
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-coordinate calcium peroxide and oxide complexes†
Stefan
Thum
 ,
Marcel A.
Schmidt
,
Jens
Langer
and
Sjoerd
Harder
*
,
Marcel A.
Schmidt
,
Jens
Langer
and
Sjoerd
Harder
*
Inorganic and Organometallic Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@fau.de
First published on 4th July 2025
Abstract
A CaI synthon with superbulky β-diketiminate ligands (BDI*) and a N22− dianion, [(BDI*)Ca]2(N2), is key to the synthesis of binuclear (BDI*)Ca(μ2-O2)Ca(BDI*) and (BDI*)Ca(μ2-O)Ca(BDI*) complexes. The Ca oxide complex is particularly unstable in solution and was only accessible by a solvent-free reaction between solid [(BDI*)Ca]2(N2) and N2O gas.
From a historical perspective, the alkaline earth (Ae) metal oxides beryllia, magnesia, lime, strontia and baria are among the oldest known group 2 metal compounds. In fact, most of the alkaline earth metal's names originate from these rock salt minerals. Solid MgO or CaO are often used as supports for catalysts1 but also alone can be catalytically active.2,3 Their ionic nature combines a Lewis acidic Ae2+ cation with a Brønsted basic O2− anion, which is a perfect combination for small molecule activation by an FLP-type mechanism.4 As the reactive cationic and anionic centres should be accessible for substrates, low-coordination numbers are a requirement. This was most recently elegantly demonstrated by Stasch and coworkers who showed that a hydrocarbon-soluble Mg oxide complex is able to split the strong H–H bond (Scheme 1a).5 A THF-solvate of this Mg oxide complex, for which a first example was reported earlier by Jones,6 only reacted with H2 after prior elimination of the THF ligands.5 In fact, addition of THF to the mixed Mg hydride/hydroxide complex, the product of H2 activation, reversed the reaction and led to Mg oxide formation under H2 release (Scheme 1a). The reaction of the Mg oxide complex with H2 can also be seen as a deprotonation of H2 which is known to have a very high pKa value of circa 49.7 This underscores the extreme basicity of low-coordinate Mg oxides. In contrast, the high-coordinate μ4-O or μ3-O arrangements in early main group metal complexes8–26 are much less reactive than rare low-coordinate μ2-O compounds.5,6,27
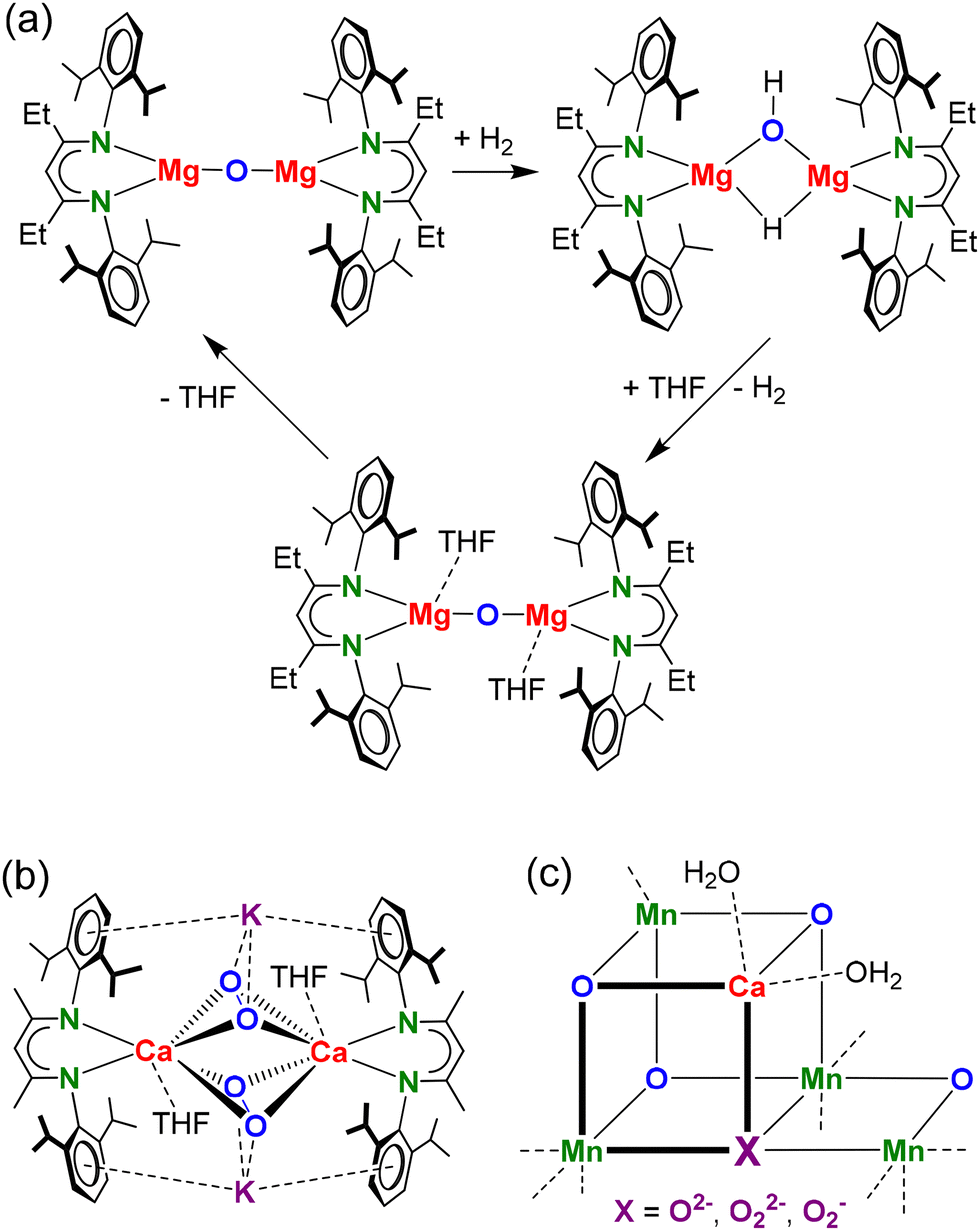 | ||
| Scheme 1 (a) Low-coordinate Mg oxide complex reacting with H2 and H2 release by addition of THF.5 (b) Heterobimetallic Ca/K peroxide complex.29 (c) Mn4CaO4X cluster in photosystem II. | ||
Herein, we report our investigations towards low-coordinate Ca oxide and peroxide complexes. Considering that the reactivity of Ae metal complexes rapidly increases with metal size,28 we anticipate that the isolation of low-coordinate Ca oxide or peroxide complexes will be challenging. Lewinsky and coworkers recently isolated a first example of a heterobimetallic Ca/K peroxide complex29 (Scheme 1b) which, although not intended, incorporated K+ originating from KCl salt eliminated in an previous salt-metathesis step. This demonstrates the difficulty in making low-coordinate (per)oxide complexes.
Our interest in Ca oxide and peroxide chemistry is also motivated by the prominent role of the Ca2+ cation in the O2 evolving Photosystem II which is used by nature for water splitting: 2H2O → 4H+ + 4e− + O2. The Lewis-acidic Ca2+ cation is essential for catalytic activity of the central Mn4CaO5 cluster by stabilizing the oxide anion and transient peroxide species30 (Scheme 1c) and also serves as a “taxi-stand” for incoming H2O before moving into the active site.31
The key to well-defined Ca (per)oxide complexes is a recently isolated Ca dinitrogen complex, [(BDI*)Ca]2(N2) (I, Scheme 2, BDI* = HC[C(Me)N-(DIPeP)]2, DIPeP = 2,6-(CHEt2)-phenyl).32 This complex can be obtained in good yield, either ether-free or as THF or THP (tetrahydropyran) adduct, and was shown to react as a CaI synthon by release of N2 and 2e−.32–35 Reaction of dry air with a suspension of ether-free I in hexanes at −85 °C led to a rapid color change from red-brown to yellow but all attempts to crystallize the product failed. However, after the addition of a few drops of THP the Ca peroxide complex 1 started to crystallize rapidly (yield: 53%). Alternatively, the complex can also be prepared by oxidation of I-THP with dry air. The raw product of this latter reaction is nearly pure (>95%, Fig. S9, ESI†). 1H and 13C NMR spectra of 1 (Fig. S1–S3, ESI†) show one benzylic quintet and one Me-backbone signal, indicative of high average symmetry. 1H NMR monitoring shows that a benzene-d6 solution of 1 decomposes at 65 °C (Fig. S10, ESI†). We were not able to crystallize the decomposition product but, as observed by Lewinsky and coworkers,29 we propose formation of a HOO− complex. This is in agreement with appearance of signals around −0.5 ppm indicating deprotonation of alkyl arms.
 | ||
| Scheme 2 Synthesis of low-coordinate Ca peroxide (1) and Ca oxide (2) complexes. | ||
Calcium peroxide 1 crystallized as a centrosymmetric, dinuclear complex in which the peroxide dianion O22− bridges the pentacoordinated Ca2+ centres in a μ2-η2:η2-fashion (Fig. 1a). The Ca–O distances (2.1911(8)–2.2132(9) Å) are shorter than those in the only other known Ca peroxide complex (Scheme 1b: 2.315(2)–2.328(2) Å)29 which shows a higher coordination number at the Ca nuclei and additional O22−⋯K+ interactions. The Ca–O distances in 1 are comparable to those reported for [(DIPPBDI)Ca(THF)]2(OH)2 (2.224(2) Å and 2.218(2) Å).36 The peroxide anion in 1 shows an O–O bond distance of 1.593(2) Å which is exceptionally long when compared to the only other reported Ca peroxide complex (Scheme 1b, O–O: 1.550(3) Å)29 or to a Ca/V peroxide complex (O–O: 1.466(1)–1.482(1) Å).37 Hill and coworkers reported pyridine and DMAP stabilized Mg peroxide complexes with longer O–O distances than that in 1 (1.625(1)–1.638(5) Å).38 Samarium peroxide complexes show shorter O–O distances ranging from 1.509(4)–1.538 Å.39,40
 | ||
| Fig. 1 Crystal structures of 1 (a) and 2 (b); H atoms and CHEt2 groups omitted for clarity. (c) The HOMO−3 and HOMO−4 for Ca oxide complex 2. | ||
The remarkable reactivity of a low-coordinate Mg oxide complex5 prompted us to isolate a similar but even more reactive Ca oxide complex. Reactions of I or I-THP in alkanes with N2O are fast but, independent of the temperature (−70 °C/+20 °C), highly unselective. This was concluded from appearance of several 1H NMR signals for the CH backbone in the BDI* ligand (Fig. S11, ESI†). Cooling the concentrated reaction mixture led to crystallization of a decomposition product in which the backbone Me group has been deprotonated (II, Scheme 2). The same complex was formed upon decomposition of I and has been fully characterized.32 A solid-state reaction turned out to be more successful. Crystalline I-THP was cooled to −70 °C and the protective N2 atmosphere was replaced with N2O. Slowly warming the solid to 0 °C led to a colour change from red-brown to off-white. Rapid recrystallization from pentane at −25 °C allowed for isolation of [(BDI*)Ca(THP)]2(μ2-O) (2) in 27% yield. Its high reactivity and very high solubility both contribute to the moderate yield.
Dissolving crystalline 2 in benzene-d6 or methylcyclohexane-d14 led to immediate decomposition in several species. A solution of 2 in methylcyclohexane-d14 decomposed even at −80 °C. The low temperature 1H NMR spectrum showed several CH-backbone signals and signals at negative ppm values (Fig. S14, ESI†) which indicate the typical deprotonation of a –CHEt2 arm. Similar alkyl deprotonations occurred upon attempted isolation of a (BDI)MgO− anion.18 This illustrates the highly basic character of the oxide anion in 2.
The crystal structure of 2 (Fig. 1b) shows a centrosymmetric dinuclear complex with perfectly linear Ca–(μ2-O)–Ca bridging. Coordination of THP results in 4-coordinate Ca centres. Attempts to synthesize and crystallize ether-free Ca oxide complexes with 3-coordinate Ca centres failed, possibly due to extreme reactivity. The low coordination number for the bridging μ2-O results in very short Ca–O distances of 2.0478(3) Å. Calcium complexes with μ3-O or μ4-O oxide ligands show much longer Ca–O distances: 2.223(9)–2.292(8) Å,14 2.187(2)–2.265(8) Å,11 or 2.128(2)–2.138(2) Å.12
Alkane solutions of the Ca oxide complex 2 react even at −80 °C instantaneous with H2 but due to its instability in solution such reactivity studies gave only complicated mixtures of products. A similar observation was made for reaction with CO2. Although 2 is reasonably stable in the solid crystalline state, even solid-state reactions of crystalline 2 with H2 or CO2 led to unselective product formation. Note that the similar Mg oxide complex only reacts with H2 when ether-free (Scheme 1a) whereas the Ca oxide complex also reacts in presence of THP. This underscores the extreme reactivity of calcium complexes with μ2-bridging oxide ligands.
A DFT study sheds light on bonding and electron distribution in 1 and 2. The optimized structures (B3PW91-GD3BJ/def2tzvp//B3PW91-GD3BJ/def2svp) fit reasonably well with the experimentally determined molecular structures (Fig. S17 and S18, ESI†), indicating a sufficient level of theory.
The formation of the Ca oxide or peroxide complexes from the ether-free CaI synthon I is thermodynamically highly favoured. Oxidation of [(BDI*)Ca]2(N2) (I) with N2O to form [(BDI*)Ca](μ2-O) was calculated to be exergonic by ΔG298K = –109.8 kcal mol−1 (Fig. S25, ESI†). Reaction of I with O2 to form a Ca peroxide complex is even more exergonic (ΔG298K = –150.0 kcal mol−1). This underscores the highly reducing nature of CaI synthon I caused by the facile 2e-oxidation of the bridging dianion: N22− → N2 + 2e−.
Natural Population Analysis (NPA) confirms essentially ionic Ca–(O2) or Ca–O bonds with highly positive charges on the Ca centres (1: +1.79, 2: +1.79) and negative charges on the bridging (per)oxide dianions (O22−: –1.78, O2−: –1.74) (Fig. S19 and S20, ESI†). Ionic bonding is in agreement with Atoms-In-Molecules (AIM) analysis which shows low electron densities ρ(r) and positive Laplacian ∇2ρ(r) in the Ca–O bond-critical-points (bcp's); Fig. S21 and S22 (ESI†). The Wiberg Bond Index (WBI) of 0.99 for the O–O bond in the O22− anion confirms single bond character for the peroxide anion in 1. AIM analysis also shows that bridged O22− in 1 and O2− in 2 are involved in weak O⋯H–C bonding with organic fragments of the BDI* ligand (Fig. S21 and S22, ESI†). These non-classical hydrogen bonds are typical for low-coordinate ligands.41
An interesting aspect of bonding in the central Ca–(μ2-O)–Ca of 2 is Ca d-orbital participation which is currently controversially discussed. While the HOMO, HOMO−1 and HOMO−2 are mainly centred on the O2− and BDI* ligands (Fig. S24, ESI†), the HOMO−3 combines 84.8% O(2pz) with 7.9% Ca(3dxz) character (Fig. 1c). For HOMO−4 following contributions are calculated: 77.9% O(2pz) with 11.2% Ca(3dxz).
In summary, first Ca (per)oxide complexes with low-coordinate μ2-O2 or μ2-O dianions are accessible from the CaI synthon [(BDI*)Ca(THP)]2(N2) and O2 or N2O, respectively. The very high reactivity and instability of the Ca oxide complex in solution required a solid-state synthesis. The low-coordination number of the peroxide and oxide dianions result in numerous non-classical C–H⋯O hydrogen bonds but also cause instability. Facile decomposition likely proceeds through deprotonation of the ligand's alkyl groups, impeding selective reactivity. However, the reactivity of the low-coordinate Ca peroxide complex 1 as an oxidizing reagent is currently under investigation.
We acknowledge Dr C. Färber and J. Schmidt for assistance with NMR analyses and A. Roth for CHN analyses. Dr J. Mai, L. Klerner and T. Vilpas are acknowledged for assistance with DFT calculations. We thank the Deutsche Forschungsgemeinschaft for funding (HA 3218/11-1).
Conflicts of interest
There are no conflicts to declare.Data availability
X-ray crystallographic data have been deposited in the Cambridge Crystallographic Data Centre with reference numbers: CCDC 2427514–2427515. Complex syntheses and analyses, NMR spectra, crystallographic details including ORTEP representations, details for the DFT calculations including XYZ-files have been included as part of the ESI.†Notes and references
- N. M. Julkapli and S. Bagheri, Rev. Inorg. Chem., 2016, 36, 1–41 CAS.
- V. R. Chintareddy and M. Lakshmi Kantam, Catal. Surv. Asia, 2011, 15, 89–110 CrossRef CAS.
- W. Widayat, T. Darmawan, H. Hadiyanto and R. A. Rosyid, J. Phys.: Conf. Ser., 2017, 877, 012018 CrossRef.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- S. Thompson, S. Burnett, R. Ferns, T. van Mourik, A. P. McKay, A. M. Z. Slawin, D. B. Cordes and A. Stasch, J. Am. Chem. Soc., 2025, 147, 5247–5257 CrossRef CAS PubMed.
- R. Lalrempuia, A. Stasch, A. Stasch and C. Jones, Chem. Sci., 2013, 4, 4383–4388 RSC.
- K. Abdur-Rashid, T. P. Fong, B. Greaves, D. G. Gusev, J. G. Hinman, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2000, 122, 9155–9171 CrossRef CAS.
- G. Stucky and R. E. Rundle, J. Am. Chem. Soc., 1964, 86, 4821–4825 CrossRef CAS.
- A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, Angew. Chem., Int. Ed., 1998, 37, 3180–3183 CrossRef CAS PubMed.
- H. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Organometallics, 2024, 43, 879–888 CrossRef CAS PubMed.
- R. Fischer, H. Görls and M. Westerhausen, Inorg. Chem. Commun., 2005, 8, 1159–1161 CrossRef CAS.
- C. Ruspic and S. Harder, Organometallics, 2005, 24, 5506–5508 CrossRef CAS.
- M. Westerhausen, M. Gärtner, R. Fischer, J. Langer, L. Yu and M. Reiher, Chem. – Eur. J., 2007, 13, 6292–6306 CrossRef CAS PubMed.
- S. Krieck, H. Görls and M. Westerhausen, J. Organomet. Chem., 2009, 694, 2204–2209 CrossRef CAS.
- S. Yadav, V. S. V. S. N. Swamy, R. Gonnade and S. S. Sen, ChemistrySelect, 2016, 1, 1066–1071 CrossRef CAS.
- B. Wei, L. Liu, W. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2017, 56, 9188–9192 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, D. T. Nguyen, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2023, 60, 1016–1019 RSC.
- J. Maurer, L. Klerner, J. Mai, H. Stecher, S. Thum, M. Morasch, J. Langer and S. Harder, Nat. Chem., 2025, 17, 703–709 CrossRef CAS PubMed.
- N. C. Mösch-Zanetti, M. Ferbinteanu and J. Magull, Eur. J. Inorg. Chem., 2002, 950–956 CrossRef.
- A. M. Drummond, L. T. Gibson, A. R. Kennedy, R. E. Mulvey, C. T. O’Hara, R. B. Rowlings and T. Weightman, Angew. Chem., Int. Ed., 2002, 41, 2382–2384 CrossRef CAS PubMed.
- A. R. Kennedy, J. G. MacLellan and R. E. Mulvey, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m302–m303 CrossRef PubMed.
- R. Fischer, H. Görls and M. Westerhausen, Organometallics, 2007, 26, 3269–3271 CrossRef CAS.
- H. Vitze, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1614 CrossRef CAS PubMed.
- A. D. Obi, N. C. Frey, D. A. Dickie, C. E. Webster and R. J. Gilliard, Angew. Chem., Int. Ed., 2022, 61, e202211496 CrossRef CAS PubMed.
- J. Mai, J. Maurer, J. Langer and S. Harder, Nat. Synth., 2023, 3, 368–377 CrossRef.
- C. Ritschel, C. Donsbach and C. Feldmann, Chem. – Eur. J., 2024, 30, e202400418 CrossRef CAS PubMed.
- S. Schnitzler, T. P. Spaniol and J. Okuda, Inorg. Chem., 2016, 55, 12997–13006 CrossRef CAS PubMed.
- S. Harder and J. Langer, Nat. Rev. Chem., 2023, 7, 843–853 CrossRef PubMed.
- A. Kornowicz, T. Pietrzak, K. Korona, M. Terlecki, I. Justyniak, A. Kubas and J. Lewiński, J. Am. Chem. Soc., 2024, 146, 18938–18947 CrossRef CAS PubMed.
- D. Lionetti and T. Agapie, Nature, 2014, 513, 495–496 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Dalton Trans., 2018, 47, 14381–14387 RSC.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- S. Thum, O. P. E. Townrow, J. Langer and S. Harder, Chem. Sci., 2025, 16, 4528–4536 RSC.
- S. Thum, J. Mai, N. Patel, J. Langer and S. Harder, ChemistryEurope, 2025, e202500080 Search PubMed.
- S. Thum, J. Mai, M. A. Schmidt, J. Langer and S. Harder, Chem. Sci., 2025, 16, 12058–12067 RSC.
- C. Ruspic, S. Nembenna, A. Hofmeister, J. Magull, S. Harder and H. W. Roesky, J. Am. Chem. Soc., 2006, 128, 15000–15004 CrossRef CAS PubMed.
- T. Higuchi, A. Uchida and M. Hashimoto, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1494–1497 CrossRef CAS PubMed.
- M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and C. Weetman, Dalton Trans., 2011, 40, 12500–12509 RSC.
- B. Neumüller, F. Weller, T. Gröb and K. Dehnicke, Z. Anorg. Allg. Chem., 2002, 628, 2365–2371 CrossRef.
- N. J. C. Van Velzen and S. Harder, Organometallics, 2018, 37, 2263–2271 CrossRef CAS.
- S. Harder, Chem. – Eur. J., 1999, 1852–1861 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2427514 and 2427515. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc03340c |
| This journal is © The Royal Society of Chemistry 2025 |