
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wet-chemical approach to the synthesis of iron whitlockite (Ca18Fe2(HPO4)2(PO4)12) via a dissolution–precipitation process under hydrothermal conditions†
Diana
Griesiute
a,
Jonas
Stadulis
a,
Agne
Kizalaite
 a,
Andris
Antuzevics
ab,
Arita
Dubnika
cd,
Dominika
Zakutna
e,
Vaclav
Tyrpekl
e,
Chen-Ying
Su
fg,
Hsu-Wei
Fang
fg and
Aleksej
Zarkov
*a
a,
Andris
Antuzevics
ab,
Arita
Dubnika
cd,
Dominika
Zakutna
e,
Vaclav
Tyrpekl
e,
Chen-Ying
Su
fg,
Hsu-Wei
Fang
fg and
Aleksej
Zarkov
*a
aInstitute of Chemistry, Vilnius University, Naugarduko 24, LT-03225 Vilnius, Lithuania. E-mail: aleksej.zarkov@chf.vu.lt
bInstitute of Solid State Physics, University of Latvia, Kengaraga 8, LV-1063 Riga, Latvia
cInstitute of Biomaterials and Bioengineering, Faculty of Natural Sciences and Technology, Riga Technical University, LV-1007 Riga, Latvia
dBaltic Biomaterials Centre of Excellence, Headquarters at Riga Technical University, LV-1658 Riga, Latvia
eDepartment of Inorganic Chemistry, Charles University, 128 00 Prague, Czech Republic
fHigh-Value Biomaterials Research and Commercialization Center, National Taipei University of Technology, Taipei 10608, Taiwan
gDepartment of Chemical Engineering and Biotechnology, National Taipei University of Technology, Taipei 10608, Taiwan
First published on 16th June 2025
Abstract
In the present work, iron whitlockite (Fe-WH, Ca18Fe2(HPO4)2(PO4)12) powder was successfully synthesized by a wet-chemical approach. The synthesis was performed through a dissolution–precipitation process under hydrothermal conditions. The final product was obtained in a time-efficient manner via a phase conversion from CaHPO4·2H2O at 230 °C in just 1 h. Structural properties of the obtained material were comprehensively characterized by X-ray diffraction analysis and FTIR, Raman, and EPR spectroscopy. It was determined that in the as-prepared material, Fe ions mainly exist in a reduced divalent state in accordance with the peculiarities of the WH crystal structure. Magnetic studies also revealed paramagnetic behavior of Fe-WH in the temperature range from 5 to 300 K. Upon annealing, the Fe-WH structure decomposed, forming Fe-doped β-Ca3(PO4)2 and Ca2P2O7. Thermally induced decomposition was accompanied by the oxidation of Fe2+ to Fe3+. The biocompatibility of the synthesized material was assessed by in vitro cytotoxicity experiments with the MC3T3-E1 preosteoblastic cell line. The investigated Fe-WH powder did not show a cytotoxic effect on the cells at all studied concentrations, demonstrating its high biocompatibility.
1. Introduction
Calcium phosphates (CPs) are a family of materials widespread in nature, both in biological and geological systems.1 These materials are of particular importance for humans since the hard tissues of human beings are made of CPs, namely calcium hydroxyapatite (HA).2 The chemical similarity with biological calcified tissues makes synthetic CPs highly biocompatible, which in turn leads to the use of these materials in medicine for various applications, including bone regeneration and reconstruction, gene delivery, bioimaging, etc.2,3In literature, the name “whitlockite” is commonly referred to as magnesium whitlockite (Mg-WH, Ca18Mg2(HPO4)2(PO4)12), which can be considered as Mg-substituted CP. This mineral can be found both in the human body and in nature.4,5 The WH of biological origin is assumed to be pure Mg-WH; however, there is a lack of data on the chemical analysis of this biomineral; therefore, the presence of other divalent ions in its structure cannot be excluded. The geological WH found in nature also contains some other elements besides Mg2+ ions, such as Na+, Sr2+, Fe2+, Mn2+etc.4
In recent decades, synthetic Mg-WH has attracted attention with the perspective of using this material in regenerative medicine. As a result, various synthetic approaches were demonstrated for preparing Mg-WH in the form of powders,6–10 granules,11 and composites.12,13 Further biological studies indicated that in some cases, Mg-WH-based materials demonstrate superior properties compared to those of the most commonly used CPs such as HA or tricalcium phosphate (TCP, Ca3(PO4)2).13–18
More recently, studies on the synthesis and application of transition metal (TM) WH were published, reporting the preparation and application of Zn-, Cu-, Co-, and Mn-WH ((Ca18M2(HPO4)2(PO4)12), M = Zn2+, Cu2+, Co2+, Mn2+).19–25 In these materials, the WH structure is stabilized by relatively small first-row TM ions instead of Mg2+. One more member of divalent TM ions is Fe2+; however, according to our knowledge, there are no papers describing the synthesis of iron whitlockite (Fe-WH, Ca18Fe2(HPO4)2(PO4)12) powders by wet chemical approaches. It is worth noting that Fe is redox-active, which can lead to the spontaneous oxidation of Fe2+ to Fe3+ in the reaction environment,26 bringing additional difficulties to the synthesis of Fe-WH, where Fe ions should be present in their reduced oxidation state. According to our knowledge, currently, there is only one published article reporting the synthesis and characterization of deuterated Fe-WH powder.27 In that work, the target material was prepared by a two-step procedure, which includes a high-temperature solid-state synthesis of Ca9Fe(PO4)7 (Fe oxidation state is +3) with its further transformation to Fe-WH at elevated temperature in a reducing atmosphere.
Iron is not only an essential element in the human body, involved in numerous physiological processes, but also possesses magnetic properties, which can be employed for different biomedical applications. Particularly, magnetic Fe-substituted CPs or CP-based composites attracted scientific interest due to their potential application in magnetic hyperthermia, drug delivery, magnetic resonance imaging (MRI), etc.28–34 Various Fe-substituted CPs and their composites were prepared and characterized in recent decades. For instance, Iafisco et al.33 employed a superparamagnetic composite of Fe-doped HA and Fe3O4 as a delivery system for the anticancer drug doxorubicin. The application of magnetic field resulted in an increased release of the drug. A similar composite was demonstrated as a suitable agent for MRI, opening new prospects for the design of theranostic agents for personalized medical applications.28 Duraisamy et al.30 fabricated a multifunctional NiFe2O4-calcium fluorapatite composite with magnetic and antibacterial properties. Under tumor-mimicking conditions, the prepared composite achieved a hyperthermia temperature of ca. 43 °C; moreover, it was considered to be a promising drug delivery agent. Ribeiro et al.32 determined HA substituted with mixed-valent Fe ions as pH-sensitive and biodegradable peroxidase-like nanozymes for cancer chemodynamic therapy and MRI. Taking into account the aforementioned properties of Fe-containing phosphates and their composites, Fe-WH can be considered as a more beneficial candidate for biomedical applications such as magnetic hyperthermia, drug delivery, or MRI, when compared to Mg-WH.
Our work aims to study Fe-WH as a member of the CP family, which is poorly investigated. First of all, we were interested in the feasibility of forming this material by a wet-chemical process in an aqueous medium. Further comprehensive characterization of structural, morphological, magnetic, and cytotoxic properties was performed, opening new horizons for using Fe-WH for biomedical applications.
2. Experimental
2.1. Synthesis
The synthesis of Fe-WH powder was performed through a dissolution-precipitation process under hydrothermal conditions. Dicalcium phosphate dihydrate (DCPD, CaHPO4·2H2O, 99.1%, Eurochemicals) and iron sulfate heptahydrate (FeSO4·7H2O, ≥99.5%, Roth) were used as starting materials. In a typical procedure, appropriate amounts of CaHPO4·2H2O and FeSO4·7H2O were dissolved in a mixture of 30 mL of deionized water and 30 mL of 1M phosphoric acid (H3PO4, 75%, Roth) in a 90 mL Teflon liner. The molar Ca-to-Fe ratio in the above mixture was equal to 9, and the total metal ion concentration was 0.42 mol L−1. After the complete dissolution of precursors, the pH value of the obtained mixture was adjusted to 6.2 with an ammonia solution (NH4OH, 25%, Roth) under constant mixing on a magnetic stirrer. The increase in the pH value resulted in the instantaneous formation of precipitates. Next, the obtained mixture was sealed in a stainless steel container and placed into an oven preheated to 230 °C for 1 h. The reaction time was calculated as a period from the placement of the container into the oven to the moment it was removed to room temperature. After the reaction, the precipitates were vacuum filtered, washed with deionized water, and dried at 80 °C in the oven.2.2. Characterization
Powder X-ray diffraction (XRD) data of the synthesized powders were obtained using a Rigaku MiniFlex II diffractometer (Cu-Kα, λ = 1.5419 Å) working in the Bragg–Brentano (θ/2θ) geometry. The data were collected within the 10–100° 2θ angle range with a speed of 1° min−1. The FullProf suite was employed for structural refinement (FullProf suite software version September-2020). Fourier transform infrared (FTIR) spectra were taken in the 400–4000 cm−1 range (30 scans) with a Bruker ALPHA-FTIR spectrometer. Raman spectra were recorded using a combined Raman and scanning near-field optical microscope WiTec Alpha 300 R equipped with a 532 nm excitation laser source. The measurements were performed with 600 g mm−1 diffraction grating, pixel resolution of <3 cm−1, the integration time was 1 s, the number of scans was 50 and the approximate laser power of 15 mW. The morphology of the synthesized specimens was studied by scanning electron microscopy (SEM) using a Hitachi SU-70 microscope. Magnetic susceptibility data for Fe-WH were collected using a Quantum Design MPMS3 SQUID magnetometer. The data collection involved a temperature sweep ranging from 5 K to 300 K at a magnetic field strength of 100 Oe under both zero-field cooled (ZFC) and field-cooled (FC) conditions. Magnetic hysteresis data were collected at 5 K and 300 K over a range of −20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe to 20000 Oe. Data collection was performed either in vibrating sample magnetometry (VSM) mode with 5 mm scan lengths and 2 s averaging or in direct current (DC) scan mode with 30 mm scan lengths and 5 s averaging. A Bruker ELEXSYS-II E500 CW-EPR spectrometer was used for electron paramagnetic resonance (EPR) investigations of the samples. The spectra acquisition settings were: room temperature, 9.839 GHz (X-band) or 34.00 GHz (Q-band) microwave frequency, 1 mW microwave power, and 0.4 mT magnetic field modulation amplitude. Elemental composition of the Fe-WH powder was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a Perkin-Elmer Optima 7000 DV spectrometer. The sample was dissolved in 5% nitric acid (HNO3, Rotipuran® Supra 69%, Carl Roth) and diluted to appropriate volume. Calibration solutions were prepared by an appropriate dilution of the stock standard solutions (single-element ICP standards 1000 mg L−1, Carl Roth).
000 Oe to 20000 Oe. Data collection was performed either in vibrating sample magnetometry (VSM) mode with 5 mm scan lengths and 2 s averaging or in direct current (DC) scan mode with 30 mm scan lengths and 5 s averaging. A Bruker ELEXSYS-II E500 CW-EPR spectrometer was used for electron paramagnetic resonance (EPR) investigations of the samples. The spectra acquisition settings were: room temperature, 9.839 GHz (X-band) or 34.00 GHz (Q-band) microwave frequency, 1 mW microwave power, and 0.4 mT magnetic field modulation amplitude. Elemental composition of the Fe-WH powder was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a Perkin-Elmer Optima 7000 DV spectrometer. The sample was dissolved in 5% nitric acid (HNO3, Rotipuran® Supra 69%, Carl Roth) and diluted to appropriate volume. Calibration solutions were prepared by an appropriate dilution of the stock standard solutions (single-element ICP standards 1000 mg L−1, Carl Roth).
2.3. In vitro cytotoxicity studies
The cytotoxicity of the Fe-WH powder was evaluated using the MC3T3-E1 preosteoblast cell line. For the extract test, 4500 cells per well were seeded in a 96-well plate with 100 μL of cell culture medium, which consisted of 89% Alpha Modified Eagle Medium (α-MEM), 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin (P/S). The plates were incubated overnight at 37 °C with 5% CO2 (New Brunswick™ S41i CO2 Incubator Shaker, Eppendorf, Hamburg, Germany).Fe-WH powder was suspended in 5 mL of fresh cell culture medium to achieve a 10 mg mL−1 concentration. After 24 and 48 h, the entire solution was collected and replaced with another 5 mL of fresh cell culture medium. The collected solution was filtered through a 0.2 μm syringe filter and then diluted with fresh medium to concentrations of 1 mg mL−1, 0.1 mg mL−1, and 0.01 mg mL−1. These extract dilutions were immediately added to the preincubated cells (100 μL per well). Untreated cells served as the positive control, while cells treated with a 5% dimethylsulfoxide (DMSO) solution in the medium served as the negative control. Each sample and control conditions were tested in six replicates.
Cytotoxicity of the Fe-WH extracts was assessed using a Cell Counting Kit-8 (CCK-8) assay. Samples were incubated for 24 h for both time points, after which 10 μL of CCK-8 solution was added to each well and incubated for 1 h at 37 °C with 5% CO2. Absorbance at 450 nm was measured using an Infinite M Nano microplate reader (Tecan, Männedorf, Switzerland).
3. Results and discussion
The as-precipitated powder (without hydrothermal treatment) was analyzed by means of XRD analysis, and the crystalline phase was identified as DCPD (Fig. S1†), which is in good agreement with the previous data on the dissolution-precipitation synthesis of Zn-WH.20 However, a broad signal centered at ca. 30° suggests that the amorphous component also co-exists in the sample. The XRD analysis of the as-prepared Fe-WH did not indicate the presence of secondary crystalline phases (Fig. 1a). All diffraction peaks matched very well with the standard XRD data of Mg-WH (#04-009-3397). There were no peaks corresponding to any impurity such as monetite or HA, which are commonly obtained secondary phases during the wet synthesis of WH materials.20,35 Rietveld refinement was further performed using a rhombohedral structure with a space group R3c (#161) adopted from Mg-WH as a model. The results demonstrated a good agreement between the calculated and experimental data; the cell parameters were calculated as a = 10.341(85) Å and c = 37.130(24) Å. These values are close to those of Mg-, Zn- and Cu-WH synthesized by the wet chemical approach.6,20,23 Overall, the lattice parameters of isostructural Mg-WH and divalent TM-WH are quite similar, which can be explained by the closeness of the ionic radii of Mg2+ and TM2+ ions.36 In all cases, a small cation (Mg2+ or TM2+) occupies the octahedral Ca(5) site, which is reasonable since this site has the smallest coordination number in the WH crystal structure. In our case, it was also determined that Fe2+ ions occupy the Ca(5) site (Fig. S2†). The agreement of the lattice parameters of our synthesized Fe-WH and literature data4,20,37 can be considered as an indirect confirmation of the predominant divalent state of Fe ions. The parameters reported by Belik et al.27 for the deuterated analog of Fe-WH, namely Ca18Fe2(DPO4)2(PO4)12 were slightly larger.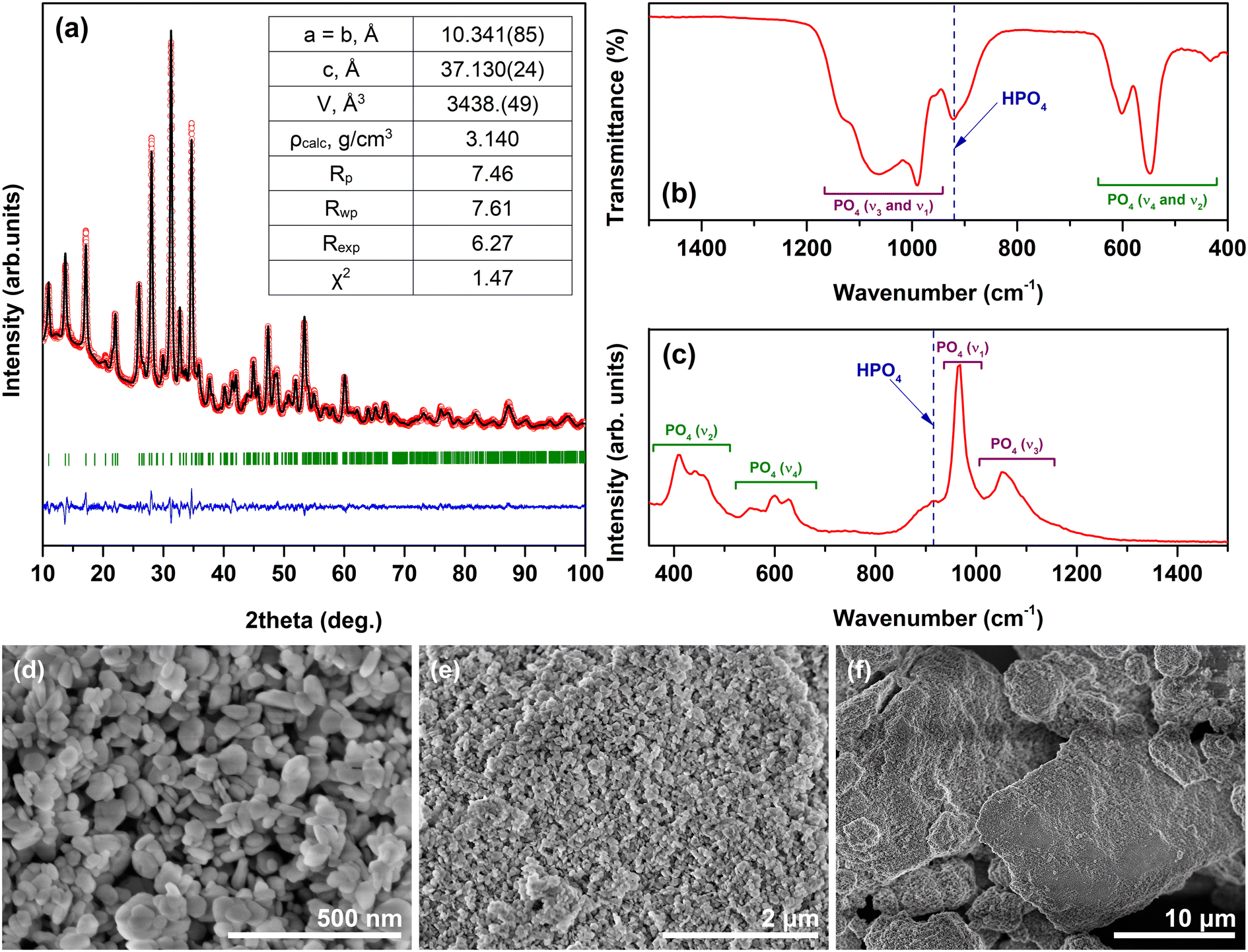 | ||
| Fig. 1 XRD pattern (a); FTIR spectrum (b); Raman spectrum (c); and SEM micrographs (d–f) of the Fe-WH powder. | ||
FTIR and Raman spectroscopy are powerful tools for identifying the WH structure since these techniques allow observing the absorption band corresponding to the HPO42− group. The presence of this spectral marker confirms the formation of WH but excludes related structures such as Fe-doped β-TCP or Ca9Fe(PO4)7, which do not contain this structural unit.38 The FTIR spectrum of the Fe-WH powder is given in the representative range of 400–1500 cm−1 (Fig. 1b) since the materials with the WH structure do not show any bands in the rest part of the spectrum up to 4000 cm−1. Three groups of absorption bands can be highlighted: the bands located in the range from ca. 1200 to 945 cm−1 correspond to ν3 and ν1 stretching modes of the phosphate tetrahedra; while the bands ranging from 500 to 650 cm−1 and the band centered at 430 cm−1 are ascribed to ν4 and ν2 bending modes, respectively;19,39 finally, the band centered at 920 cm−1 represents the HPO42− unit.6,7 A remarkable feature of the observed FTIR spectrum is the width of the band at 920 cm−1, which was significantly broader compared to that observed for Mg- and TM-WH reported in previous works.6,20,23 Corresponding bands were also observed in the Raman spectrum in the ranges of approximately 370–510 cm−1 (ν2), 530–655 cm−1 (ν4), and 1100–1200 cm−1 (ν3) (Fig. 1c); the most intense band peaked at 965 cm−1 is attributed to the ν1 symmetric stretching mode.40 The band assigned to the HPO42− group located at ca. 920 cm−1 is clearly visible in the spectrum;41,42 however, it is not perfectly resolved due to overlapping with the ν1 band. Similarly, like in the FTIR spectrum, the bands in the Raman spectrum are very broad compared to previously reported data on both synthetic20 and especially natural WH.42 A possible reason for the broadening could be the low crystallinity of the synthesized material. The relatively low intensity of the ν1 band also could be attributed to the low crystallinity. Alternatively, the presence of Fe3+ defects in the crystal lattice of Fe-WH could lead to the distortion of crystal structure and broadening of the bands. Overall, the results of both spectroscopic techniques are in good agreement and confirm the formation of the Fe-WH structure.
The SEM images of the Fe-WH powder depict the formation of well-defined plate-like particles varying in size from approximately 40 to 100 nm (Fig. 1d). The polygonal shape of the particles is characteristic of WH materials; however, the formation of a particular shape depends on many parameters, including the synthetic approach, concentration of starting materials, the ratio of metal cations, etc.7,9,19,20 The SEM micrographs taken under lower magnification indicate the aggregation of individual particles to large agglomerates of micrometric dimensions (Fig. 1e and f).
The elemental analysis of the Fe-WH powder was performed by means of ICP-OES to determine the chemical composition of the synthesized material (Table 1). The theoretical values were calculated according to the Ca18Fe2(HPO4)2(PO4)12 formula, which assumes that Fe ions are exclusively divalent. Overall, the experimental data agree very well with the theoretical data; however, the experimentally determined Ca/Fe ratio was slightly higher than that of the theoretical data, while the determined (Ca + Fe)/P molar ratio was slightly lower compared to that of the theoretical data. This observation is compatible with the hypothesis that a minor part of the Fe ions is oxidized to the trivalent state; although, the discrepancies are insignificant. Moreover, the presence of minor amorphous impurities, influencing the composition of the bulk material, cannot be excluded.
| Theoretical Ca/Fe molar ratio | Determined Ca/Fe molar ratio | Theoretical (Ca + Fe)/P molar ratio | Determined (Ca + Fe)/P molar ratio |
|---|---|---|---|
| 9.00 | 9.16 | 1.43 | 1.40 |
We were also interested in the feasibility of synthesizing a mixed-valent Fe-WH containing comparable/significant amounts of both Fe2+ and Fe3+ ions. For this purpose, a mixture of divalent and trivalent iron precursors was used, namely FeSO4·7H2O and Fe(NO3)3·9H2O. A series of powders was synthesized, varying the ratio of Fe3+/Fe2+ in the initial reaction mixture from 0 to 100 mol% with a step of 20 mol%. The results demonstrated that the presence of Fe3+ ions moved the phase equilibrium toward the formation of HA (Fig. S3†). The presence of 20 mol% of Fe3+ already resulted in the formation of biphasic material consisting of Fe-WH and HA, while HA was a dominant phase when a higher amount of Fe3+ was used. These results indicate that by varying the iron precursors, the phase composition of the final product can be tuned; however, the co-existence of a significant amount of both Fe3+ and Fe2+ in the crystal structure of Fe-WH was impossible to achieve. On the other hand, previous works demonstrated that the WH structure is stabilized exclusively by relatively small divalent ions. To our knowledge, there are no reports on preparing the WH structure containing only Ca2+ and trivalent ions. By contrast, a successful synthesis of Fe2+/Fe3+-doped HA was previously reported in the literature, indicating a better ability of the HA crystal structure to adopt aliovalent ions compared to WH.26,43
Magnetic properties of the synthesized Fe-WH powder were investigated by measuring the inverse magnetic susceptibility 1/χ vs. T and the dependence of magnetization on the applied field. The 1/χ vs. T curves obtained in both FC and ZFC modes are presented in Fig. 2a. The linear dependence was observed in both cases, reflecting a paramagnetic behavior of Fe-WH in the temperature range from 300 K down to 5 K. No difference between FC and ZFC curves was observed. The effective magnetic moment, μeff = (8C)1/2, was calculated as 5.07 μB (μB, Bohr magneton) per Fe2+ ion, which is only slightly higher compared to the theoretical value of 4.90 μB. The calculated μeff value can be assumed as a confirmation of the presence of Fe ions in a divalent state. The results agree with the data reported by Belik et al.27 for Ca18Fe2(DPO4)2(PO4)12, who calculated the μeff value as 4.97 μB. The M vs. H curves, taken at 5 K and 300 K, also show an expected paramagnetic behavior demonstrating magnetization values of 6.7 and 0.19 emu g−1 at the magnetic field of 20 kOe at 5 K and 300 K, respectively (Fig. 2b).
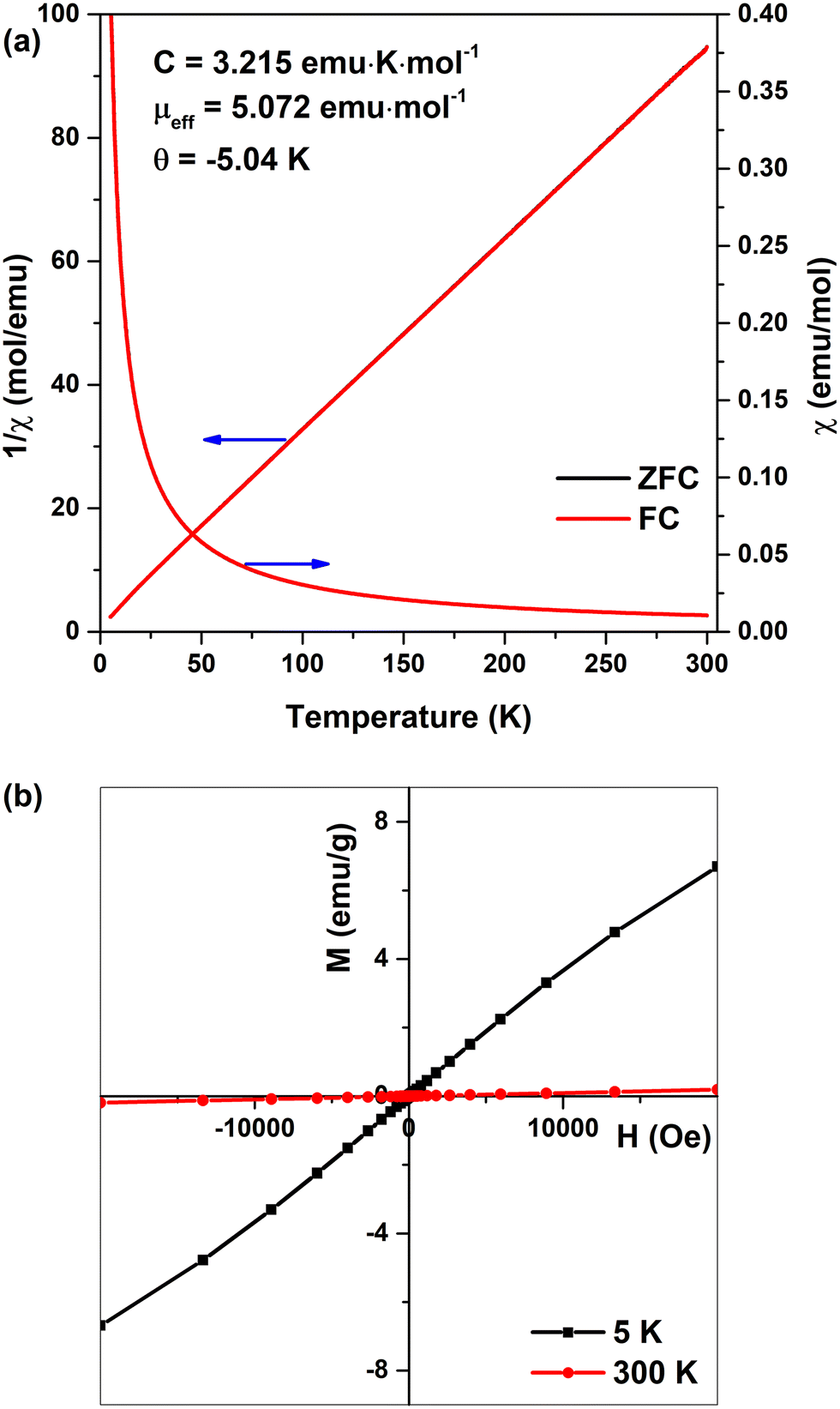 | ||
| Fig. 2 Magnetic susceptibility and inverse magnetic susceptibility of Fe-WH vs. T (a); M vs. H curves of Fe-WH at 5 K and 300 K (b). | ||
Tuning of the magnetic properties of Fe-WH potentially could be achieved by the fabrication of Fe-WH composites with iron oxides. This approach was previously demonstrated by Tampieri et al.,26 who investigated the composite of Fe-doped HA and iron oxides. It was shown that hyperthermia curves of undoped HA-Fe3O4 and Fe-doped HA-Fe3O4 differ significantly. The difference was ascribed to the potential interaction between two magnetic phases compared to a single magnetic material in HA-Fe3O4.
Both Mg- and TM-WH are known to be thermally unstable.6,19,23,44 Upon annealing, these materials gradually decompose, forming ion-substituted β-TCP and Ca2P2O7 (CPP). In terms of chemical bonds, this phase transformation leads to the formation of the P–O–P bond presenting in the CPP structure instead of the decomposed HPO42− structural unit. The detailed study on the thermally-induced degradation of Mg-WH revealed a broad temperature range of the co-existence of 3 crystalline phases: WH, β-TCP, and CPP.6 Moreover, it was shown that the information obtained from the XRD analysis is limited, since it allows only for determining the starting degradation temperature by detecting the appearance of the secondary CPP phase. This technique does not detect the end of the phase transformation due to the identical XRD patterns of WH and β-TCP.
It should be noted that in the case of Mg-, Zn-, or Cu-stabilized WH, the oxidation state of all composing elements is supposed to be the same after the thermal treatment; however, the situation is different in the case of redox-active Fe. At least two scenarios should be considered for the possible mechanism of thermally-induced degradation of Fe-WH in the air atmosphere. The first scenario assumes the oxidation of Fe2+ to Fe3+ accompanied by the transformation of HPO42− to exclusively PO43− and H2. In this case, the reaction should occur according to eqn (1), and the oxidation of Fe achieves the charge compensation. For this reaction, the XRD analysis will not show the appearance of new diffraction peaks.
| Ca18Fe2(HPO4)2(PO4)12 → 2Ca9Fe(PO4)7 + H2 | (1) |
The representative XRD patterns of Fe-WH powders annealed at different temperatures are given in Fig. 3a (all XRD patterns for annealing temperatures from 500 °C to 1200 °C are shown in Fig. S4†). Annealing up to 600 °C did not produce new diffraction peaks; however, the XRD pattern of the powder annealed at 700 °C already contained some reflections corresponding to CPP. The temperature of the formation of CPP agrees well with the results obtained for Mg- and other TM-WH.6,23 The annealing at higher temperatures showed no further significant changes except the narrowing of the diffraction peaks.
 | ||
| Fig. 3 XRD patterns (a) and FTIR spectra (b) of Fe-WH powders annealed at different temperatures. | ||
As a rule, the decomposition of the WH structure can be detected in FTIR spectra by simultaneous reduction of the intensity of the HPO42−-related band and the appearance of the P–O–P absorption band centered at 726 or 754 cm−1, characteristic of CPP.45 For Mg-, Zn-, and Cu-WH, these processes occur gradually and start at around 600–700 °C.6,20,23Fig. 3b shows that the FTIR spectrum of Fe-WH considerably changes already after annealing at 500 °C. The absorption band ascribed to HPO42− (ca. 920 cm−1) disappeared. On the other hand, two new bands centered at 877 and 930 cm−1 were observed instead. Such behavior was not previously observed for Mg-, Zn- or Cu-WH. The reason for the drastic spectral change could be associated with the change in the oxidation state of Fe2+ ions. The appearance of the absorption bands centered at 726 and 756 cm−1 was observed only at 700 °C. These bands indicate the simultaneous formation of both α- and β-CPP polymorphs.45 At higher annealing temperatures, only β-CPP was detected up to 1200 °C. The band observed at 877 cm−1 gradually shifted to the lower wavenumbers, peaking at 864 cm−1 for 900 °C.
Doping of a closely related β-TCP structure with aliovalent Fe3+ or Ga3+ ions modified the FTIR spectrum of β-TCP.38,46,47 The absorption bands at 873 cm−1 and 871 cm−1 appeared with an increase of the dopant concentration in β-TCP prepared at 800 °C and 900 °C, respectively.38,46 Overall, the spectrum of Fe-WH annealed at 500 °C is very similar to that of Fe3+-doped β-TCP in terms of the shape and band positions.38,46 These observations suggest that annealing at 500 °C causes the oxidation of Fe2+ to Fe3+; however, it does not induce the transformation of HPO42− structural unit to P2O74−, which occurs starting from ca. 700 °C. Thus, the degradation pathway could be assumed to be an intermediate between the abovementioned mechanisms. Previously, it was demonstrated that 1H and 31P solid-state nuclear magnetic resonance is the most powerful tool in monitoring the phase transformation from Mg-WH to β-TCP;6 unfortunately, in the case of Fe-WH, the use of this technique is impossible due to the presence of Fe ions.
EPR spectroscopy analysis of the investigated samples is presented in Fig. 4. The EPR spectra cover a broad magnetic field range and depend on the sample treatment temperature. Iron in the Fe3+ state is detectable in conventional EPR measurements at room temperature.38,48–50 X-band EPR spectra, normalized to sample mass (Fig. 4a), can be used to compare the relative Fe3+ ion content in the samples. A Fe3+ signal can be detected in the as-prepared sample, possibly showing the presence of a minor amount of Fe3+ in the Fe-WH crystal lattice or a minor contribution of Fe3+-rich impurity. EPR spectra double integral (DI) values, which are proportional to the number of paramagnetic centers, exhibit an increasing tendency up to 1100 °C annealing temperature. This increase in signal intensity is likely due to the oxidation of Fe2+ to Fe3+ during sample annealing in an air atmosphere.
 | ||
| Fig. 4 X-band (a) and normalized Q-band (b) EPR spectra of the Fe-WH powders. Inset: double integral (DI) intensities of EPR spectra as a function of the sample annealing temperature. | ||
Annealing also produces changes in the local structure of Fe3+ ions, which is evidenced by the variations in EPR signal shapes. This effect is illustrated in Fig. 4b, where Q-band EPR spectra are shown in a normalized representation. As EPR transitions of paramagnetic ions are sensitive to the coordination geometry with neighboring ligands, EPR spectra are helpful in monitoring structural transformations of materials. The EPR spectra of the annealed samples resemble those of Fe3+-doped β-TCP,38,49 which is consistent with the conclusion inferred from XRD and FTIR analysis that Fe-WH decomposes into β-TCP.
The cell viability of Fe-WH was evaluated in adherence to ISO 10993-5:2009 guidelines. Analysis indicated that none of the tested Fe-WH extract dilutions exhibited cytotoxic effects following a 24-hour incubation period at both 24 and 48 h timepoints (Fig. 5).
 | ||
| Fig. 5 Fe-WH extract effect on MC3T3-E1 cell proliferation. | ||
Two main observations can be highlighted from the examination of cell viability data. First, at the 24 h timepoint, slightly lower cell viability was observed for a higher concentration of Fe-WH (1 mg mL−1). Second, the observed pattern suggests that cell viability declines across the lower Fe-WH dilutions (1 and 0.1 mg mL−1) following a 48-hour incubation period. Nevertheless, the value remains consistently and noticeably above 70%, indicating that cytotoxic effects are not pronounced. A recent study by Predoi et al.51 on amorphous iron-calcium phosphate nanoparticles showed that osteoblastic cell viability was dependent on the incubation period, and iron-containing CP coating promoted the proliferation and adhesion of the cells after 72 h of incubation. Furthermore, Wei et al.52 studied the effect of collagen-coated composite of iron oxide nanospheres and Sr-substituted HA on the MC3T3-E1 preosteoblast cell line, showing increased gene expression and, thus, cell differentiation in the presence of the nanospheres. Cell viability and upregulation of the osteogenic and angiogenic genes are linked with the Fe concentration and release kinetics from the material since concentrations from 10 to 100 μM enhance angiogenesis, but excessive amounts can cause toxicity and ferroptosis.53 Unfortunately, other studies on the biological properties of Fe-WH are currently absent for comparison; however, our results demonstrate the potential of Fe-WH for further studies in the biomedical field.
4. Conclusions
Iron whitlockite (Fe-WH, Ca18Fe2(HPO4)2(PO4)12) powder was successfully synthesized by a wet chemical approach under hydrothermal conditions in just 1 h. According to the XRD analysis, the obtained material was phase-pure, without any crystalline impurities. Rietveld refinement confirmed that Fe2+ ions occupy the Ca(5) site, which agrees well with the peculiarities of the WH crystal structure. The magnetic studies confirmed that in the as-prepared Fe-WH, Fe ions mostly exist in a reduced divalent state; however, the Fe3+ signal in the as-prepared sample was detected by EPR, possibly showing the presence of a minor amount of Fe3+ in the Fe-WH crystal lattice. The magnetic studies also revealed a paramagnetic behavior of Fe-WH in the temperature range from 5 to 300 K. Upon annealing, the Fe-WH structure decomposed, forming Fe-doped β-Ca3(PO4)2 and Ca2P2O7. Thermally induced decomposition was accompanied by the oxidation of Fe2+ to Fe3+. The biocompatibility of the synthesized material was assessed by in vitro cytotoxicity experiments and did not show a cytotoxic effect on the cells at all studied concentrations, demonstrating its high biocompatibility.Data availability
The data supporting this study have been included within the article and the corresponding ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Research Council of Lithuania (LMTLT), agreement No S-MIP-23-85. The authors acknowledge the access to the infrastructure and expertise of the BBCE – Baltic Biomaterials Centre of Excellence (European Union's Horizon 2020 research and innovation programme under grant agreement No. 857287). Prof. S. L. Stoll, Dr. O. Stewart (Georgetown University) and Dr. T. Murauskas (Vilnius University) are highly acknowledged for their help with material characterization.References
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 Search PubMed.
- W. Habraken, P. Habibovic, M. Epple and M. Bohner, Mater. Today, 2016, 19, 69–87 Search PubMed.
- M. Gu, W. Li, L. Jiang and X. Li, Acta Biomater., 2022, 148, 22–43 CrossRef CAS PubMed.
- F. Capitelli, F. Bosi, S. C. Capelli, F. Radica and G. Della Ventura, Crystals, 2021, 11, 225 CrossRef CAS.
- F. A. Shah, Acta Biomater., 2021, 125, 72–82 CrossRef CAS PubMed.
- A. Kizalaite, V. Klimavicius, V. Balevicius, G. Niaura, A. N. Salak, J. C. Yang, S. H. Cho, T. Goto, T. Sekino and A. Zarkov, CrystEngComm, 2023, 25, 4370–4379 RSC.
- C. Wang, K.-J. Jeong, H. J. Park, M. Lee, S.-C. Ryu, D. Y. Hwang, K. H. Nam, I. H. Han and J. Lee, J. Colloid Interface Sci., 2020, 569, 1–11 CrossRef CAS PubMed.
- A. Afonina, A. Kizalaite, A. Zarkov, A. Drabavicius, T. Goto, T. Sekino, A. Kareiva and I. Grigoraviciute-Puroniene, Ceram. Int., 2022, 48, 32125–32130 CrossRef CAS.
- H. L. Jang, H. K. Lee, K. Jin, H.-Y. Ahn, H.-E. Lee and K. T. Nam, J. Mater. Chem. B, 2015, 3, 1342–1349 RSC.
- H. L. Jang, K. Jin, J. Lee, Y. Kim, S. H. Nahm, K. S. Hong and K. T. Nam, ACS Nano, 2014, 8, 634–641 CrossRef CAS PubMed.
- W.-B. Lee, C. Wang, J.-H. Lee, K.-J. Jeong, Y.-S. Jang, J.-Y. Park, M. H. Ryu, U.-K. Kim, J. Lee and D.-S. Hwang, ACS Appl. Bio Mater., 2020, 3, 7762–7768 CrossRef CAS PubMed.
- X. Zhang, W. Liu, J. Liu, Y. Hu and H. Dai, ACS Biomater. Sci. Eng., 2021, 7, 3321–3331 CrossRef CAS PubMed.
- S. Pouraghaei Sevari, J. K. Kim, C. Chen, A. Nasajpour, C.-Y. Wang, P. H. Krebsbach, A. Khademhosseini, S. Ansari, P. S. Weiss and A. Moshaverinia, ACS Appl. Mater. Interfaces, 2021, 13, 35342–35355 CrossRef CAS PubMed.
- H. L. Jang, G. B. Zheng, J. Park, H. D. Kim, H.-R. Baek, H. K. Lee, K. Lee, H. N. Han, C.-K. Lee, N. S. Hwang, J. H. Lee and K. T. Nam, Adv. Healthcare Mater., 2016, 5, 128–136 CrossRef CAS PubMed.
- D. Zhou, C. Qi, Y.-X. Chen, Y.-J. Zhu, T.-W. Sun, F. Chen and C.-Q. Zhang, Int. J. Nanomed., 2017, 12, 2673–2687 CrossRef CAS PubMed.
- H. D. Kim, H. L. Jang, H.-Y. Ahn, H. K. Lee, J. Park, E.-S. Lee, E. A. Lee, Y.-H. Jeong, D.-G. Kim, K. T. Nam and N. S. Hwang, Biomaterials, 2017, 112, 31–43 CrossRef CAS PubMed.
- H. Cheng, R. Chabok, X. Guan, A. Chawla, Y. Li, A. Khademhosseini and H. L. Jang, Acta Biomater., 2018, 69, 342–351 CrossRef CAS PubMed.
- M. Hu, F. Xiao, Q.-F. Ke, Y. Li, X.-D. Chen and Y.-P. Guo, Chem. Eng. J., 2019, 359, 1–12 CrossRef CAS.
- A. Kizalaite, I. Grigoraviciute-Puroniene, D. R. C. Asuigui, S. L. Stoll, S. H. Cho, T. Sekino, A. Kareiva and A. Zarkov, ACS Biomater. Sci. Eng., 2021, 7, 3586–3593 CrossRef CAS PubMed.
- A. Kizalaite, V. Klimavicius, J. Versockiene, E. Lastauskiene, T. Murauskas, R. Skaudzius, T. Yokoi, M. Kawashita, T. Goto, T. Sekino and A. Zarkov, CrystEngComm, 2022, 24, 5068–5079 RSC.
- M. Wang, J. Yao, S. Shen, C. Heng, Y. Zhang, T. Yang and X. Zheng, Nano Res., 2023, 16, 757–770 CrossRef CAS.
- T. Konishi and S. Watanabe, Phosphorus Res. Bull., 2022, 38, 18–24 CrossRef CAS.
- D. Griesiute, A. Kizalaite, A. Dubnika, V. Klimavicius, V. Kalendra, V. Tyrpekl, S. H. Cho, T. Goto, T. Sekino and A. Zarkov, Dalton Trans., 2024, 53, 1722–1734 RSC.
- S. Y. Stefanovich, B. I. Lazoryak, A. M. Antipin, A. S. Volkov, A. I. Evdokimov, O. A. Gurbanova, O. V. Dimitrova and D. V. Deyneko, Z. fur Krist. - Cryst. Mater., 2023, 238, 301–309 CrossRef CAS.
- C. Heng, W. Liu, X. Zheng, X. Ma, J. Hui and D. Fan, Colloids Surf., B, 2023, 222, 113120 CrossRef CAS PubMed.
- A. Tampieri, T. D'Alessandro, M. Sandri, S. Sprio, E. Landi, L. Bertinetti, S. Panseri, G. Pepponi, J. Goettlicher, M. Bañobre-López and J. Rivas, Acta Biomater., 2012, 8, 843–851 CrossRef CAS PubMed.
- A. A. Belik, F. Izumi, S. Y. Stefanovich, B. I. Lazoryak and K. Oikawa, Chem. Mater., 2002, 14, 3937–3945 CrossRef CAS.
- A. Adamiano, M. Iafisco, M. Sandri, M. Basini, P. Arosio, T. Canu, G. Sitia, A. Esposito, V. Iannotti, G. Ausanio, E. Fragogeorgi, M. Rouchota, G. Loudos, A. Lascialfari and A. Tampieri, Acta Biomater., 2018, 73, 458–469 CrossRef CAS PubMed.
- B. Srinivasan, E. Kolanthai, N. E. A. Kumaraswamy, R. R. Jayapalan, D. S. Vavilapalli, L. H. Catalani, G. S. Ningombam, N. S. Khundrakpam, N. R. Singh and S. N. Kalkura, J. Phys. Chem. B, 2019, 123, 5506–5513 CrossRef CAS PubMed.
- K. Duraisamy, A. Gangadharan, K. S. Martirosyan, N. K. Sahu, P. Manogaran and G. Easwaradas Kreedapathy, ACS Appl. Bio Mater., 2023, 6, 104–116 CrossRef CAS PubMed.
- T. M. Oliveira, F. C. B. Berti, S. C. Gasoto, B. Schneider, M. A. Stimamiglio and L. F. Berti, Front. Med., 2021, 3, 700266 Search PubMed.
- T. P. Ribeiro, B. Salgado, J. Pinto, P. C. Silva, J. A. M. Santos, J. A. Moreira, F. J. Monteiro and M. S. Laranjeira, Mater. Today Chem., 2024, 35, 101861 CrossRef CAS.
- M. Iafisco, C. Drouet, A. Adamiano, P. Pascaud, M. Montesi, S. Panseri, S. Sarda and A. Tampieri, J. Mater. Chem. B, 2016, 4, 57–70 RSC.
- H. Inam, S. Sprio, M. Tavoni, Z. Abbas, F. Pupilli and A. Tampieri, Int. J. Mol. Sci., 2024, 25, 2809 CrossRef CAS PubMed.
- A. Afonina, A. Dubauskas, V. Klimavicius, A. Zarkov, A. Kareiva and I. Grigoraviciute, Ceram. Int., 2023, 49, 38157–38164 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- R. Gopal, C. Calvo, J. Ito and W. K. Sabine, Can. J. Chem., 1974, 52, 1155–1164 CrossRef CAS.
- L. Sinusaite, A. Popov, A. Antuzevics, K. Mazeika, D. Baltrunas, J.-C. Yang, J. L. Horng, S. Shi, T. Sekino, K. Ishikawa, A. Kareiva and A. Zarkov, Mater. Sci. Eng., C, 2020, 112, 110918 CrossRef CAS PubMed.
- R. G. Carrodeguas and S. De Aza, Acta Biomater., 2011, 7, 3536–3546 CrossRef CAS PubMed.
- P. N. de Aza, C. Santos, A. Pazo, S. de Aza, R. Cuscó and L. Artús, Chem. Mater., 1997, 9, 912–915 CrossRef CAS.
- B. Wopenka and J. D. Pasteris, Mater. Sci. Eng., C, 2005, 25, 131–143 CrossRef.
- B. L. Jolliff, J. M. Hughes, J. J. Freeman and R. A. Zeigler, Am. Mineral., 2006, 91, 1583–1595 CrossRef CAS.
- F. Pupilli, M. Tavoni, C. Drouet, A. Tampieri and S. Sprio, Open Ceram., 2024, 18, 100610 CrossRef CAS.
- V. K. Kaliannagounder, N. P. M. J. Raj, A. R. Unnithan, J. Park, S. S. Park, S.-J. Kim, C. H. Park, C. S. Kim and A. R. K. Sasikala, Nano Energy, 2021, 85, 105901 CrossRef CAS.
- D. Griesiute, E. Raudonyte-Svirbutaviciene, A. Kareiva and A. Zarkov, CrystEngComm, 2022, 24, 1166–1170 RSC.
- R. K. Singh, M. Srivastava, N. K. Prasad, S. Awasthi, A. Dhayalan and S. Kannan, Mater. Sci. Eng., C, 2017, 78, 715–726 CrossRef CAS PubMed.
- C. Mellier, F. Fayon, V. Schnitzler, P. Deniard, M. Allix, S. Quillard, D. Massiot, J.-M. Bouler, B. Bujoli and P. Janvier, Inorg. Chem., 2011, 50, 8252–8260 CrossRef CAS PubMed.
- B. Gabbasov, M. Gafurov, A. Starshova, D. Shurtakova, F. Murzakhanov, G. Mamin and S. Orlinskii, J. Magn. Magn. Mater., 2019, 470, 109–117 CrossRef CAS.
- D. Griesiute, L. Sinusaite, A. Kizalaite, A. Antuzevics, K. Mazeika, D. Baltrunas, T. Goto, T. Sekino, A. Kareiva and A. Zarkov, CrystEngComm, 2021, 23, 4627–4637 RSC.
- F. Murzakhanov, G. V. Mamin, S. Orlinskii, M. Goldberg, N. V. Petrakova, A. Y. Fedotov, P. Grishin, M. R. Gafurov and V. S. Komlev, ACS Omega, 2021, 6, 25338–25349 CrossRef CAS PubMed.
- D. Predoi, S. L. Iconaru, S. C. Ciobanu, S.-A. Predoi, N. Buton, C. Megier and M. Beuran, Coatings, 2021, 11, 186 CrossRef CAS.
- X. Wei, X. Zhang, Z. Yang, L. Li and H. Sui, Arabian J. Chem., 2021, 14, 102984 CrossRef CAS.
- M. A. Saghiri, A. Asatourian, J. Orangi, C. M. Sorenson and N. Sheibani, Crit. Rev. Oncol. Hematol., 2015, 96, 129–142 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ce00207a |
| This journal is © The Royal Society of Chemistry 2025 |