rep. Phys. Sci., 2024, 5, 102228 CrossRef CAS.
- D. Wang, L.-J. Jhang, R. Kou, M. Liao, S. Zheng, H. Jiang, P. Shi, G.-X. Li, K. Meng and D. Wang, Nat. Commun., 2023, 14, 1895 CrossRef CAS PubMed.
- X. Shi, Z. Zeng, M. Sun, B. Huang, H. Zhang, W. Luo, Y. Huang, Y. Du and C. Yan, Nano Lett., 2021, 21, 9325–9331 CrossRef CAS PubMed.
- H. Kim, Y. E. Park, K. Park and S. Kim, J. Power Sources, 2024, 621, 235284 CrossRef CAS.
- Y. Luo, S. Pan, J. Tian, Y. Liang, H. Zhong, R. Ma, J. Gu, Y. Wu, H. Zhang, H. Lin, W. Huang, Y. Deng, Y. Su, Z. Gong, J. Huang, Z. Hu and Y. Yang, Adv. Mater., 2024, 36, 2413325 CrossRef CAS PubMed.
- T. Jin, K. Liang, J.-H. Yu, T. Wang, Y. Li, T.-D. Li, S. P. Ong, J.-S. Yu and Y. Yang, Nano Lett., 2024, 24, 6625–6633 CrossRef CAS PubMed.
- Y. Zhang, T. Liu, Q. Zhang, X. Zhang, S. Wang, X. Wang, L. Li, L.-Z. Fan, C.-W. Nan and Y. Shen, J. Mater. Chem. A, 2018, 6, 23345–23356 RSC.
- A. Sakuda, Y. Sato, A. Hayashi and M. Tatsumisago, Energy Technol., 2019, 7, 1900077 CrossRef CAS.
- G. Zhou, H. Chen and Y. Cui, Nat. Energy, 2022, 7, 312–319 CrossRef CAS.
- X. Zhu, W. Jiang, L. Wang and J. Lu, Adv. Energy Mater., 2024, 14, 2304244 CrossRef CAS.
- Y. Wang, D. Lu, J. Xiao, Y. He, G. J. Harvey, C. Wang, J.-G. Zhang and J. Liu, Energy Storage Mater., 2019, 19, 80–87 CrossRef.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- A. Bhargav, S. V. Patil and Y. Fu, Sustainable Energy Fuels, 2017, 1, 1007–1012 RSC.
- W. Guo, A. Bhargav, J. D. Ackerson, Y. Cui, Y. Ma and Y. Fu, Chem. Commun., 2018, 54, 8873–8876 RSC.
- G. F. Dewald, S. Ohno, J. G. C. Hering, J. Janek and W. G. Zeier, Batteries Supercaps, 2021, 4, 183–194 CrossRef CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172–1198 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- X. Li, J. Liang, J. T. Kim, J. Fu, H. Duan, N. Chen, R. Li, S. Zhao, J. Wang, H. Huang and X. Sun, Adv. Mater., 2022, 34, 2200856 CrossRef CAS PubMed.
- M. Zhao, X.-Y. Li, X. Chen, B.-Q. Li, S. Kaskel, Q. Zhang and J.-Q. Huang, eScience, 2021, 1, 44–52 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00043b |
| This journal is © The Royal Society of Chemistry 2025 |