
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutonomous laboratories in China: an embodied intelligence-driven platform to accelerate chemical discovery
Jinpeng
Li†
a,
Chuxuan
Ding†
a,
Daobin
Liu†
 *a,
Linjiang
Chen
a and
Jun
Jiang
*ab
*a,
Linjiang
Chen
a and
Jun
Jiang
*ab
aState Key Laboratory of Precision and Intelligent Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: ldbin@ustc.edu.cn; jiangj1@ustc.edu.cn
bHefei National Laboratory, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 13th June 2025
Abstract
The emergence of autonomous laboratories—automated robotic platforms integrated with rapidly advancing artificial intelligence (AI)—is poised to transform research by shifting traditional trial-and-error approaches toward accelerated chemical discovery. These platforms combine AI models, hardware, and software to execute experiments, interact with robotic systems, and manage data, thereby closing the predict-make-measure discovery loop. However, key challenges remain, including how to efficiently achieve autonomous high-throughput experimentation and integrate diverse technologies into cohesive systems. In this perspective, we identify the fundamental elements required for closed-loop autonomous experimentation: chemical science databases, large-scale intelligent models, automated experimental platforms, and integrated management/decision-making systems. Furthermore, with the advancement of AI models, we emphasize the progress from simple iterative-algorithm-driven systems to comprehensive intelligent autonomous systems powered by large-scale models in China, which enable self-driving chemical discovery within individual laboratories. Looking ahead, the development of intelligent autonomous laboratories into a distributed network holds great promise for further accelerating chemical discoveries and fostering innovation on a broader scale.
1 Introduction
The scale of chemical research spans from the microscopic realm of atoms and molecules to the macroscopic domain of material systems, making accurate prediction and comprehensive description inherently challenging. Traditional research paradigms, which primarily rely on exhaustive trial-and-error approaches, struggle to navigate the vast chemical space and often fail to uncover the mechanisms underlying materials. Furthermore, the search for optimal formulations and processes often converges on local optima, thereby limiting global exploration. The complexity and high-dimensionality of chemical systems further impede the elucidation of structure–property relationships, exacerbating the gap between fundamental research and practical application. Consequently, the innovation of traditional research paradigms is urgently required.Since the term ‘artificial intelligence (AI)’ was first coined by McCarthy in 1956, it has become a key driver of transformative developments in science.1,2 The 2024 Nobel Prizes in physics3,4 and chemistry5,6 both highlighted advancements in AI, recognizing its transformative role in modeling complex physical systems and predicting biochemical structures. A landmark in the application of AI is its ability to efficiently handle heterogeneous data, enabling the interpretation and understanding of complex datasets in genomics, proteomics, metabolomics2,7 and spectroscopy.8,9 By deciphering high-dimensional correlations within these datasets, AI can accelerate high-precision simulations to elucidate structure–property relationships,10,11 further achieving more efficient predictions of highly anticipated targets.5,6,12,13 AlphaFold 2 (ref. 5 and 13) represents a groundbreaking advancement in protein structure prediction, utilizing deep neural networks and self-attention mechanisms to achieve high-precision results. The updated AlphaFold 3 (ref. 6) enables joint structure prediction of complexes, significantly enhancing the accuracy of biomolecular interaction modeling and offering transformative potential for drug design and disease diagnosis. In parallel, DeepMind developed the GNoME intelligent model14,15 for crystal structure prediction, which has expanded the number of known stable materials nearly tenfold to 421![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Beyond predicting material properties, the recommendation of synthesis strategies for targeted materials is also in high demand. For example, several AI-assisted tools for molecular synthesis have been developed to optimize experimental workflows, including AiZynthFinder,16 AIDDISON17 and Chematica (now known as SYNTHIA™).18 Cernak et al.19 conducted retrosynthetic studies on 12 COVID-19 antiviral drugs using SYNTHIA™, which identified simpler and more efficient synthesis routes for 11 of them, thus significantly alleviating pressure on existing supply chains. To date, the number of research fields integrated with AI are rapidly evolving and hold immense untapped potential.
000. Beyond predicting material properties, the recommendation of synthesis strategies for targeted materials is also in high demand. For example, several AI-assisted tools for molecular synthesis have been developed to optimize experimental workflows, including AiZynthFinder,16 AIDDISON17 and Chematica (now known as SYNTHIA™).18 Cernak et al.19 conducted retrosynthetic studies on 12 COVID-19 antiviral drugs using SYNTHIA™, which identified simpler and more efficient synthesis routes for 11 of them, thus significantly alleviating pressure on existing supply chains. To date, the number of research fields integrated with AI are rapidly evolving and hold immense untapped potential.
An effective AI-driven approach relies on large amounts of high-quality, structured data as its foundation to develop robust prediction models. However, the majority of available data, particularly experimental data, suffers from significant issues such as non-standardization, fragmentation, and poor reproducibility.20,21 In this context, automated robotic platforms are being rapidly developed to generate high-quality experimental data in a standardized and high-throughput manner while minimizing manual effort.22–25 More importantly, these platforms can fully leverage their advantages when integrated with AI algorithms. Such integration not only automates routine tasks but also enables complex decision-making processes, optimization of synthesis methods, and even planning of experimental workflows.15,25–31 A pioneering study was conducted by Cooper et al.,27 who developed a mobile chemist capable of autonomously conducting high-throughput photocatalyst selection, outperforming humans through the application of Bayesian optimization. They further designed a fully autonomous solid-state workflow involving three multipurpose robots for powder X-ray diffraction (PXRD) experiments.28 The “Chemputer” system, developed by Cronin et al., integrates literature analysis, protocol customization, organic synthesis, and characterization, demonstrating extraordinary capability in automatic synthesis.29 The closed-loop self-driving laboratory, developed by the Aspuru-Guzik group, implements a design-make-test-analyze cycle to accelerate the discovery of new organic semiconductor laser materials.30 The A-Lab, developed by DeepMind,15,31 utilizes computational tools, literature data, machine learning, and active learning to plan and interpret the outcomes of experiments performed by robotics, addressing the challenges associated with handling and characterizing solid inorganic powders. Therefore, autonomous laboratories that integrate automated robotic platforms with AI are capable of conducting experiments that were once deemed unfeasible and thus may expand the frontiers of scientific exploration.
In this perspective, we first summarize the fundamental elements required for autonomous laboratories to satisfy the complex demands of autonomous experimentation. The discussion primarily focuses on the current state of autonomous laboratories in China, where development has progressed from simple iterative-algorithm-driven systems to comprehensive intelligent autonomous systems powered by large-scale models. It is worth noting that most autonomous laboratories are established to tackle specific challenges and operate in isolation, with limited inter-lab communication and data sharing. To this end, we explore the future prospects of these distributed autonomous laboratories, emphasizing the adoption of coordinated strategies, such as cloud-based systems, to achieve seamless data and resource integration across laboratories.
2 Fundamental elements of autonomous laboratories
Autonomous laboratories are advanced robotic platforms equipped with embodied intelligence, enabling them to execute experiments, interact with robotic systems, and manage data.26 These capabilities allow them to effectively close the predict-make-measure discovery loop.32 To achieve fully autonomous, self-driving laboratories, it is essential to integrate several fundamental elements: chemical science databases, large-scale intelligent models, automated experimental platforms, and management and decision systems. These elements work synergistically to create a seamless, closed-loop research environment (Fig. 1). | ||
| Fig. 1 The four fundamental elements of autonomous laboratories. | ||
2.1 Chemical science database
The chemical science database is a cornerstone of autonomous laboratories, serving as the backbone for managing and organizing diverse chemical data. By integrating, processing, and structuring multimodal data into an AI-powered framework, the database provides essential support for experimental design, prediction, and optimization.Multimodal data form the backbone of chemical science databases, encompassing information ranging from synthesis planning to property prediction. These data resources include structured entries from proprietary databases (e.g., Reaxys and SciFinder) and open-access platforms (e.g., ChEMBL33 and PubChem34), as well as unstructured data extracted from scientific literature, patents, and experimental reports. The extraction of unstructured data is extensively achieved using Natural Language Processing (NLP) techniques.35 Consequently, toolkits such as ChemDataExtractor,36 ChemicalTagger,37 and OSCAR4,38 which leverage named entity recognition (NER), have been developed for the extraction of chemical reactions, compounds, and properties from textual documents. Image recognition further enhances the robotic understanding of chemical diagrams and molecular structures.39 Together, these methods represent complementary approaches to converting unstructured data into formats directly usable by robotic systems.
Following data mining, databases are constructed by intelligent methods to efficiently store, manage, and facilitate the retrieval of processed data for subsequent analysis and decision-making. The processed data can be further organized and represented in the form of knowledge graphs (KGs), which provide a structured representation of data and have been widely applied in various domains. Canonical methods for KG construction primarily focus on extracting logical rules based on semantic patterns.40 With the advancement of AI, methods for KG construction based on large language models (LLMs) have recently gained widespread adoption, demonstrating superior performance and enhanced interpretability for human understanding.41,42 Furthermore, to address issues such as contextual noise and knowledge hallucination,43 in a recent study, a general KG construction framework, named SAC-KG, is proposed that leverages LLMs as skilled automatic constructors for domain KGs.44
2.2 Large-scale intelligent model
Interpretable predictive models and advanced algorithms are crucial components of the autonomous laboratory workflow. They enable efficient data processing, accurate outcome prediction, and informed decision-making at each experimental stage. By leveraging data from previous experiments, predictive models can forecast the results of proposed experiments more effectively. For instance, in a study by Moosavi et al.,45 involving genetic algorithm (GA)-guided robotic platform optimized crystallinity and phase purity in metal–organic frameworks, they explored a nine-parameter space through 90 experiments across three generations. A random forest model, trained iteratively on prior data, predicted outcomes and excluded experiments likely to yield suboptimal results. GAs46 are particularly effective for handling large numbers of variables and are widely applied in the discovery of novel catalysts and their synthesis optimization.47,48Beyond GAs, the SNOBFIT algorithm49 improves search efficiency by combining local and global search strategies. It has been successfully applied to optimize chemical reactions in continuous flow reactors.50 Another widely used method in autonomous laboratories is Bayesian optimization, which minimizes the number of trials needed to achieve convergence.27,31,51–53 The performance of Bayesian optimization is highly dependent on the choice of the surrogate model, with Gaussian processes (GPs) and random forests (RFs) being the most common for regression tasks.20,54 The Phoenics algorithm, based on the Bayesian neural network (BNN), achieves faster convergence than GPs and RFs.55 It has been integrated into ChemOS (a versatile software package) for several automated platforms, including the Ada self-driving laboratory for thin-film materials,53 and a mobile robotic chemist by Burger et al.27 for optimizing aqueous photocatalysts.
Alongside advanced algorithms, another critical aspect of optimizing workflows for intelligent models is the iterative experiment-theory feedback loop. Automated theoretical calculations, such as density functional theory (DFT),56,57 provide valuable prior knowledge and bridge the gap between theory and experiment. This data fusion enhances adaptive learning capabilities, allowing models to continuously update and refine their predictions. As a result, these intelligent models drive the development of closed-loop iterative automation processes.51,52
2.3 Automated experimental platform
Automated robotic platforms are essential for executing self-driving, high-throughput experiments and generating high-quality, standardized experimental data. In addition, these platforms can take over time-consuming and repetitive tasks traditionally performed by humans,26 allowing researchers to focus on proposing new theories or mechanisms, thereby contributing to the optimization of the workforce structure in society. The experimental protocol is a key foundational resource to plan experiments in autonomous laboratories. This protocol encompasses procedural templates from past experiments and configurations of automated experimental stations within the lab.30 Serving as a systematic framework, it outlines the necessary steps and procedures for carrying out experiments effectively. To formalize these protocols and convert them into machine-executable actions, several approaches have been developed. For example, synthesis action sequences encode detailed instructions required for robotic systems to conduct reactions. These sequences can be transformed into machine-executable actions using methods such as pattern matching combined with expert-defined heuristics29 or deep learning-based sequence-to-sequence models.58Once experimental protocols are received via an API, robotic systems execute the required actions with high precision. Autonomous experimental robots are equipped with advanced capabilities to perform complex tasks independently. They typically feature dexterous robotic arms (single27,52 or dual24,59) with a high degree of freedom for precise manipulation, mobile platforms for enhanced versatility, and sensing systems such as IR projectors (for depth sensing),51 laser scanners (for point cloud generation),27 and RGB sensors (for object recognition)51,52 to achieve accurate perception. To navigate and operate efficiently in dynamic environments, these robots employ high-precision localization and mapping methods, including SLAM,27,51 six-point localization (for pose determination),27 ArUco labels (for visual marker tracking)52 and so on.
Robotic systems are seamlessly integrated with automation software and real-time feedback mechanisms, enabling the optimization of experimental workflows and significantly enhancing reproducibility.60,61 When combined with automated workstations, these systems can execute complex, dexterous experiments and manage entire workflows—spanning synthesis, characterization, and testing—with minimal human intervention. For instance, Lunt et al.28 designed a solid-state workflow incorporating a PXRD instrument, two grinding stations, and a Chemspeed liquid dispensing platform to conduct PXRD experiments efficiently. Similarly, Cooper et al.27 developed a robotic system with eight workstations to identify photocatalyst mixtures for hydrogen production, ultimately achieving formulations six times more active than the initial ones. Further advancing the field, Zhu et al.52 implemented a system with fourteen workstations, featuring dedicated regions for auto-synthesis, auto-characterization, and auto-performance testing. This comprehensive system enables fully automated experimental processes, accelerating catalyst discovery and optimization.
2.4 Management and decision system
The management and decision system is a sophisticated, multi-layered framework designed to integrate and coordinate critical components for task distribution and workflow orchestration in autonomous laboratories.62 This system unifies chemical science databases, large-scale intelligent models, and automated experimental equipment into a single platform, enabling a closed-loop iteration between experimental execution and intelligent prediction.26,63 Standardized communication protocols are essential for flexibly connecting the mentioned software and hardware modules while facilitating seamless interaction with human researchers. For example, the experimental protocols must be translated into machine-executable commands at a more basic level, such as JSON, XML, or other specific experimental scripting languages,30,51,64 to serve as instruction sets for experimental execution. A classic example is the Chempiler system within the Chemputer architecture,29,65 which uses the GraphML format to map paths between source and target flasks and employs the ChASM scripting language to code synthetic procedures, ensuring precise control over all implemented machine operations.Once the instruction sets are transmitted, large-scale models and advanced algorithms can efficiently steer the decision-making system, enabling the optimization and seamless coordination of experimental workflows for greater efficiency and adaptability. After automated experimental platforms generate vast amounts of data, effective data management becomes crucial to ensure both the integrity and usability of the data. Cloud computing infrastructures and big data techniques have emerged as viable solutions for storing and processing large datasets, offering the flexibility and scalability necessary to handle extensive volumes of information.66 Additionally, techniques such as dimensionality reduction and anomaly detection can be applied to reduce the size of datasets while emphasizing valuable data points, thereby facilitating more efficient data analysis.54
The graphical user interface (GUI) serves as a user-friendly, interactive way to control experimental workflows and communicate with the decision system for researchers, as seen in ChemIDEs.29 This interface simplifies the complexity of the underlying processes, providing an intuitive way to visualize data, monitor ongoing experiments, and access real-time analytics.52,64,67 Additionally, the GUI enhances collaboration and reproducibility through features such as experiment logging, protocol sharing, and version control. This enables researchers to quickly assess the status of experiments and make informed decisions without needing to dive into technical details.
3 The current autonomous laboratories in China and future prospects
In the past few years, China has seen significant progress in the development of autonomous laboratories, showcasing the growing integration of AI and robotics in scientific research. Initially, autonomous labs focused on enhancing experimental efficiency and accuracy with single robotic arms and automated equipment. However, as AI models advanced, these labs evolved into autonomous platforms driven by iterative algorithms, which led to the emergence of autonomous experiments driven by a computational ‘brain’ and eventually to fully integrated end-to-end intelligent systems powered by large-scale models. This section highlights key advancements in AI-driven automation in laboratories and, based on current trends, looks ahead to the future of intelligent autonomous systems.3.1 Autonomous platform driven by iterative algorithms
The creation of advanced automation machinery and improvements in robotic systems marked the beginning of China's automation laboratories. Laboratory investigations are now faster, more accurate, and reproducible, thanks to the automation of chemical synthesis and material handling.68 In 2018, Zhu et al. initially reported AIR-Chem, an intelligent robot system for chemistry, in China.69 The system consists of multiple mobile robots for sampling, injection, and synthesis, along with a computer vision (CV) module for real-time monitoring of the synthesis process (Fig. 2a). It operates remotely via cloud computing, autonomously performs the entire experimental process, and adjusts experimental conditions using a gradient descent algorithm. To validate the system's feasibility, the automated synthesis and real-time characterization of CsPbBr3 quantum dots (QDs) have been carried out. Through optimization via iterative algorithms, an improved nucleation theory for perovskite QDs has been proposed. | ||
| Fig. 2 Some autonomous platforms driven by iterative algorithms. (a) AIR-Chem. (b) High-throughput thin-layer chromatography. (c) Automated synthesis and in situ characterization platform of colloidal nanocrystals. (d) AI-Chemist. | ||
Recent advancements in automation technology and the rapid development of AI have significantly accelerated the generation of experimental data. Furthermore, these advancements have enabled the screening and optimization of reaction conditions, facilitating the creation of machine learning models that can predict reaction yields. Mo et al. constructed an automated system for high-throughput thin-layer chromatography (TLC) analysis (Fig. 2b). By utilizing a large amount of data collected under standardized conditions, they built a machine learning (ML) model that associates the structure of organic compounds with their polarity (reflected by the retention factor (Rf)). This model can accurately predict the polarity of organic compounds in various solvent combinations, providing effective guidance for selecting purification conditions and quickly generating and analyzing high-quality TLC data.70 Xu et al. used a self-built high-throughput automated platform to screen a series of metal catalysts and solvents, discovering that [Ir(COD)Cl]2 can achieve the first selective cross-dimerization of sulfonamides, with high yield and good stereoselectivity.71 Additionally, through a comprehensive exploration of the reaction space (600 reactions), they developed a ML model (XGB-MAF) that can predict reaction yields, demonstrating the utility and generalizability of this iridium-catalyzed cross-dimerization method. Fang et al. developed a fully automated system that integrates high-throughput catalyst synthesis, online spectral detection, and photocatalytic reaction condition screening. The system utilizes liquid-core waveguide (LCW) technology to design and build a novel microfluidic photocatalytic microreactor, which can complete ultrafast photocatalytic reactions in seconds and achieve ultra-large-scale screening of up to 10000 reactions per day, providing solid data support for AI applications.72
Additionally, the ongoing integration of machine learning models with automated experimental platforms has enabled the controllable synthesis and reverse design of materials. Zhao et al.73,74 developed a robotic platform capable of controllably synthesizing colloidal nanocrystals with unique physicochemical properties (Fig. 2c). This platform automates the synthesis, in situ characterization, and external validation using initial synthesis parameters determined through data mining of existing literature. This makes it possible to precisely synthesize nanocrystals with the morphologies that are required. Furthermore, they achieved reverse design of colloidal nanocrystal morphology by discovering connections between morphology and structure-directing agents through the training of ML models on an ever-expanding experimental database. Jiang et al.75 reported an AI-guided robotic chemist capable of independently completing the entire process of constructing, characterizing, and testing chiral films. Through experimental absorption spectra and structural/process parameters, a ML model capable of accurately predicting chiral optical activity was constructed, along with an inverse design ML model that can generate chiral films with target chiral optical properties covering the entire visible spectrum (Fig. 2d). This expands the potential of using AI-Chemist to discover and optimize new materials. However, for ML model-driven automated platforms to achieve accurate predictions and reverse design of materials, they often require a large amount of reliable data as training sets, which to some extent limits the application and promotion of this method.
3.2 Iterative autonomous experiments driven by a computational ‘brain’
The automated platform driven by iterative algorithms is essentially a black-box optimization technique that uses experimental data for training to obtain the relationship between design variables and the observed values of the objective function, thereby enabling learning and decision-making. This optimization process lacks systematic prior knowledge and has low efficiency in exploring chemical space, and the interpretability of the optimization results is poor.48 As an essential tool for comprehending molecular behavior and forecasting chemical properties, computational chemistry is ideally suited for prior knowledge in optimization procedures. Computational chemistry can greatly increase the speed and scalability of chemical space exploration by incorporating machine learning.76First-principles computational simulations can obtain microscopic information that is difficult to acquire in the real world, such as adsorption energy and electronic structures. The macroscopic properties of materials often depend on their microscopic characteristics. Therefore, combining machine learning models with first-principles calculations can provide pre-trained models and theoretical support for experiments, thereby guiding the experimental process and accelerating the iteration of materials. Yin et al.77 established an approach that utilizes ML-accelerated theoretical calculations, enabling collaboration between experiments and theory for screening small-sized ordered alloy catalysts. By calculating the solubility and chemical ordering of a third metal element in a PtCo ordered alloy system, as well as the adsorption of related intermediates, they found that the introduction of Cu or Ni into the PtCo alloy is beneficial for the thermodynamic driving force to transition from disorder to order. In contrast, the introduction of Mn and Fe inhibits the disorder-to-order transition of the alloy. Moreover, the synthesized PtCoNi and PtCoCu alloys exhibited excellent oxygen reduction reaction (ORR) performance. This makes it possible to quickly discover potential ordered alloys with high thermodynamic driving force and good performance from the vast design space. Zhang et al.78 used high-throughput DFT calculations to obtain the formation energy Ef and surface stress εsurf of high-entropy intermetallic compounds (HEICs) with different compositions. Based on the obtained 538 DFT datasets, they used crystal graph convolutional neural networks to construct a ML model capable of predicting εsurf and Ef with high accuracy. By further calculating several chemical properties of HEICs, it was found that the difference in the atomic radius and mixing enthalpy were considered key chemical characteristics that respectively influence εsurf and Ef and are expected to become new descriptors for developing HEICs with excellent ORR performance. Wang et al.,79 based on the adhesion energies of 178 metal-oxide interfaces obtained from experiments and 14 highly independent and important physical features obtained through symbolic regression and cross-validation, employed an interpretable ML model to conduct a comprehensive search of over 30 billion mathematical expressions. This led to the development of a physical model that can describe the metal-support interaction (MSI) and accurately predict the adhesion energy and contact angle of metal-oxide interfaces (Fig. 3a). Furthermore, through extensive experiments involving 10 metals and 16 oxides, they formulated and validated principles for the strong metal–metal interactions that occur during encapsulation. These theories have greatly advanced the design and development of supported metal catalysts. Li et al.,80 based on the linear scaling relationship of energy during the sintering process of metal nanoparticles (NPs), obtained a representative set of 323 metal-support pairs. By simulating the sintering kinetics of these metal support pairs, they discovered that the sintering kinetics exhibit a Sabatier principle with respect to MSI. Both excessively strong and weak MSI can lead to the sintering of metal NPs. They also found that the sintering initiation temperature of metal NPs with appropriate MSI is about half of the bulk metal melting temperature of typical NPs (∼3 nm), which is consistent with the long-reported empirical Tammann temperature. In addition, based on the revealed Sabatier principle and scaling relationships, high-throughput screening of carrier combinations with different energies was conducted, resulting in carriers that increase the sintering temperature. This has greatly advanced the design of ultra-stable supported metal NP catalysts.
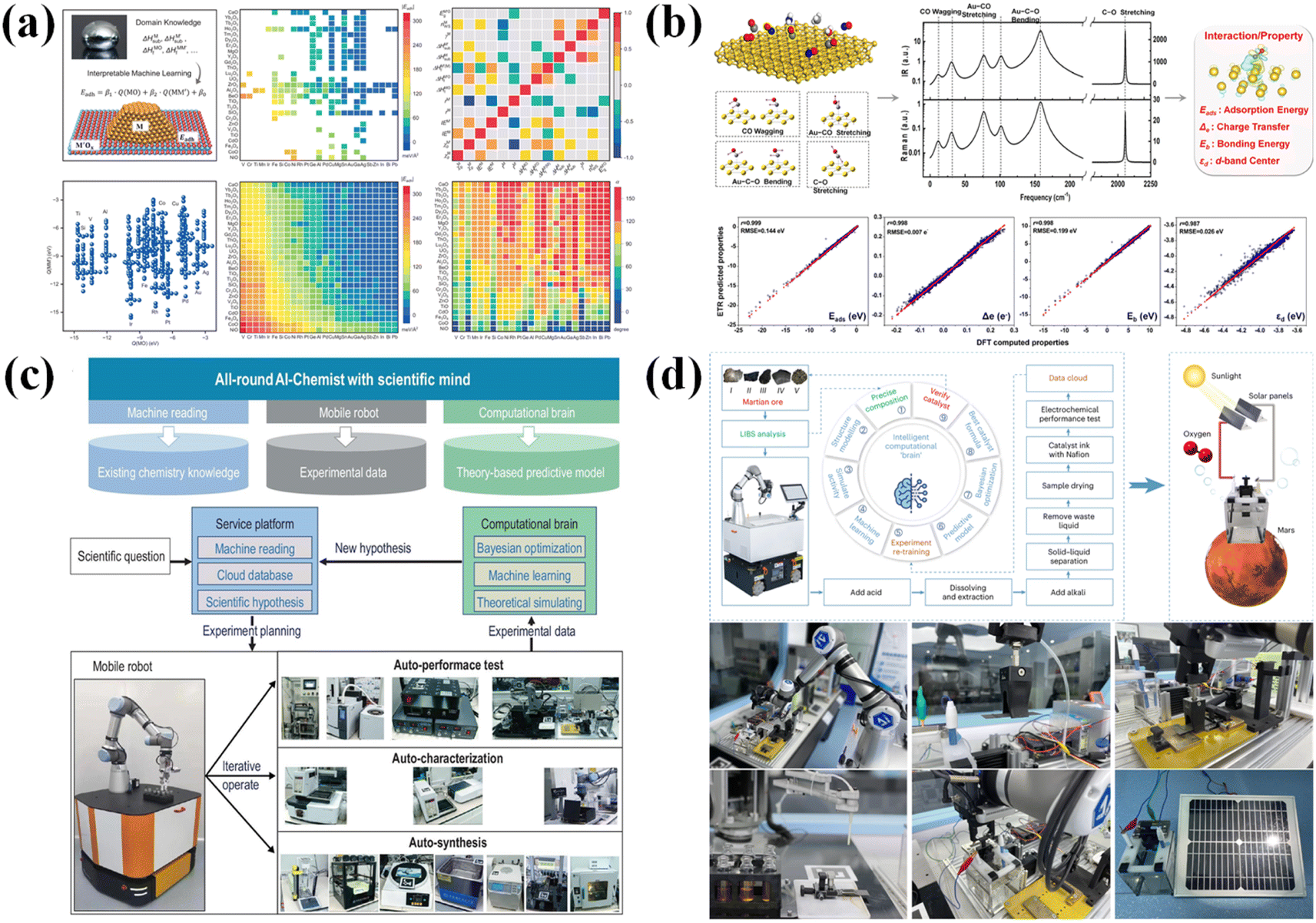 | ||
| Fig. 3 Some ML models and autonomous platforms driven by a computational ‘brain’. (a and b) ML models driven by theoretical calculation. (c and d) All-round AI-chemistry laboratory. | ||
In addition to first-principles calculations, the material structures and properties obtained through spectroscopic characterization can also serve as prior knowledge in optimization procedures. Wang et al.81 proposed a method to establish a connection between surface–adsorbate interaction characteristics and spectral signals through an ML approach (Fig. 3b). By using the infrared and Raman signals of carbon monoxide and nitrogen monoxide adsorbed on metal surfaces as descriptors, important characteristics including adsorption energy and charge transfer degree were quantitatively determined, with good accuracy and transferability. This significantly broadens the application range of traditional in situ spectroscopic techniques in high-throughput screening. Li et al.82 proposed an ML model that uses infrared spectroscopy to monitor the evolution of adsorbate–surface interaction behavior. Taking the C–C coupling process in catalytic reactions as an example, the convolutional neural network was used to identify and extract spectral features, depicting the atomic structure and chemical interactions in the catalytic system. This resulted in obtaining key energy barriers and corresponding structural information, and the predicted promotion trend of CO–CO dimerization closely matched previous literature, demonstrating the ability to accurately track dynamic transformations of metal surfaces. It highlights the practicality and versatility of this machine learning model in tracking the evolution of complex structures. Zhang et al.83 proposed a machine learning descriptor of Chemical Information Molecular Graph (CIMG) to represent chemical reactions. The CIMG constructs a structured graph by encoding nuclear magnetic resonance (NMR) chemical shifts as vertex features, bond dissociation energies as edge features, and solvent/catalyst information as global features. The method based on the CIMG can effectively predict and recommend full synthesis routes for catalysts/solvents, representing a novel data-driven approach to automated retrosynthesis planning that does not rely entirely on historical synthesis data. Cui et al.84 quantitatively predicted how various electric fields would affect catalytic performance using the vibrational spectral signals of carbon dioxide adsorbed on metal single-atom catalyst molecules as descriptors. The adsorption patterns and energies of carbon dioxide molecules on 27 distinct metal single-atom catalysts in varied orientations and at varied intensities were theoretically investigated using metal-doped graphitic C3N4 (g-C3N4) catalysts as an example. In order to measure the facilitative effect of the electric field on CO2 catalytic conversion, a spectral characteristic model was developed using ML techniques to associate infrared/Raman spectral descriptors with adsorption energy/charge transfer. In the meantime, inverse prediction of electric field strength from spectra was achieved by mining catalytic insights into the link between spectra and adsorption patterns based on the attention mechanism. This study introduces a novel quantitative method for controlling electrocatalytic reactions and monitoring spectra using machine learning.
Completely automated systems powered by intelligent brains have emerged as a result of the ongoing integration of AI and theoretical computations. A pioneering effort in this field was undertaken by Zhu et al.,52 who built an all-round AI-chemistry laboratory. The architecture of the AI-Chemist consists of three modules, including a machine-reading module to extract chemical knowledge from literature, a mobile robot module to perform experiments, and a computational brain module to generate physics/theory-based predictive models. Therefore, this system can achieve a closed-loop iterative process of reading relevant literature, conducting theoretical calculations to form preliminary experimental plans, designing experimental plans, executing automated experiments, analyzing the obtained experimental data, training machine learning models, and making decisions to generate new plans (Fig. 3c). This greatly reduces the time human chemists spend on experiments, changing the way new materials are discovered and manufactured. The same team85 expanded on this work by demonstrating a robotic AI chemist for intelligent optimization and automated synthesis of oxygen evolution reaction (OER) catalysts made from Martian meteorites. Martian ore pretreatment, synthesis and characterization testing of catalytic materials, and iterative catalytic formula tuning were all carried out without the need for human interaction. The system determined the ideal catalyst formula from more than three million potential compositions using an ML model trained on first-principles calculations (nearly 30000 theoretical datasets) and experimental observations (243 experimental datasets) (Fig. 3d). With a low overpotential of 445.1 mV and stability for more than 550000 s at a current density of 10 mA cm−2, the improved catalyst demonstrated exceptional performance. Even in extraterrestrial settings, this work demonstrates the promise of AI-driven systems for automated chemical synthesis and materials discovery.
3.3 End-to-end intelligent autonomous systems powered by large-scale models
The overall input-to-output nature of machine learning methods can form a powerful single-purpose tool, capable of quickly generating experimental plans and obtaining optimal experimental solutions. However, the goal of fully autonomous end-to-end synthesis reaction design and development is still to be achieved. First, an end-to-end intelligent automation platform requires a large-scale model with strong generalization ability and wide applicability. It is difficult to achieve ML methods trained solely on data obtained from theoretical calculations and experiments. The emergence of LLMs based on ChatGPT tools in 2022 has made such large-scale models possible. Agent-based LLMs can accommodate vast amounts of knowledge and information and possess strong human–computer interaction capabilities, enabling them to make flexible decisions based on complex and non-standardized inputs. Researchers have also developed large-scale models tailored for chemistry to enhance reasoning capabilities in autonomous laboratories. For example, SynAsk, an LLM-powered organic chemistry platform created by AIChemEco Inc., was presented by Zhang et al.86 Through domain-specific data refinement and integration with a chain-of-thought methodology, SynAsk provides easy access to advanced chemical tools and a comprehensive knowledge base in a question-and-answer style (Fig. 4a). A basic chemistry knowledge base, molecular information retrieval, reaction performance prediction, retrosynthesis prediction, and chemical literature acquisition are some of the features offered by this platform. This creative approach creates a paradigm unique to organic chemistry that makes research and discoveries in the field easier by integrating external resources with fine-tuning procedures.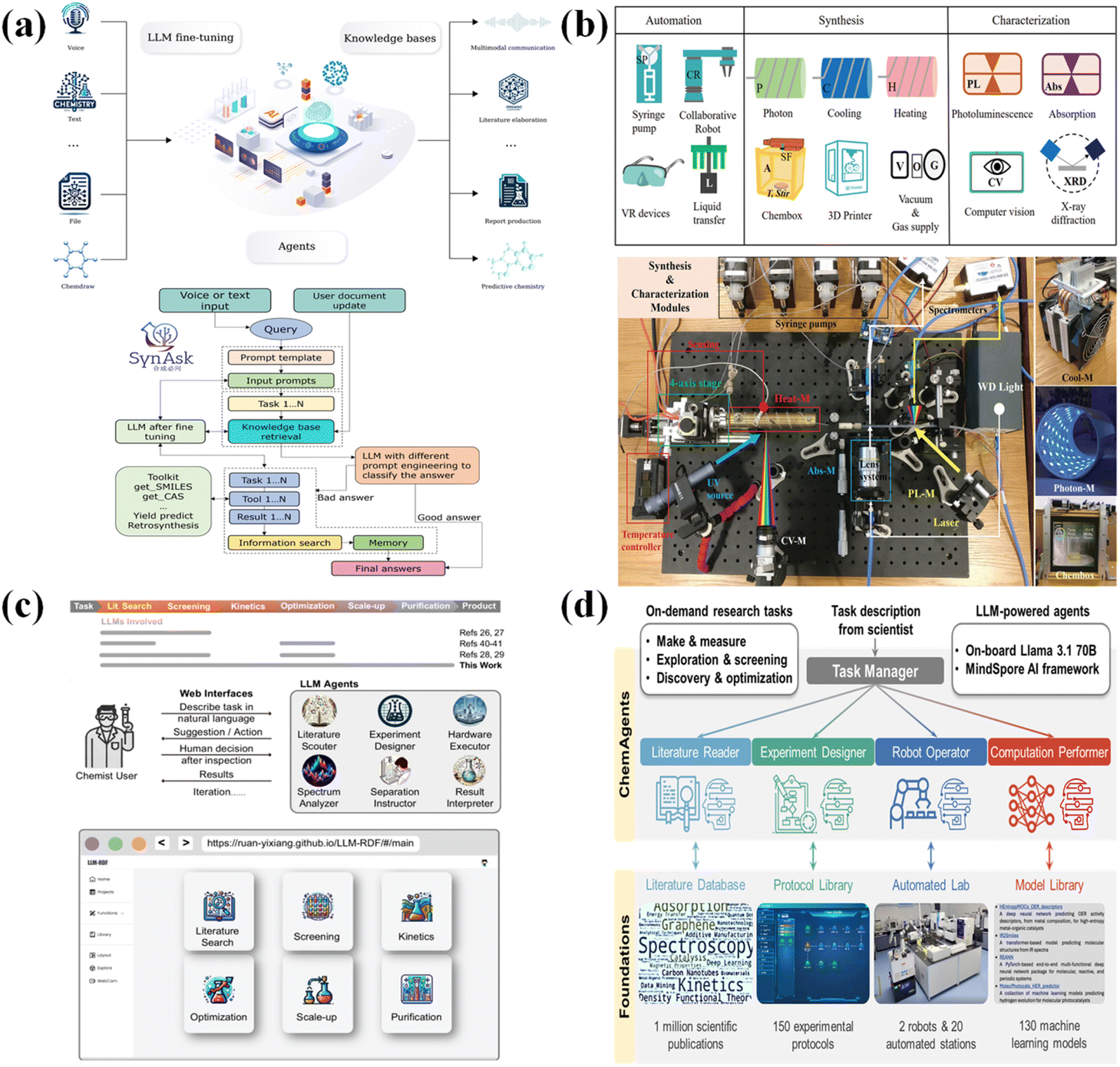 | ||
| Fig. 4 Some intelligent management systems and end-to-end intelligent autonomous platforms. (a) SynAsk. (b) MAOS. (c) LLM-RDF. (d) ChemAgents. | ||
Moreover, a complete chemical experiment workflow typically includes three stages: synthesis, characterization, and performance testing. To create an end-to-end intelligent automation platform capable of handling experimental protocols generated by large-scale models, an intelligent management decision system for scheduling instruments, analyzing feedback data, and optimizing synthesis plans is essential. The design, architecture, and hardware/software systems of a robotic AI-chemist platform that combines chemical synthesis, characterization, and performance testing were published by Xiao et al.87 Its ability to substitute a human chemist in actual experimental operations is demonstrated by the fact that the robotic AI chemist was trained to perform photocatalysis experiments. Similarly, a materials acceleration operating system (MAOS) with a distinct language and compiler architecture was created by Li et al.88 For autonomous materials synthesis, properties research, and self-optimized quality assurance, the MAOS combines virtual reality (VR), cooperative robots, and a reinforcement learning (RL) scheme. Following VR training, the MAOS can function on its own, saving money on labor and time (Fig. 4b).
Ultimately, with the continuous development of intelligent large-scale models and decision-making systems, end-to-end intelligent platforms driven by large-scale models are gradually being realized. To illustrate the adaptability and effectiveness of LLM-based agents throughout the whole chemical synthesis process, Ruan et al.89 established a unified LLM-based reaction development framework (LLM-RDF) (Fig. 4c). They demonstrated how LLM agents can support end-to-end synthesis development by using aerobic alcohol oxidation to aldehyde—an emerging sustainable aldehyde synthesis protocol—as a model transformation. Using state-of-the-art LLM technology, this work presents a feasible route toward autonomous end-to-end chemical synthesis. Furthermore, a hierarchical multi-agent system dubbed ChemAgents, which is based on an on-board Llama-3-70B LLM, powers a robotic AI chemist, according to Song et al.90 With little assistance from humans, this system can carry out intricate, multi-step studies. It functions by means of a task manager agent that communicates with human researchers and manages four specialized agents: the robot operator, which controls a cutting-edge automated lab; the experiment designer, which makes use of a vast protocol library; the computation performer, which makes use of a flexible model library; and the literature reader, which accesses a comprehensive literature database (Fig. 4d). A major step toward completely automated chemical discovery is made possible by the combination of various agents and resources, which allow the system to plan, carry out, and optimize experiments on its own.
3.4 The evolving intelligent autonomous systems
To summarize, while most autonomous laboratory platforms can address specific issues, they currently lack inter-lab communication and data sharing, highlighting the need for further development. Looking ahead, cloud platforms can be adopted for seamless resource allocation and sharing, overcoming geographical and temporal constraints and further establishing advanced nationwide or global networks of an intelligent scientist system (Fig. 5). These networks would integrate intelligent systems to conduct end-to-end autonomous research, achieving high levels of cognitive and operational integration through the fusion of AI models and robotic processes. | ||
| Fig. 5 Operational workflow of an intelligent scientist system. | ||
Once scientists submit requests for material innovation, advanced scientific large models intelligently recommend research strategies and preparation solutions, including candidate materials and synthesis schemes. Human-machine collaborative systems should be developed to optimize the analysis of scientific problems through cognitive intelligence, enabling scientists to further refine and optimize experimental plans. Guided by these plans, robotic experimental cloud facilities conduct high-throughput experiments, while high-throughput computing platforms perform theoretical simulations. Notably, to standardize robotic experimental systems, it is essential to establish and promote standardized protocols for instruction sets, interface functions,91 experimental templates, and intelligent equipment.92 This process drives robotic experimental systems and computer simulations, generating high-quality, multi-domain, multi-modal, and standard data that are fed back into AI models for optimization and refinement. Driven by multimodal large models, the system iteratively optimizes processes, integrating aligned theoretical and experimental data into comprehensive scientific big data. Based on these data, knowledge and logic enhanced models are trained, combining scientific expertise with machine learning techniques to predict global optima and optimize material creation and problem-solving.
Additionally, communication and data sharing between laboratories require not only technical compatibility across various software and hardware platforms but also adherence to data privacy, security, and regulatory policies. Open-source frameworks such as LabTwin, DigCat (Digital Catalysis Platform) and the EU's “AI-on-Demand” initiative serve as pioneering examples of secure data sharing on cloud platforms. Therefore, the development of an intelligent scientist system must incorporate secure data-sharing technologies—such as blockchain or federated learning—to enable lawful and protected resource exchange, thereby reducing barriers to interdisciplinary and cross-domain collaboration. At the same time, specialized domain-models, targeting specific scientific challenges, are developed from model training results and can be securely and commercially shared via cloud platforms, fostering the growth of an intelligent scientist system.
The concept of an intelligent scientist system envisions the establishment of centralized platforms that consolidate and analyze extensive datasets, develop advanced intelligent models, and refine scientific methodologies and technologies. These platforms, serving as the intellectual nucleus of the system, will orchestrate a network of distributed innovation facilities, supporting scientists in achieving specific, targeted scientific breakthroughs. This integrated framework will fundamentally transform the form of scientific research by combining the centralized, resource-intensive development of scientific intelligence with decentralized, localized experimental operations that drive innovation. Such a structure will lower the barriers to interdisciplinary and cross-domain collaboration, enabling researchers and scientists across both academia and industry to engage in highly specialized experimentation and personalized scientific inquiry.
4 Conclusion
In this perspective, we identified the essential elements required for closed-loop autonomous experiments: chemical science databases, large-scale AI models, automated experimental platforms, and integrated management/decision systems, and reviewed China's significant progress from simple iterative algorithm-driven systems to comprehensive intelligent autonomous systems supported by large-scale models, making autonomous chemical discovery within a single laboratory possible. However, communication and data sharing between laboratories remain limited, highlighting the necessity for further development. Looking ahead, cloud platforms can be used to achieve seamless resource allocation and sharing, overcoming geographical and temporal limitations and further establishing advanced intelligent scientist system networks at the national and even global levels. These networks will integrate intelligent systems for end-to-end autonomous research, achieving high-level cognitive and operational integration through the fusion of AI models and robotic processes. Such a structure will lower the barriers to interdisciplinary and cross-domain collaboration, enabling researchers and scientists from academia and industry to engage in highly specialized experiments and personalized scientific exploration.Glossary
| Term | Definition |
|---|---|
| Sabatier principle | States that the best catalytic activity is obtained when the interaction between catalysts and reactants is balanced |
| Symbolic regression | A regression analysis method based on symbolic expressions |
| Blockchain | A decentralized data storage and transmission technology |
| Federated learning | A decentralized machine learning technique that allows several people to work together to build a common model without sharing local data |
Data availability
As this is a perspective article, no primary research results, data, software or code have been included.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. J. acknowledges the Strategic Priority Research Program of the Chinese Academy of Sciences for funding (Grant XDB0450302). J. J. acknowledges the National Natural Science Foundation of China (Grants 22025304 and 22033007) and the CAS Project for Young Scientists in Basic Research (Grant YSBR-005) for funding. The AI-driven experiments, simulations and model training were performed on the robotic AI-Scientist platform of Chinese Academy of Science. We gratefully acknowledge the USTC Center for Micro and Nanoscale Research and Fabrication for providing experimental resources and the USTC supercomputing center, as well as the Hefei Advanced Computing Center, for providing computational resources.Notes and references
- J. McCarthy, M. L. Minsky, N. Rochester and C. E. Shannon, A Proposal for the Dartmouth Summer Research Project on Artificial Intelligence, August 31, 1955, AI Magazine, 2006, 27(4), 12 Search PubMed.
- Editorial, AI will transform science—now researchers must tame it, Nature, 2023, 621, 658 CrossRef PubMed.
- J. J. Hopfield, Neural Networks and Physical Systems with Emergent Collective Computational Abilities, Proc. Natl. Acad. Sci. U. S. A., 1982, 79(8), 2554–2558 CrossRef CAS PubMed.
- D. H. Ackley, G. E. Hinton and T. J. Sejnowski, A Learning Algorithm for Boltzmann Machines, Cogn. Sci., 1985, 9(1), 147–169 Search PubMed.
- J. Jumper, R. Evans and A. Pritzel, et al., Highly Accurate Protein Structure Prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- J. Abramson, J. Adler and J. Dunger, et al., Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3, Nature, 2024, 630(8016), 493–500 CrossRef CAS PubMed.
- P. S. Reel, S. Reel, E. Pearson, E. Trucco and E. Jefferson, Using Machine Learning Approaches for Multi-Omics Data Analysis: A Review, Biotechnol. Adv., 2021, 49, 107739 CrossRef CAS PubMed.
- Y. Chong, Y. Huo and S. Jiang, Machine Learning of Spectra-Property Relationship for Imperfect and Small Chemistry Data, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(20), 10–14 CrossRef PubMed.
- S. Jiang, X. Wang and Y. Chong, et al., Spectra-Based Machine Learning for Predicting the Statistical Interaction Properties of CO Adsorbates on Surface, J. Phys. Chem. Lett., 2024, 15(9), 2400–2404 CrossRef CAS PubMed.
- Z. Lin, H. Akin and R. Rao, et al., Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model, Science, 2023, 379(6637), 1123–1130 CrossRef CAS PubMed.
- Z. Qiao, W. Nie, A. Vahdat, T. F. Miller and A. Anandkumar, State-Specific Protein–Ligand Complex Structure Prediction with a Multiscale Deep Generative Model, Nat. Mach. Intell, 2024, 6(2), 195–208 CrossRef.
- J. Zhang, M. Lang, Y. Zhou and Y. Zhang, Predicting RNA Structures and Functions by Artificial Intelligence, Trends Genet., 2024, 40(1), 94–107 CrossRef CAS PubMed.
- Z. Yang, X. Zeng, Y. Zhao and R. Chen, AlphaFold2 and Its Applications in the Fields of Biology and Medicine, Signal Transduction Targeted Ther., 2023, 8(1), 115 CrossRef PubMed.
- A. Merchant, S. Batzner and S. S. Schoenholz, et al., Scaling Deep Learning for Materials Discovery, Nature, 2023, 624(7990), 80–85 CrossRef CAS PubMed.
- M. Peplow, Google AI and Robots Join Forces to Build New Materials, Nature, 2023, 10, 1–7 CrossRef.
- L. Saigiridharan, A. K. Hassen and H. Lai, et al., AiZynthFinder 4.0: Developments Based on Learnings from 3 Years of Industrial Application, J. Cheminf., 2024, 16(1), 57 Search PubMed.
- A. Rusinko, M. Rezaei and L. Friedrich, et al., AIDDISON: Empowering Drug Discovery with AI/ML and CADD Tools in a Secure, Web-Based SaaS Platform, J. Chem. Inf. Model., 2024, 64(1), 3–8 CrossRef CAS PubMed.
- T. Klucznik, B. Mikulak-Klucznik and M. P. McCormack, et al., Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 2018, 4(3), 522–532 CAS.
- Y. Lin, Z. Zhang and B. Mahjour, et al., Reinforcing the Supply Chain of Umifenovir and Other Antiviral Drugs with Retrosynthetic Software, Nat. Commun., 2021, 12(1), 7327 CrossRef CAS PubMed.
- V. Hassija, V. Chamola and A. Mahapatra, et al., Interpreting Black-Box Models: A Review on Explainable Artificial Intelligence, Cognit. Comput, 2024, 16(1), 45–74 CrossRef.
- J. A. Esterhuizen, B. R. Goldsmith and S. Linic, Interpretable Machine Learning for Knowledge Generation in Heterogeneous Catalysis, Nat. Catal., 2022, 5(3), 175–184 CrossRef.
- S. Chatterjee, M. Guidi, P. H. Seeberger and K. Gilmore, Automated Radial Synthesis of Organic Molecules, Nature, 2020, 579(7799), 379–384 CrossRef CAS PubMed.
- A. A. Volk, R. W. Epps and D. T. Yonemoto, et al., AlphaFlow: Autonomous Discovery and Optimization of Multi-Step Chemistry Using a Self-Driven Fluidic Lab Guided by Reinforcement Learning, Nat. Commun., 2023, 14(1), 1403 CrossRef CAS PubMed.
- M. Walker, G. Pizzuto, H. Fakhruldeen and A. I. Cooper, Go with the Flow: Deep Learning Methods for Autonomous Viscosity Estimations, Digital Discovery, 2023, 2(5), 1540–1547 RSC.
- J. Yang and M. Ahmadi, Empowering Scientists with Data-Driven Automated Experimentation, Nat. Synth, 2023, 2(6), 462–463 CrossRef CAS.
- M. Abolhasani and E. Kumacheva, The Rise of Self-Driving Labs in Chemical and Materials Sciences, Nat. Synth., 2023, 2(6), 483–492 CrossRef CAS.
- B. Burger, P. M. Maffettone and A. I. Cooper, et al., A Mobile Robotic Chemist, Nature, 2020, 583(7815), 237–241 CrossRef CAS PubMed.
- A. M. Lunt, H. Fakhruldeen and A. I. Cooper, et al., Modular, multi-robot integration of laboratories: an autonomous workflow for solid-state chemistry, Chem. Sci., 2024, 15(7), 2456–2463 RSC.
- S. Hessam, M. Craven, A. I. Leonov, G. Keenan and L. Cronin, A Universal System for Digitization and Automatic Execution of the Chemical Synthesis Literature, Science, 2020, 370(6512), 101–108 CrossRef PubMed.
- M. Seifrid, R. Pollice, A. Aguilar-Granda, Z. M. Chan, K. Hotta, C. T. Ser, J. Vestfrid, T. C. Wu and A. Aspuru-Guzik, Autonomous chemical experiments: challenges and perspectives on establishing a self-driving lab, Acc. Chem. Res., 2022, 55, 2454–2466 CrossRef CAS PubMed.
- N. J. Szymanski, B. Rendy and Y. Fei, et al., An Autonomous Laboratory for the Accelerated Synthesis of Novel Materials, Nature, 2023, 624(7990), 86–91 CrossRef CAS PubMed.
- G. Tom, S. P. Schmid and S. G. Baird, et al., A Self-Driving Laboratories for Chemistry and Materials Science, Chem. Rev., 2024, 124(16), 9633–9732 CrossRef CAS PubMed.
- A. Gaulton, L. J. Bellis and A. P. Bento, et al., ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery, Nucleic Acids Res., 2012, 40(1), 1100–1107 CrossRef PubMed.
- S. Kim and E. E. Bolton, A Large-Scale Public Chemical Database for Drug Discovery, Open Access Databases and Datasets for, Drug Discovery , 2024, 39–66 Search PubMed.
- J. Guo, A. S. Ibanez-Lopez and H. Gao, et al., Automated Chemical Reaction Extraction from Scientific Literature, J. Chem. Inf. Model., 2022, 62(9), 2035–2045 CrossRef CAS PubMed.
- M. C. Swain and J. M. Cole, ChemDataExtractor: A Toolkit for Automated Extraction of Chemical Information from the Scientific Literature, J. Chem. Inf. Model., 2016, 56(10), 1894–1904 CrossRef CAS PubMed.
- L. Hawizy, D. M. Jessop, N. Adams and P. Murray-rust, ChemicalTagger: A Tool for Semantic Text-Mining in Chemistry, J. Cheminf., 2011, 3, 1–13 Search PubMed.
- D. M. Jessop, S. E. Adams, E. L. Willighagen, L. Hawizy and P. Murray-rust, OSCAR4: A Flexible Architecture for Chemical Text-Mining, J. Cheminf., 2011, 3(1), 41 CAS.
- M. Shafiq and Z. Gu, Deep Residual Learning for Image Recognition: A Survey Proc, IEEE Comput. Soc. Conf. Comput. Vis. Pattern Recogn., 2022, 12(18), 8972 CAS.
- J. Zhan and H. Zhao, Span Model for Open Information Extraction on Accurate Corpus, Proc. AAAI Conf. Artif. Intell., 2020, 34(5), 9523–9530 Search PubMed.
- J. Han, N. Collier, W. Buntine, E. Shareghi, PiVe: Prompting with Iterative Verification Improving Graph-Based Generative Capability of LLMs, arXiv, 2023, preprint, arXiv:2305.12392, DOI:10.48550/arXiv.2305.12392.
- B. Swanson, K. W. Mathewson, B. Pietrzak, S. Chen and M. Dinalescu, Story Centaur: Large Language Model Few Shot Learning as a Creative Writing Tool, Proc. 16th Conf Eur. Chapter Assoc, Comput. Linguist., Syst. Demonstr., 2021, 244–256 Search PubMed.
- Z. Ji, N. Lee and R. Frieske, et al., Survey of Hallucination in Natural Language Generation, ACM Comput. Surv., 2023, 55(12), 1–38 CrossRef.
- H. Chen, X. Shen, Q. Lv, J. Wang, X. Ni and J. Ye, SAC-KG: Exploiting Large Language Models as Skilled Automatic Constructors for Domain Knowledge Graph, arXiv, 2024, preprint, arXiv:2410.02811, DOI:10.48550/arXiv.2410.02811.
- S. M. Moosavi, A. Chidambaram, L. Talirz and M. Haranczyk, Capturing Chemical Intuition in Synthesis of Metal-Organic Frameworks, Nat. Commun., 2019, 10(1), 539 CrossRef CAS PubMed.
- W. F. Maier, K. Stöwe and S. Sieg, Combinatorial and High-Throughput Materials Science, Angew. Chem., Int. Ed., 2007, 46(32), 6016–6067 CrossRef CAS PubMed.
- P. Nikolaev, D. Hooper and F. Webber, et al., Autonomy in Materials Research: A Case Study in Carbon Nanotube Growth, Nat. Publ. Gr., 2016, 2(1), 1–6 Search PubMed.
- N. J. Szymanski, Y. Zeng, H. Huo, C. J. Bartel, H. Kim and G. Ceder, Toward Autonomous Design and Synthesis of Novel Inorganic Materials, Mater. Horiz., 2021, 8(8), 2169–2198 RSC.
- W. Huyer and A. Neumaier, SNOBFIT – Stable Noisy Optimization by Branch and Fit, ACM Trans. Math. Software, 2008, 35(2), 1–25 CrossRef.
- A. D. Clayton, J. A. Manson, C. J. Taylor, T. W. Chamberlain, B. A. Taylor, G. Clemens and R. A. Bourne, Algorithms for the Self-Optimisation of Chemical Reactions, React. Chem. Eng., 2019, 4(9), 1545–1554 RSC.
- Q. Zhu, Y. Huang and D. Zhou, et al., Automated Synthesis of Oxygen-Producing Catalysts from Martian Meteorites by a Robotic AI Chemist, Nat. Synth, 2024, 3(3), 319–328 CrossRef CAS.
- Q. Zhu, F. Zhang and Y. Huang, et al., An All-Round AI-Chemist with a Scientific Mind, Natl. Sci. Rev., 2022, 9(10), nwac190 CrossRef CAS PubMed.
- B. P. MacLeod, F. G. L. Parlane and T. D. Morrissey, et al., Self-Driving Laboratory for Accelerated Discovery of Thin-Film Materials, Sci. Adv., 2020, 6(20), eaaz8867 CrossRef CAS PubMed.
- Y. Himeur, M. Elnour and F. Fadli, et al., AI-Big Data Analytics for Building Automation and Management Systems: A Survey, Actual Challenges and Future Perspectives, Artif. Intell. Rev., 2023, 56(6), 4929–5021 CrossRef PubMed.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ChemOS: Orchestrating Autonomous Experimentation, Sci. Robot., 2018, 3(19), eaat5559 CrossRef PubMed.
- B. J. Shields, J. Stevens and J. Li, et al., Bayesian Reaction Optimization as a Tool for Chemical Synthesis, Nature, 2021, 590(7844), 89–96 CrossRef CAS PubMed.
- S. Sun, A. Tiihonen and F. Oviedo, et al., A Data Fusion Approach to Optimize Compositional Stability of Halide Perovskites, Matter, 2021, 4(4), 1305–1322 CrossRef CAS.
- A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller and T. Laino, Automated Extraction of Chemical Synthesis Actions from Experimental Procedures, Nat. Commun., 2020, 11(1), 3601 CrossRef CAS PubMed.
- Y. Jiang, H. Fakhruldeen and A. I. Cooper, et al., Autonomous Biomimetic Solid Dispensing Using a Dual-Arm Robotic Manipulator, Digital Discovery, 2023, 2(6), 1733–1744 RSC.
- V. Sans, L. Porwol, V. Dragone and L. Cronin, A Self Optimizing Synthetic Organic Reactor System Using Real-Time in-Line NMR Spectroscopy, Chem. Sci., 2015, 6(2), 1258–1264 RSC.
- A. Bédard, A. Adamo and K. C. Aroh, et al., Reconfigurable System for Automated Optimization of Diverse Chemical Reactions, Science, 2018, 361(6408), 1220–1225 CrossRef PubMed.
- Y. Ruan, S. Lin and Y. Mo, AROPS: A Framework of Automated Reaction Optimization with Parallelized Scheduling, J. Chem. Inf. Model., 2023, 63(3), 770–781 CrossRef CAS PubMed.
- T. Dai, S. Vijayakrishnan and A. I. Cooper, et al., Autonomous Mobile Robots for Exploratory Synthetic Chemistry, Nature, 2024, 635(8040), 890–897 CrossRef PubMed.
- A. I. Leonov, A. J. S. Hammer and S. Lach, et al., An Integrated Self-Optimizing Programmable Chemical Synthesis and Reaction Engine, Nat. Commun., 2024, 15(1), 1240 CrossRef CAS PubMed.
- S. Steiner, J. Wolf and S. Glatzel, et al., Organic Synthesis in a Modular Robotic System Driven by a Chemical Programming Language, Science, 2019, 363(6423), eaav2211 CrossRef CAS PubMed.
- I. A. T. Hashem, I. Yaqoob and N. B. Anuar, et al., The Rise of “Big Data” on Cloud Computing: Review and Open Research Issues, Inf. Syst., 2015, 47, 98–115 CrossRef.
- X. Du, L. Lüer and T. Heumueller, et al., Elucidating the Full Potential of OPV Materials Utilizing a High-Throughput Robot-Based Platform and Machine Learning, Joule, 2021, 5(2), 495–506 CrossRef CAS.
- J. Yang and M. Ahmadi, Empowering scientists with data-driven automated experimentation, Nat. Synth., 2023, 2, 462–463 CrossRef CAS.
- J. Li, Y. Lu, Y. Xu, C. Liu, Y. Tu, S. Ye, H. Liu, Y. Xie, H. Qian and X. Zhu, AIR-Chem: Authentic Intelligent Robotics for Chemistry, J. Phys. Chem. A, 2018, 122, 9142–9148 CrossRef CAS PubMed.
- H. Xu, J. Lin and F. Mo, et al., High-throughput discovery of chemical structure-polarity relationships combining automation and machine-learning techniques, Chem, 2022, 8, 3202–3214 CAS.
- Y. Xu, Y. Gao and K. Liao, et al., High-Throughput Experimentation and Machine Learning-Assisted Optimization of Iridium-Catalyzed Cross-Dimerization of Sulfoxonium Ylides, Angew. Chem., Int. Ed., 2023, 62, e202313638 CrossRef CAS PubMed.
- J. M. Lu, H. F. Wang and Q. Fang, et al., Roboticized AI-assisted microfluidic photocatalytic synthesis and screening up to 10000 reactions per day, Nat. Commun., 2024, 15, 8826 CrossRef CAS PubMed.
- L. Xing, Z. Chen and H. Zhao, et al., Robotic platform for accelerating the high-throughput study of silver nanocrystals in sensitive/selective Hg2+ detection, Chem. Eng. J., 2023, 466, 143225 CrossRef CAS.
- H. Zhao, W. Chen and J. Jiang, et al., A robotic platform for the synthesis of colloidal nanocrystals, Nat. Synth., 2023, 2, 505–514 CrossRef CAS.
- Y. Xie, S. Feng and J. Jiang, et al., Inverse design of chiral functional films by a robotic AI-guided system, Nat. Commun., 2023, 14, 6177 CrossRef CAS PubMed.
- A. Aldossary, J. A. Campos-Gonzalez-Angulo and S. Pablo-García, et al., In Silico Chemical Experiments in the Age of AI: From Quantum Chemistry to Machine Learning and Back, Adv. Mater., 2024, 36, 2402369 CrossRef CAS PubMed.
- P. Yin, X. Niu and S. B. Li, et al., Machine-learning-accelerated design of high-performance platinum intermetallic nanoparticle fuel cell catalysts, Nat. Commun., 2024, 15, 415 CrossRef CAS PubMed.
- L. Zhang, X. Zhang and C. Chen, et al., Machine Learning-Aided Discovery of Low-Pt High Entropy Intermetallic Compounds for Electrochemical Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2024, 63, e202411123 CrossRef CAS PubMed.
- T. Wang, J. Hu and W.-X. Li, et al., Nature of metal-support interaction for metal catalysts on oxide supports, Science, 2024, 386, 915–920 CrossRef CAS PubMed.
- S. Hu and W.-X. Li, Sabatier principle of metal-support interaction for design of ultrastable metal nanocatalysts, Science, 2024, 374(6573), 1360–1365 CrossRef PubMed.
- X. Wang, J. Jiang and Y. Luo, et al., Quantitatively Determining Surface-Adsorbate Properties from Vibrational Spectroscopy with Interpretable Machine Learning, J. Am. Chem. Soc., 2022, 144, 16069–16076 CrossRef CAS PubMed.
- L. Yang, Z. Zhao, J. Jiang and S. Ye, et al., Monitoring C–C coupling in catalytic reactions via machine-learned infrared spectroscopy, Natl. Sci. Rev., 2025, 12(2), nwae389 CrossRef PubMed.
- B. Zhang, X. Zhang, W. Du, Z. Song, J. jiang and L. Yi, Chemistry-informed molecular graph as reaction descriptor for machine-learned retrosynthesis planning, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2212711119 CrossRef CAS PubMed.
- C. X. Cui, J. Jiang and Y. Luo, et al., Quantitative Insight into the Electric Field Effect on CO2 Electrocatalysis via Machine Learning Spectroscopy, J. Am. Chem. Soc., 2024, 146(50), 34551–34559 CrossRef CAS PubMed.
- Q. Zhu, J. Jiang and Y. Luo, et al., Automated synthesis of oxygen-producing catalysts from Martian meteorites by a robotic AI chemist, Nat. Synth., 2024, 3, 319–328 CrossRef CAS.
- C. Zhang, Q. Lin and K. Liao, et al., SynAsk: unleashing the power of large language models in organic synthesis, Chem. Sci., 2025, 16, 43–56 RSC.
- H. Y. Xiao, J. Jiang and Y. Luo, et al., AI-chemist for chemistry synthesis, property characterization, and performance testing, Sci. Sin.: Chim., 2023, 53, 9–18 Search PubMed.
- J. Li, Y. Tu and X. Zhu, et al., Toward “On-Demand” Materials Synthesis and Scientific Discovery through Intelligent Robots, Adv. Sci., 2020, 7, 1901957 CrossRef CAS PubMed.
- Y. Ruan, Q. Zhang and Y. Mo, et al., An automatic end-to-end chemical synthesis development platform powered by large language models, Nat. Commun., 2024, 15, 10160 CrossRef CAS PubMed.
- T. Song, J. Jiang and Y. Luo, et al., A Multiagent-Driven Robotic AI Chemist Enabling Autonomous Chemical Research On Demand, J. Am. Chem. Soc., 2025, 147(15), 12534–12545 CrossRef CAS PubMed.
- R. Rauschen, M. Guy and J. E. Hein, et al., Universal Chemical Programming Language for Robotic Synthesis Repeatability, Nat. Synth, 2024, 3, 488–496 CrossRef CAS.
- M. Sim, M. G. Vakili and F. Strieth-Kalthoff, et al., ChemOS 2.0: An Orchestration Architecture for Chemical Self-Driving Laboratories, Matter, 2024, 7(9), 2959–2977 CrossRef CAS.
Footnote |
| † These authors contributed equally: J. L., C. D., and D. L. |
| This journal is © The Royal Society of Chemistry 2025 |