
Coordinated assembly of alkali and alkaline earth metals with the perfluorinated [AlF6] group to design deep-ultraviolet zero-order waveplate materials†
Shaohua
Liu
ab,
Lili
Zhang
 *ab,
Ke
Li
c,
Xiaotian
Ma
cd,
Xifa
Long
cd and
Yun
Yang
*cd
*ab,
Ke
Li
c,
Xiaotian
Ma
cd,
Xifa
Long
cd and
Yun
Yang
*cd
aXinjiang Laboratory of Phase Transitions and Microstructures in Condensed Matter Physics, College of Physical Science and Technology, Yili Normal University, Yining, Xinjiang 835000, China
bYili Engineering Research Center of Green Silicon-based Materials, Yining, Xinjiang 83500, China
cResearch Center for Crystal Materials, CAS State Key Laboratory of Functional Materials and Devices for Special Environments; Xinjiang Key Laboratory of Functional Crystal Materials; Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, 40-1 South Beijing Road, Urumqi 830011, China. E-mail: yangyun@ms.xjb.ac.cn
dCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 31st March 2025
Abstract
The capability of zero-order waveplates to manipulate the polarization of fundamental-frequency light has garnered significant attention in light of the rapid advancements in laser technology. In particular, zero-order waveplate materials in the deep-ultraviolet range (DUV; λ < 200 nm) are in urgent and short supply. In this study, three new aluminum fluorides, AlF3 (P6/mmm), BaAlF5 (I4/m), and Li2KAl2F9 (C2/m), have been successfully designed and synthesized through a strategy that combines the perfluorinated [AlF6] group with alkali and alkaline-earth metals. The results of experimental and theoretical calculations indicate that three new aluminum fluorides have a short cutoff edge (λ < 200 nm) and small birefringence (0.0006–0.0056@1064 nm). In addition, the AlF3 (P6/mmm) has the shortest wavelength of 125 nm based on theoretical calculations, which is comparable to the shortest wavelength of the commercially available MgF2. However, its birefringence of 0.0006@1064 nm is about 20 times lower than that of the MgF2 (0.012@546 nm), making it highly advantageous for fabricating deep-ultraviolet zero-order waveplate materials. Further microscopic analysis reveals that the [AlF6] group exhibits a substantial band gap of 9.17 eV and small polarizability anisotropy, indicating that the aluminum fluorides are potential candidates for designing suitable compounds for DUV zero-order waveplate materials.
Introduction
Waveplate materials, serving as crucial devices for phase modulation and polarization state transformation of light waves, find extensive applications in the fields of polarization measurement, laser technology, and optical imaging.1–4 Zero-order waveplates are widely acknowledged to possess several advantages over multistage ones, including lower sensitivity, higher temperature stability, and effective reception angle.5–7 However, the processing of zero-order wave plate materials has long been hindered by the inverse relationship between thickness and birefringence.6,7 Meanwhile, with the continuous development of laser technology, its application wavelength range has expanded from the visible region to the ultraviolet (UV) and deep-ultraviolet (DUV) bands.8–14 The current commercial crystals for DUV and UV applications are MgF2 and quartz, which pose challenges in fabrication and processing due to their big birefringence as zero-order waveplate materials.15–18 Balancing a smaller birefringence and a wide wavelength range, particularly in the DUV band, presents an urgent challenge that needs to be addressed.The materials for DUV zero-order waveplates should possess a small birefringence (Δn < 0.01) and DUV (λ < 200 nm) transparency range.19–21 According to the design requirements for zero-order waveplate materials, the building blocks must exhibit small polarizability anisotropy and large HOMO–LUMO gaps, such as non-π-conjugated tetrahedral groups.22–26 Furthermore, rigid octahedral motifs exhibit identical properties.27–30 For instance, the hybridization of Al and O atoms occurs via sp3, sp3d, and sp3d2 orbitals to form [AlOn] (n = 4, 5, 6) polyhedral groups, which also belong to non-π-conjugated groups possessing low optical anisotropy and do not impact the bandgap due to the absence of d–d and f–f electron transitions in Al3+ ions.31–36 The introduction of fluorine atoms into the [AlOmFn] (m + n = 4, 5, 6) polyhedra enables modulation, where substituting highly electronegative fluorine for oxygen atoms contributes to a blue-shift in the UV absorption edge by inducing ionization of the Al–F bonds.37–47 Particularly when the fluorine atoms entirely substitute for the oxygen atoms, such as the [AlF6] group, the blue shift of the UV absorption edge becomes more pronounced. Therefore, the incorporation of fluorine atoms into conventional non-π-conjugated oxypolyhedral structures not only preserves their low optical anisotropy but also facilitates the enhancement of their microscopic HOMO–LUMO bandgap.48–50
Based on the above ideas, a synergistic combinatorial approach is employed to design DUV zero-order waveplate materials by incorporating perfluorinated [AlF6] octahedra, which exhibit a large HOMO–LUMO energy gap and low polarizability anisotropy, with alkali and alkaline-earth metals lacking d–d or f–f transitions. Through systematic experiments, three aluminum fluorides, AlF3, BaAlF5, and Li2KAl2F9, were successfully synthesized using the high temperature melt method. Theoretical calculations demonstrate that all three materials exhibit large energy band gaps (8.85–9.89 eV) and small birefringence (0.0006–0.0056@1064 nm). This indicates that aluminum fluorides are potential candidates for DUV zero-order waveplate materials.
Experimental section
Experimental reagents
Lithium fluoride (LiF, 99%), potassium fluoride (KF, 99%), aluminum fluoride (AlF3, 99%), ammonium hexafluoroaluminate (H12AlF6N, 98%), potassium fluoroaluminate (KAlF4, CP), lead fluoride (PbF2, 99.9%), barium carbonate (BaCO3, AR), tetrafluoroboric acid (HBF4, AR), and boron oxide (B2O3, 99.9%) were obtained from Aladdin Chemical Industry Co., Ltd. The reagents were used as received.Syntheses
The crystals of AlF3 (P6/mmm), BaAlF5 (I4/m), and Li2KAl2F9 (C2/m) can be obtained by the high temperature solution method in a vacuum system. The AlF3 (P6/mmm) crystals were synthesized from a mixture of B2O3 (0.107 g) and KAlF4 (0.093 g) (molar ratio 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The BaAlF5 (I4/m) crystals were synthesized from a mixture of B2O3 (0.078 g), PbF2 (0.069 g), Ba(BF4)2 (0.029 g) and AlF3 (0.024 g) (molar ratio 12:1:3:3). The Li2KAl2F9 (C2/m) crystals were synthesized from a mixture of B2O3 (0.076 g), LiF (0.010 g), KF (0.043 g) and H12AlF6N (0.071 g) (molar ratio 3:1:2:1). B2O3 and PbF2 were utilized in the crystal synthesis process to serve as fluxes. The starting material Ba(BF4)2 was synthesized by reacting BaCO3 with an excess amount of HBF4. The stoichiometric starting reagents were put into a quartz tube (Φ10 mm × 120 mm) after grinding uniformly and then a vacuum was created inside the tube (1 × 10−3 Pa). The encapsulated quartz tube was placed into a programmable temperature furnace. The temperature was maintained at 700 °C for 24 hours, and then reduced to 600 °C at a rate of 1.0 °C h−1, followed by a further reduction to 500 °C at a rate of 2.0 °C h−1, and finally it was allowed to cool to room temperature. The crystals of the three aluminum fluorides inside the quartz tubes were taken out for structural determination.
:3). The BaAlF5 (I4/m) crystals were synthesized from a mixture of B2O3 (0.078 g), PbF2 (0.069 g), Ba(BF4)2 (0.029 g) and AlF3 (0.024 g) (molar ratio 12:1:3:3). The Li2KAl2F9 (C2/m) crystals were synthesized from a mixture of B2O3 (0.076 g), LiF (0.010 g), KF (0.043 g) and H12AlF6N (0.071 g) (molar ratio 3:1:2:1). B2O3 and PbF2 were utilized in the crystal synthesis process to serve as fluxes. The starting material Ba(BF4)2 was synthesized by reacting BaCO3 with an excess amount of HBF4. The stoichiometric starting reagents were put into a quartz tube (Φ10 mm × 120 mm) after grinding uniformly and then a vacuum was created inside the tube (1 × 10−3 Pa). The encapsulated quartz tube was placed into a programmable temperature furnace. The temperature was maintained at 700 °C for 24 hours, and then reduced to 600 °C at a rate of 1.0 °C h−1, followed by a further reduction to 500 °C at a rate of 2.0 °C h−1, and finally it was allowed to cool to room temperature. The crystals of the three aluminum fluorides inside the quartz tubes were taken out for structural determination.
Li2KAl2F9 polycrystalline samples were obtained by a solid state reaction. LiF (0.280 g), KF (0.314 g), and AlF3 (0.906 g) (molar ratio 2:1:2) were mixed, ground, and put into a quartz tube. The mixture was heated to 500 °C and this temperature was maintained for 24 hours, followed by cooling it down to room temperature. This process was repeated three times. The purity of the sample was confirmed by powder X-ray diffraction (XRD) (Fig. 1). Although a series of attempts under different experimental conditions were carried out, the pure polycrystalline samples of AlF3 (P6/mmm) and BaAlF5 (I4/m) were not obtained.
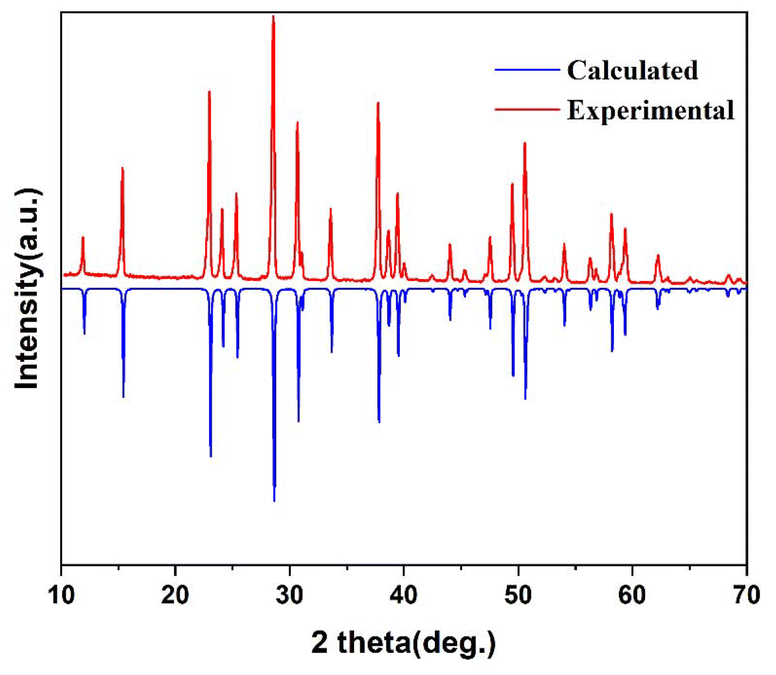 | ||
| Fig. 1 Experimental and calculated XRD patterns of Li2KAl2F9. | ||
Single-crystal XRD
High-quality crystals were selected and used for single-crystal XRD measurements. Single crystal XRD data were recorded on a Bruker D8 Venture diffractometer using Mo Kα radiation (λ = 0.71073 Å) at room temperature. The intensity, reduction, and cell refinement investigations were carried out on a Bruker SAINT.51 Crystal structures were solved by the direct methods and refined by full matrix least-squares on F2 with OLEX2 software.52 These structures were verified using the ADDSYM algorithm from PLATON.53 IUCDcheckCIF (https://checkcif.iucr.org) was used to check the consistency and integrity of the structure file in CIF format. The crystal data and refined parameters, atomic coordinates, bond lengths, and angles are provided in Tables S1 and S7.†Powder X-ray diffraction (XRD)
Powder XRD data were collected with a Bruker D2 PHASER diffractometer (Cu Kα radiation with λ = 1.5418 Å, 2θ = 10 to 70°, scan step width = 0.02°, and counting time = 1 s/step).Thermal analysis
Thermal gravimetric (TG) analysis and differential scanning calorimetry (DSC) were measured on a simultaneous NETZSCH-STA 449 F3 thermal analyzer instrument. Li2KAl2F9 polycrystalline sample was heated from 40 to 1000 °C with a heating rate of 5 °C min−1 in a Pt crucible under an atmosphere of flowing N2.Infrared (IR) spectroscopy
The IR spectroscopy was conducted using a Shimadzu IR Affinity1 spectrometer at room temperature, covering the wavelength range of 400 to 4000 cm−1. The sample and dried KBr were thoroughly mixed before the test.UV-vis-NIR diffuse reflectance spectroscopy
The UV-vis-NIR diffuse reflectance spectrum was measured at room temperature with a Shimadzu SolidSpec-3700 DUV spectrophotometer in the 200–2600 nm wavelength range.Calculation details
The electronic structure and optical properties were calculated by using the DFT method implemented in the CASTEP package.54,55 The Perdew–Burke–Ernzerhof (PBE) functional within GGA with the scheme was adopted to describe the optical performance. Because of the discontinuities in the exchange correlation energy, the GGA often understates the band gap under the norm-conserving pseudopotential (NCP).56 Therefore, the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional was chosen to provide a more accurate bandgap value.57 Based on the scissor-corrected electron structure, the linear optical properties were obtained using the Kramers–Kronig transform. The ion–electron interactions were modeled using the norm-conserving pseudopotentials for all elements. The cutoff energy was set to 900 eV for the self-consistent field (SCF) calculations, and the convergence criterion for the total energy was 1 × 10−7 eV per atom. The Monkhorst–Pack k-point meshes spanning less than 0.04 Å−1 in the Brillouin zone were used to ensure sufficient accuracy of the calculated results. From the dielectric function, the linear optical properties are obtained: ε (ω) = ε1 (ω) + iε2 (ω), where ε1 (ω) and ε2 (ω) are the real and imaginary parts of the dielectric function, respectively. The imaginary part ε2 of the dielectric function can be calculated from the electronic structure, and the real part ε1 can be calculated using the Kramers–Kronig transformation, which allows the calculation of the refractive index and birefringence (Δn).Results and discussion
Crystal structure
AlF3 crystallizes in the hexagonal central space group P6/mmm. The asymmetric unit comprises one Al atom and two F atoms. The Al atom is coordinated with six fluorine atoms forming an [AlF6]3− octahedron, with the Al–F bond lengths ranging from 1.7712(5) to 1.7798(15) Å. The [AlF6]3− octahedron is interconnected through sharing edges resulting in the [Al5F24]9− polymer (Fig. 2a). Then, the 12-membered ring pores are formed by [Al5F24]9− through the interconnection of fluorine atoms (Fig. 2b), which gives rise to a three-dimensional (3D) architecture along the c-axis direction. | ||
| Fig. 2 Crystal structures of AlF3, BaAlF5, and Li2KAl2F9. (a) [Al5F24]9− polymer. (b) 12-membered ring pore structure along the c-axis for AlF3. (c) 1∞[Al6F29]11− and 1∞[AlF5]2− chains. (d) Ba atom is embedded within the pore structure formed by the double chain along the c-axis for BaAlF5. (e) 1∞[Al6F28]10− chain. (f) Li and K atoms are embedded in the gaps of the chain along the [101] direction for Li2KAl2F9. | ||
BaAlF5 crystallizes in the tetragonal center space group I4/m. In the structure, the asymmetric unit comprises two Ba atoms, three Al atoms, and nine F atoms. The [AlF6]3− octahedron forms two distinct 1D infinite chain structures of 1∞[Al6F29]11− and 1∞[AlF5]2− through the sharing of fluorine atoms, forming a unique lattice structure along the c-axis direction (Fig. 2c and d). The Ba atom exhibits two coordination modes of [BaF8]6− and [BaF9]7− filling in the pore structure to achieve charge balance (Fig. S1†). It is noteworthy that in BaAlF5, the Ba–O–F framework interpenetrates with the Al–O–F framework, resulting in a fully integrated 3D structure.
Li2KAl2F9 exhibits a monoclinic centrosymmetric crystal structure in the space group C2/m. The asymmetric unit comprises one Al atom, two Li atoms, one K atom, and six F atoms. The [AlF6]3− octahedron forms a 1D infinite chain-like structure of 1∞[Al6F28]10−, which is neatly parallelized along the [101] direction (Fig. 2e and f). The K atom is coordinated with the F atom to generate the [KF8]7− polyhedron, which interconnects by sharing edges to form a 1D infinite chain of 1∞[KF4]3− (Fig. S2†). The Li atom exhibits two coordination modes ([LiF4]3− and [LiF5]4−) and forms the 1D infinite chains of 1∞[Li2F6]4− by sharing F atoms (Fig. S2†). The overall structure consists of three 1D infinite chains, 1∞[Al6F28]10−, 1∞[KF4]3−, and 1∞[Li2F6]4−, which finally connect through shared F atoms to form a 3D framework structure.
Bond valence calculations (BVSs) yielded bond valences for Al3+ and F− ions of 3.177 and 1.060–1.029 in AlF3; 2.917–3.175 and 0.949–1.134 in BaAlF5; 3.047 and 0.967–1.031 in Li2KAl2F9, respectively, and the detailed data calculations are shown in Tables S2, S4 and S6†, which are in agreement with normal valence consistency.
Thermal properties and optical properties
The TG-DSC curve of Li2KAl2F9 reveals prominent exothermic peaks at 587 and 620 °C (Fig. S3a†). The minor mass loss preceding the absorption peak at 587 °C is attributed to the slight volatilization of fluorine. Following the absorption peak at 620 °C, the product is gradually decomposed. To further investigate the thermal behavior of Li2KAl2F9, its polycrystalline samples were subjected to calcination experiments (Fig. S3b†). Experiments demonstrated that the samples exhibited distinct characteristics before and after calcination experiments, indicating that the compounds underwent incongruent melting.UV-vis-NIR diffuse reflectance tests were successfully performed on the synthesized Li2KAl2F9, covering the wavelength range of 200–2600 nm (Fig. S4a†). The spectral analysis reveals a cut-off edge below 200 nm. The absence of d–d and f–f electron transitions for alkali/alkaline earth metals and Al3+ ions, along with the introduction of electronegative F+ ions, results in a reduced UV cutoff edge.
The IR spectra of Li2KAl2F9 powder samples are presented in Fig. S4b†. Based on both experimental and theoretical calculations, it is demonstrated that the prominent absorption peaks observed at 660, 596, and 478 cm−1 may originate from the stretch–bend vibration of the [AlF6] unit (Fig. S4c and d†).
Theoretical calculations
The relationship between the electronic structure and optical properties is further elucidated through first-principles calculations. The energy band structure and partial density of states (PDOS) were computed for the three compounds. The calculations indicate that all three compounds possess substantial band gaps, and the GGA band gaps of AlF3 (8.853 eV), BaAlF5 (7.187 eV), and Li2KAl2F9 (7.729 eV) are shown in Fig. 3a and c. By analyzing the PDOS (Fig. 3d and f), it is evident that the dominant contribution to the valence band (VB) in all three compounds originates from F 2p orbitals, while the conduction band (CB) primarily arises from Al 3s3p, Ba 5d, K 4s and Li 2s orbitals. To accurately analyze the band gap values of the three compounds, high-precision DFT calculations of the band gap using the HSE06 hybrid functional were performed and the results are as follows: AlF3 – 9.897 eV (corresponding to 125 nm), BaAlF5 – 8.853 eV (corresponding to 140 nm), and Li2KAl2F9 – 9.598 eV (corresponding to 129 nm). It is proved that all three new compounds have large band gaps and DUV cutoff edges. The scissors operator is introduced, and the birefringence values of AlF3, BaAlF5, and Li2KAlF9 are calculated to be 0.0006, 0.0044, and 0.0056 at 1064 nm (Fig. 3g and i). The potential of the three newly discovered aluminum fluorides to serve as materials for zero-order waveplates is demonstrated. | ||
| Fig. 3 Calculated optical properties of AlF3, BaAlF5, and Li2KAl2F9. Electronic structures of (a) AlF3, (b) BaAlF5, and (c) Li2KAl2F9. Density of states of (d) AlF3, (e) BaAlF5, and (f) Li2KAl2F9. Calculated birefringence of (g) AlF3, (h) BaAlF5, and (i) Li2KAl2F9. | ||
The hybridization of Al atoms with O/F atoms via sp3 results in the formation of [AlOmFn] (m + n = 6) octahedral structures, exhibiting ten distinct coordination modes (Fig. S5†). The Gaussian 09 program package was utilized to conduct DFT calculations at the B3LYP/6-31G level, aiming to investigate the microscopic changes in F atoms following the substitution of O atoms to reflect the changes before and after substitution (Table S8†). It is found that the band gap is improved favorably when the F atom replaces the O atom, while complete substitution results in a threefold enhancement of the [AlF6] unit HOMO–LUMO band gap compared to that of the [AlO6] group (Fig. 4). The band gap of the [AlF6] unit, demonstrated by both experimental and theoretical models, exceeds 9.0 eV. By analyzing the electron localization function maps (Fig. S6a and c†), it is evident that the distribution of electron density exhibits an elliptical shape with small polarizability anisotropy. Therefore, the perfluorinated [AlF6] unit exhibits a high microscopic band gap and small anisotropic polarizability, making it an excellent building group for DUV zero-order waveplate materials.
 | ||
| Fig. 4 The HOMO–LUMO gaps of the experimental structure and theoretical model of [AlOmFn] (m + n = 6) units. | ||
Conclusions
In summary, using a synergistic combinatorial strategy of the perfluorinated [AlF6] unit (with small polarizability anisotropy and a large HOMO–LUMO band gap) and alkali/alkaline-earth metals, the three new compounds AlF3, BaAlF5, and Li2KAl2F9 were successfully synthesized. These compounds exhibit a large bandgap range of 8.85 to 9.89 eV and small birefringence ranging from 0.0006 to 0.0056 at 1064 nm. These characteristics make them promising candidates for DUV zero-order waveplates. The analysis of microscopic variations in the HOMO–LUMO band gap resulting from F atom substitution demonstrates that the [AlF6] group serves as a suitable building block for DUV applications, thereby facilitating the search for suitable DUV zero-order waveplate materials, which presents novel approaches for sourcing materials to design zero-order waveplates.Author contributions
Yun Yang and Xifa Long supervised the research and revision of the manuscript. Shaohua Liu designed and performed the experiments, wrote the first draft and revised it. Ke Li did the theoretical calculations. Lili Zhang and Xiaotian Ma assisted with the experimental work. All authors participated in the discussion.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Science and Technology Plan Project of Yili Kazakh Autonomous Prefecture (YZ2022B021), National Natural Science Foundation of China (22375215), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0880000), CAS Youth Interdisciplinary Team (JCTD-2021-18), West Light Foundation of CAS (2021-XBQNXZ-004), Outstanding Youth Science Fund Project of Natural Science Foundation of Xinjiang (2022D01E90), Xinjiang Tianshan Talent Program (2023TSYCJU0003), and Xinjiang Tian chi Talent Program, Key Training Object of Talent Project of Urumqi.References
- Y. Y. Yang, Y. Guo, Y. G. Chen, X. W. Hu, X. Zhang and X. M. Zhang, Inorg. Chem. Front., 2022, 9, 5469–5477 Search PubMed.
- Z. Y. Zhang, D. Xu, A. Tudi, Z. H. Yang, S. J. Han and S. L. Pan, Sci. China Mater., 2024, 67, 914–920 Search PubMed.
- B. Boulbry, B. Bousquet, B. Le Jeune, Y. Guern and J. Lotrian, Opt. Express, 2001, 9, 225–235 CrossRef CAS.
- J. L. Vilas, L. M. Sanchez-Brea and E. Bernabeu, Appl. Opt., 2013, 52, 1892–1896 CrossRef PubMed.
- M. Emam-Ismail, Opt. Commun., 2010, 283, 4536–4540 CrossRef CAS.
- M. Cheng, W. Q. Jin, Z. H. Yang and S. L. Pan, Inorg. Chem., 2020, 59, 13014–13018 CrossRef CAS PubMed.
- H. K. Ren, Z. X. Wu, Z. P. Wang, F. Wang, F. Q. Li, X. Sun and X. G. Xu, In: Proceedings of Second Symposium on Novel Technology of X-ray Imaging., Hefei: SPIE, 2018.
- M. Mutailipu, M. Zhang, H. P. Wu, Z. H. Yang, Y. H. Shen, J. L. Sun and S. L. Pan, Nat. Commun., 2018, 9, 3089 CrossRef PubMed.
- G. Q. Shi, Y. Wang, F. F. Zhan, B. B. Zhang, Z. H. Yang, X. L. Hou, S. L. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS.
- X. F. Wang, Y. Wang, B. B. Zhang, F. F. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 14119–14123 CrossRef CAS.
- Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 2150–2154 CrossRef CAS PubMed.
- Z. T. Yan, J. B. Fan, S. L. Pan and M. Zhang, Chem. Soc. Rev., 2024, 53, 6568–6599 RSC.
- B. B. Zhang, G. Q. Shi, Z. H. Yang, F. F. Zhang and S. L. Pan, Angew. Chem., Int. Ed., 2017, 56, 3916–3919 CrossRef CAS.
- Q. Z. Zhang, R. An, Z. H. Yang, X. F. Long, S. L. Pan and Y. Yang, Sci. China Chem., 2024, 67, 2155–2170 CrossRef CAS.
- M. J. Dodge, Appl. Opt., 1984, 23, 1980–1985 CrossRef CAS PubMed.
- Z. C. Wu, H. M. Li, Z. Y. Zhang, X. Su, H. S. Shi and Y. N. Huang, Inorg. Chem., 2024, 63, 1674–1681 CrossRef CAS PubMed.
- G. Ghosh, Opt. Commun., 1999, 163, 95–102 CrossRef.
- G. H. Li, J. H. Li and Y. Li, Appl. Opt., 1990, 29, 1870–1871 CrossRef.
- L. Grineviciute, M. Andrulevicius, A. Melninkaitis, R. Buzelis, A. Selskis, A. Lazauskas and T. Tolenis, Phys. Status Solidi A, 2017, 214, 1700764 CrossRef.
- A. V. Samoylov, V. S. Samoylov, A. P. Vidmachenko and A. V. Perekhod, J. Quant. Spectrosc. Radiat. Transfer, 2004, 88, 319–325 CrossRef.
- S. Y. Zhang, X. Wu, Y. T. Song, D. Q. Ni, B. Q. Hu and T. Zhou, J. Cryst. Growth, 2003, 252, 246–250 CrossRef.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef PubMed.
- F. Liu, P. F. Gong, Z. S. Lin and L. Kang, Coord. Chem. Rev., 2025, 526, 216349 CrossRef.
- H. Y. Sha, Y. R. Shang, Z. J. Wang, R. B. Su, C. He, X. M. Yang and X. F. Long, Adv. Opt. Mater., 2023, 11, 2300987 CrossRef.
- A. Tudi, Z. J. Li, C. W. Xie, T. Baiheti, E. Tikhonov, F. F. Zhang, S. L. Pan and Z. H. Yang, Adv. Funct. Mater., 2024, 34, 2409716 CrossRef.
- Y. C. Yan, Y. N. Chen, B. Jiang, Q. Jing and J. Zhang, New J. Chem., 2021, 45, 19883–19888 RSC.
- Z. Akdeniz and P. A. Madden, J. Phys. Chem. B, 2006, 110, 6683–6691 CrossRef.
- C. Martineau, F. Fayon, M. R. Suchomel, M. Allix, D. Massiot and F. Taulelle, Inorg. Chem., 2011, 50, 2644–2653 CrossRef.
- C. R. Morelock, J. C. Hancock and A. P. Wilkinson, J. Solid State Chem., 2014, 219, 143–147 CrossRef CAS.
- S. F. Parker, A. J. Ramirez-Cuesta and L. L. Daemen, RSC Adv., 2020, 10, 25856–25863 RSC.
- Z. Fang, X. X. Jiang, M. H. Duan, Z. Y. Hou, C. C. Tang, M. J. Xia, L. J. Liu, Z. S. Lin, F. D. Fan, L. Bai and C. T. Chen, Chemi. Eur. J., 2018, 24, 7856–7860 CrossRef CAS.
- F. J. Guo, J. Han, S. C. Cheng, S. J. Yu, Z. H. Yang and S. L. Pan, Inorg. Chem., 2019, 58, 8237–8244 CrossRef CAS.
- M. Mutailipu, M. Zhang, Z. H. Yang and S. L. Pan, Acc. Chem. Res., 2019, 52, 791–801 CrossRef CAS PubMed.
- H. K. Su, J. H. Jiao, S. B. Wang, D. H. An and M. Zhang, Dalton Trans., 2024, 53, 932–937 RSC.
- J. Sun, M. Mutailipu and S. L. Pan, Chem. Eur. J., 2021, 27, 8698–8703 CrossRef CAS PubMed.
- Z. T. Yan, D. D. Chu, Z. H. Yang, S. L. Pan and M. Zhang, Chem. Commun., 2024, 60, 15047–15050 RSC.
- S. Bai, X. D. Zhang, B. B. Zhang, L. Li and Y. Wang, Inorg. Chem., 2021, 60, 10006–10011 CrossRef CAS PubMed.
- C. Chen, D. Y. Dou, Y. J. Bai, B. B. Zhang and Y. Wang, Inorg. Chem. Front., 2024, 11, 6020–6027 RSC.
- B. Jiang, S. Shu, Z. H. Yang, F. F. Zhang, M. Zhang and S. L. Pan, Dalton Trans., 2022, 51, 3964–3969 RSC.
- J. H. Jiao, M. Zhang and S. L. Pan, Angew. Chem., Int. Ed., 2023, 62, e202217037 CrossRef CAS PubMed.
- H. K. Liu, Y. Wang, B. B. Zhang, Z. H. Yang and S. L. Pan, Chem. Sci., 2020, 11, 694–698 RSC.
- Y. Wang, J. Han, J. B. Huang, Z. H. Yang, S. Z. Huang and S. L. Pan, New J. Chem., 2020, 44, 9852–9857 RSC.
- Y. Wang, J. Han, J. B. Huang, Z. H. Yang and S. L. Pan, Inorg. Chem., 2020, 59, 810–817 CrossRef CAS.
- H. P. Wu, H. W. Yu, S. L. Pan and P. S. Halasyamani, Inorg. Chem., 2017, 56, 8755–8758 CrossRef CAS PubMed.
- Z. T. Yan, D. D. Chu, M. Zhang, Z. H. Yang and S. L. Pan, Adv. Opt. Mater., 2023, 11, 2202353 CrossRef CAS.
- Y. Yang, S. Z. Huang and S. L. Pan, J. Mater. Chem. C., 2022, 10, 11232–11238 RSC.
- H. W. Yu, J. S. Young, H. P. Wu, W. G. Zhang, J. M. Rondinelli and S. Halasyamani, Adv. Opt. Mater., 2017, 5, 1700840 CrossRef.
- M. Mutailipu, Z. H. Yang and S. L. Pan, Acc. Mater. Res., 2021, 2, 282–291 CrossRef CAS.
- Q. R. Ding, X. M. Liu, S. G. Zhao, Y. S. Wang, Y. Q. Li, L. N. Li, S. Liu, Z. S. Lin, M. C. Hong and J. H. Luo, J. Am. Chem. Soc., 2020, 142, 6472–6476 CrossRef CAS PubMed.
- G. P. Han, B. H. Lei, Z. H. Yang, Y. Wang and S. L. Pan, Angew. Chem., Int. Ed., 2018, 57, 9828–9832 CrossRef CAS PubMed.
- SAINT, Version 7.60A, Bruker Analytical X-ray Instruments, Inc., Madison, WI, 2008 Search PubMed.
- O. Dolomanov, L. Bourhis, R. Gildea, J. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Phys. Rev. B:Condens. Matter Mater. Phys., 1990, 41, 1227–1230 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. G. G. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Hamann, M. Schlüter and C. Chiang, Phys. Rev. Lett., 1979, 43, 1494–1497 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2423523–2423525. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00356c |
| This journal is © The Royal Society of Chemistry 2025 |