
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ oxidizable directing group-enabled enantioselective synthesis of chiral platform ferrocene formaldehydes†
Devendra Parganiha ,
Ashwini Dilip Dhumale,
Yagya Dutt Upadhyay,
Vishal Choudhary,
Svastik Jaiswal,
Raushan Kumar Jha and
Sangit Kumar*
,
Ashwini Dilip Dhumale,
Yagya Dutt Upadhyay,
Vishal Choudhary,
Svastik Jaiswal,
Raushan Kumar Jha and
Sangit Kumar*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-Pass Road, Bhopal, Madhya Pradesh 462066, India. E-mail: sangitkumar@iiserb.ac.in
First published on 25th June 2025
Abstract
Herein, we have developed an efficient oxidatively removable directing group approach, enabling highly enantioselective PdII-catalyzed C–H activation. Successful rationalization of difficult C–H activation over the labile β-H oxidation step led to the direct synthesis of chiral 2-alkenylated ferrocene formaldehydes with up to 71% yields and 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 er.
:2 er.
The integration of directing groups (DGs) into transition metal-catalysed C–H activation has revolutionized organic synthesis, as these directing groups not only lead to a new array of molecules but also stabilize the inherently unstable species.1 However, in situ transformation of the directing group into desired functionalities is always challenging and has not been fully addressed yet.1
Planar chiral ferrocenes,2 particularly ferrocene formaldehydes, serve as important platform building blocks for synthesizing various planar chiral catalysts and ligands, including industrially used Josiphos, Ugi's amines, PPFA, and PHOX-type ligands (Scheme 1).3 Synthesis of planar chiral ferrocene formaldehyde derivatives has always been challenging yet remains essential.4 The major limitations are the multistep conventional routes or expensive reagents for the very high enantioselective synthesis of platform ferrocene formaldehydes.4 Moreover, a PdII-enabled transient directing group approach5 for highly enantioselective C–H activation was unsuccessful for metallocenes as it led to C–H activation on the lower cp-ring.5
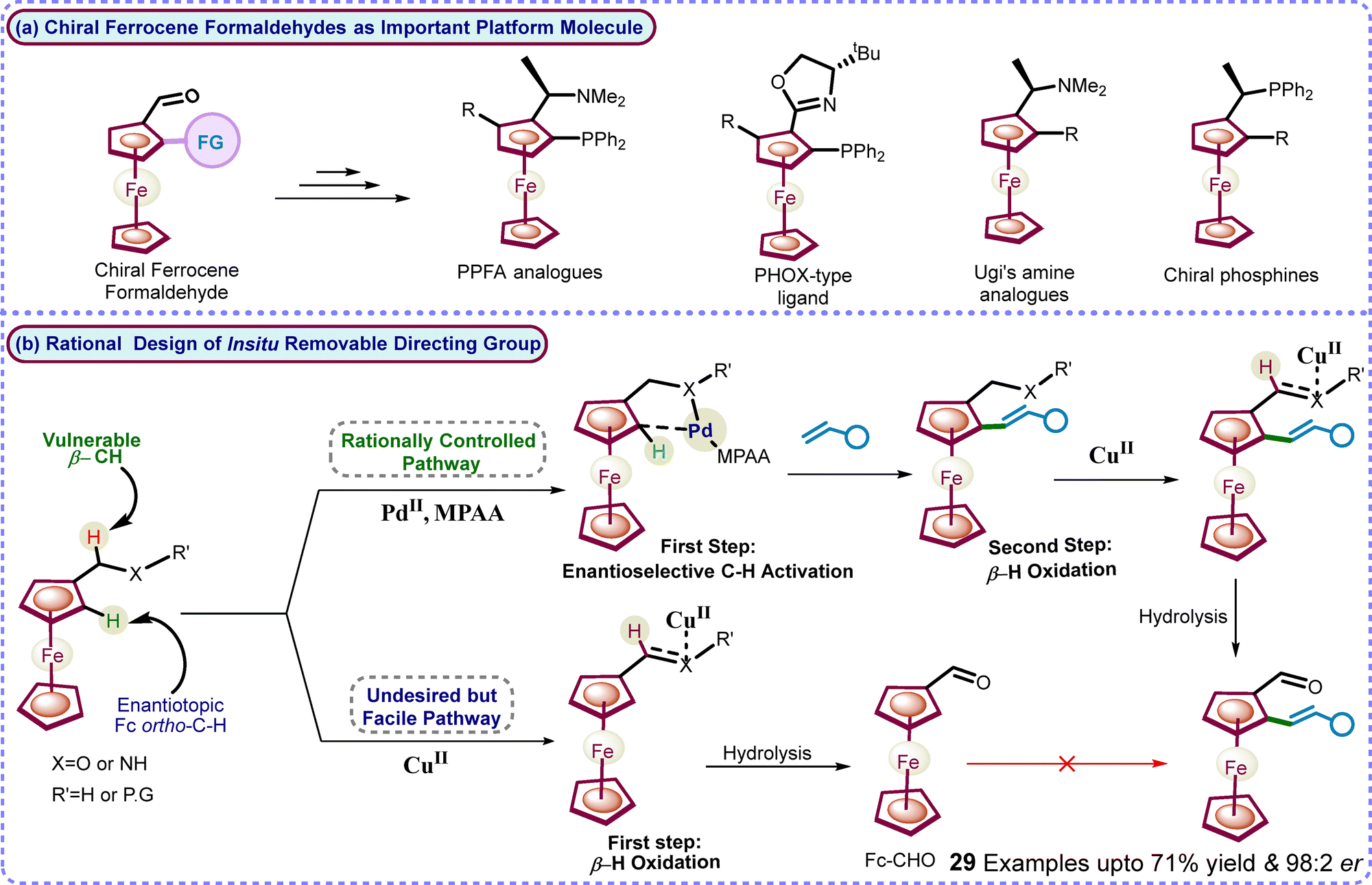 | ||
| Scheme 1 (a) Ferrocene formaldehyde as a valuable platform molecule and (b) rational design of the step-economical oxidizable directing group strategy and our work. | ||
Our group is continuously working on TM-catalyzed directing group methodologies for diverse selective C–H activation in ferrocenes.6 Furthermore, we aim to develop a highly enantioselective and efficient methodology to construct important chiral platform ferrocene formaldehydes with high enantioselectivity. Consequently, a removable directing group strategy was envisioned, leading to enantioselective C–H activation and, subsequently, in situ transformation of the directing group into important platform chiral formaldehyde functionalities (Scheme 1). Herein, we have successfully developed an in situ oxidizable directing group methodology through a sequential rationalization of unreactive enantiotopic C–H bond activation over facile β-H oxidation of protic amines. The developed in situ oxidizable directing group methodology led to the efficient synthesis of diverse new alkenylated platform chiral ferrocene formaldehydes with up to 71% yield and 98:2 er in a step-economical manner (Scheme 1).
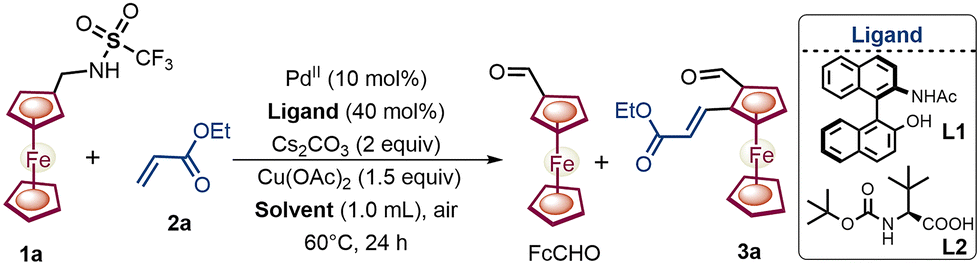
We commenced our investigation of ferrocenyl-methylamine 1a with a combination of Pd(OAc)2 as a catalyst and a recently reported chiral NOBINAc ligand6c with acrylate 2a (Table 1). In addition, an oxidant, Cu(OAc)2, with the base Cs2CO3, was added to facilitate the subsequent oxidation of the catalyst as well as the directing group. Ferrocenylamine 1a produced the desired oxidized amine product – chiral 1,2-alkenylated ferrocene formaldehyde 3a – in a poor yield of 20% with a moderate enantioselectivity of 90:10 er (Table 1, entry 1). Furthermore, various solvents were screened to control the excessive oxidation of amine 1a (Table 1, entries 2–4). Among the solvents screened, tert-amyl alcohol increased the yield of 3a to 32%, and undesirably the enantioselectivity of 3a was reduced to 70:30 er (Table 1, entry 4), which may be due to the incompatibility of the NOBINAc ligand with the polar protic tert-amyl alcohol.7 To circumvent the undesired oxidation of 3a to FcCHO and to further enhance the er, we screened a series of chiral mono-protected amino acid ligands (L2–L9; see Fig. S3 and Table S2, ESI†). To our delight, the screened MPAA ligands L2-L9 demonstrated notable efficacy, delivering yields ranging from 32% to 75% and er values from 87:13 to 98:2. Among all the ligands screened, Boc-L-tert-Leucine L2 provided the best results with amine 1a, achieving a 75% yield and 98:2 er of 3a (for details see Tables S1–S4, ESI†).
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | Ln | FcCHO (%) | 3ab (%) | erc (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Pd(OAc)2 (0.01 mmol), ligand (0.04 mmol), Cs2CO3 (0.2 mmol), Cu(OAc)2 (0.15 mmol), DMSO (0.5 mmol), dry solvent (1 mL), air, 60 °C, 24 h.b The crude yield of 3a was determined by 1H NMR with CH2Br2 as an internal standard.c The enantiomeric ratio (er) of 3a was determined by HPLC analysis.d The reaction was stirred for 30 h at 55 °C. Ligand L1 = NOBINAc and L2 = Boc-L-tert-leucine. | |||||
| 1 | THF | L1 | 80 | 20 | 90:10 |
| 2 | Toluene | L1 | 75 | 25 | 87:13 |
| 3 | DMF | L1 | 88 | 12 | 76:24 |
| 4 | tAmyl alcohol | L1 | 68 | 32 | 70:30 |
| 5 | tAmyl alcohol | L2 | 35 | 65 | 96:4 |
| 6d | tAmyl alcohol | L2 | 25 | 75 | 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 :2 |
Next, a variety of ferrocenylamines 1b–1h were tested under enantioselective C–H activation followed by oxidative deamination conditions (Fig. 1). Other sulfonyl (SO2R)-protected ferrocenylamines 1b–1e also provided the chiral product 3a in 5–55% yields, with 88:12 to 98:2 er, whereas amines 1f–1h, lacking a coordinating SO2R group, did not yield the desired alkenylated formaldehyde 3a. The screening of the various amines suggests that the low pKa of the methylene C–H bond, the coordinating ability of amine DGs, and the better leaving group ability of the –S(O)2R substituent are needed for effective enantioselective C–H activation and in situ directing group removal.8 Furthermore, the substrate scope with regard to various olefins (2a–2aa) was explored. Acrylates 2a–2t with alkyl, electron-donating and electron-withdrawing group-substituted aryl, naphthyl, and benzyl substitution yielded an array of 1,2-alkenylated ferrocene formaldehydes 3a–3t in 52–71% yields and 92:8–98:2 er (Scheme 2). Moreover, other olefins, namely, vinyl sulfonate 2u, phosphonates 2v and 2w, acrylamide 2x, and vinyl benzene 2y, also showed the amenability to afford respective chiral 1,2-alkenylated ferrocene formaldehydes 3u–3y in 48–66% yields and 92:8–97:3 er (Scheme 2). Furthermore, olefins derived from natural products L-menthol and cholesterol 2z and 2aa were also coupled diastereoselectively to afford the respective menthol- and cholesterol-containing chiral ferrocene formaldehydes 3z and 3aa in 51% and 49% yields, with 7:1 to 10:1 dr, respectively (Scheme 2). Additionally, we performed a gram-scale reaction, achieving up to 50% yield of chiral 1,2-alkenylated ferrocene formaldehyde 3a with 95:5 er. Furthermore, chiral 1,2-alkenylated ferrocene formaldehydes 3c and 3b were reduced to their respective methyl and alcohol groups, leading to molecules 4 and 5 without any reduction in enantioselectivity (3c, 97:3 er versus 4, 97:3 er, and 3b, 97:3 er versus 5, 97:3 er, Scheme 3).
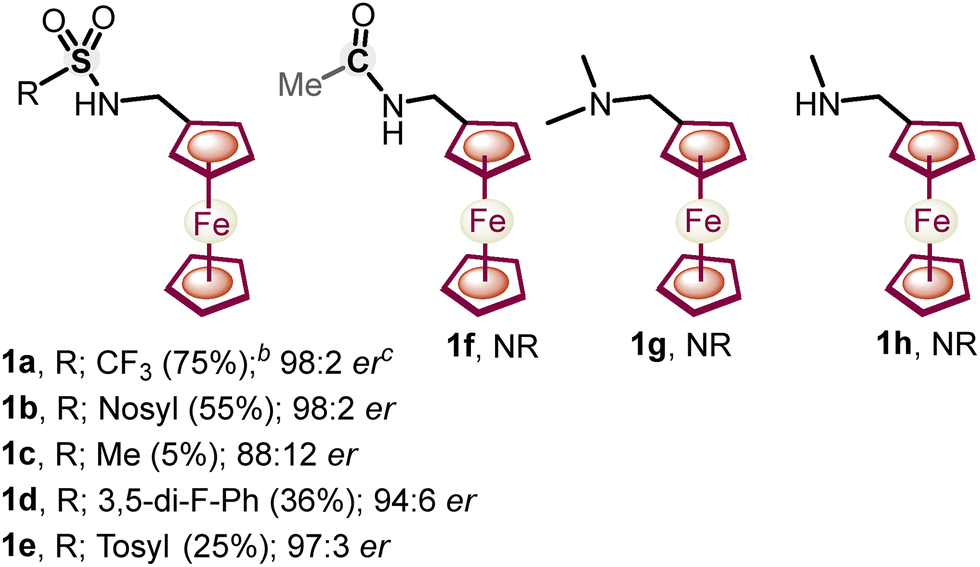 | ||
| Fig. 1 Screening of amines for enantioselective C–H activation and subsequent oxidative deamination. Reaction conditions: the same as those mentioned in Table 1, entry 6. | ||
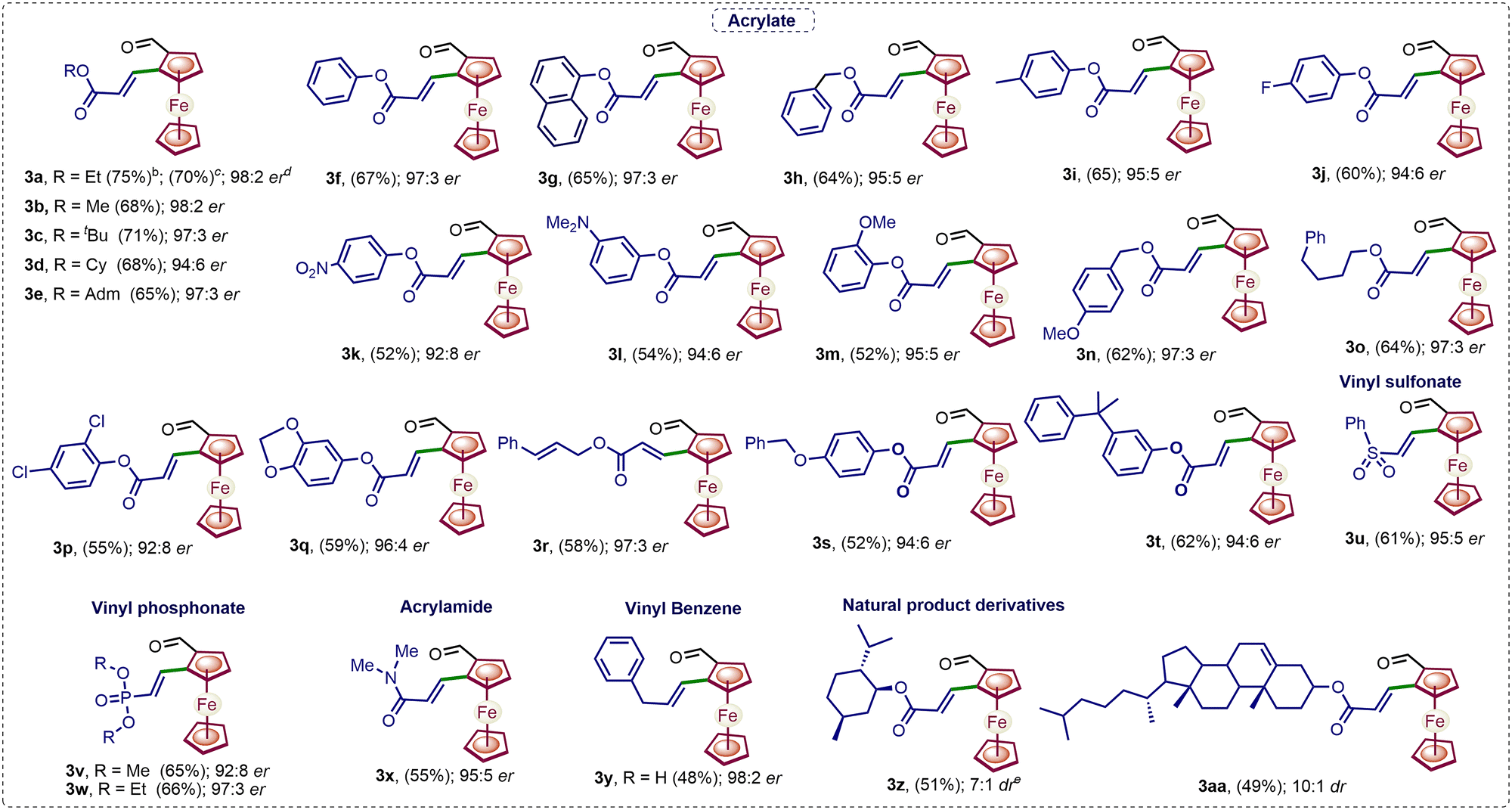 | ||
| Scheme 2 Substrate scope for enantioselective C–H alkenylation. aReaction conditions are the same as those mentioned in Table 1, entry 6. bThe crude yield of 3a was determined by 1H NMR with CH2Br2 as an internal standard. cIsolated yields of 1,2-alkenylated ferrocene formaldehydes 3a-3aa. dThe er of 3a was determined by HPLC. eThe dr ratio of the isolated product was determined by 1H NMR. | ||
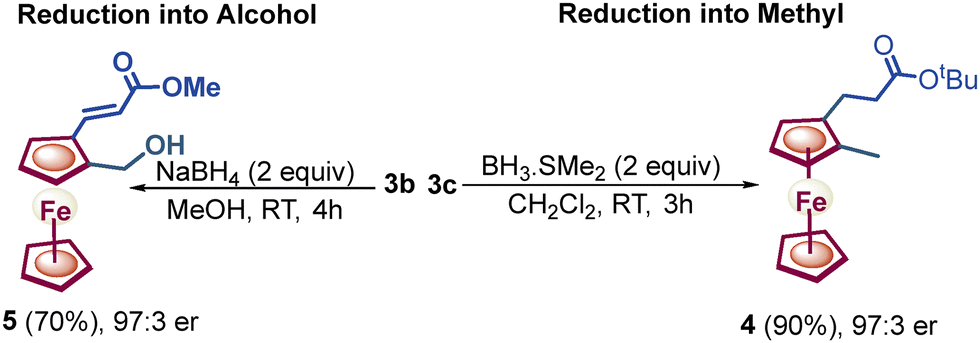 | ||
| Scheme 3 Post-derivatization of aldehydes into methyl and alcohol functionalities. | ||
Furthermore, to gain mechanistic insights into controlled amine oxidation versus enantioselective C–H activation pathways, a series of control experiments were performed (Scheme 4).
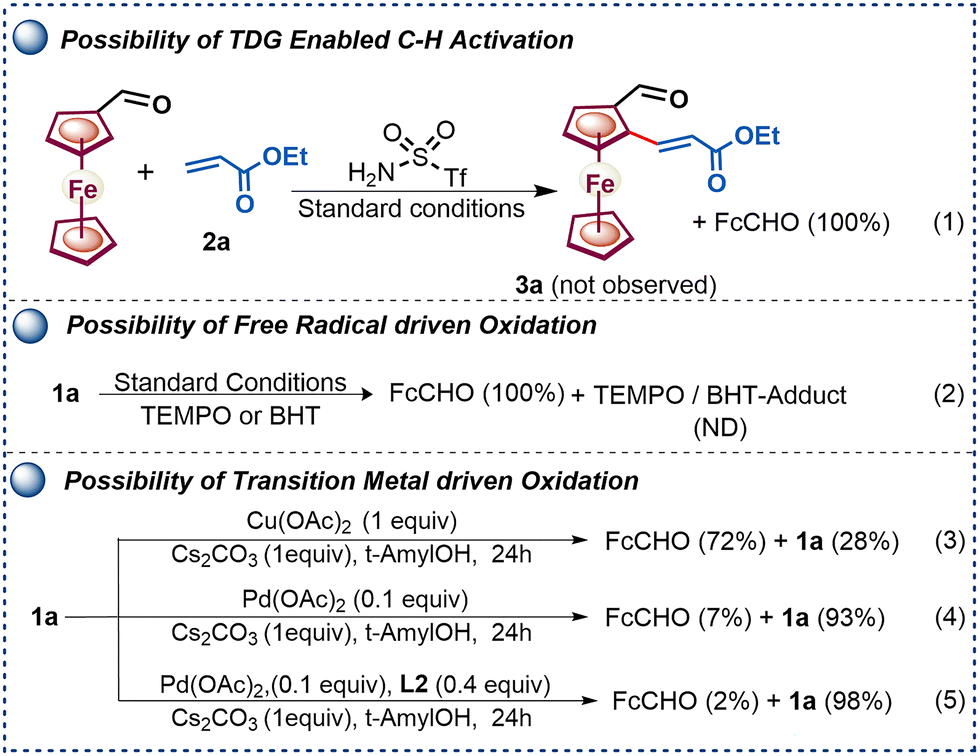 | ||
| Scheme 4 Mechanistic investigation of C–H activation versus deamination. | ||
The failure to obtain product 3a from ferrocene formaldehyde under optimized conditions (Scheme 4, eqn (1)) rules out imine involvement in the enantioselective C–H activation. Furthermore, we examined the possibility of a radical-driven process where the radical quenchers (TEMPO and BHT) failed to prevent oxidation or to form any adducts as observed by mass spectrometry (Scheme 4, eqn (2)). Next, the role of PdII and CuII in the oxidation of amine was studied by selectively varying the metal salts involved in the reaction (Scheme 4, eqn (3)–(5)). The reaction outcomes indicate that both Cu(OAc)2 and Pd(OAc)2 are capable of facilitating the respective β-H oxidation, and the presence of the base Cs2CO3 significantly accelerates the oxidation.
Additionally, it was noted that the presence of an MPAA ligand slows the β-H oxidation driven by the Pd(OAc)2 catalyst (Scheme 4, eqn (5)). Furthermore, the relative rates of C–H activation versus amine oxidation were rationalized by 1H NMR experiments, suggesting that the rate of C–H activation is 1.76 times higher than the rate of amine oxidation (see Fig. S4–S9, ESI†). Further, a ligand acceleration experiment was conducted, which revealed that the presence of ligand L2 accelerates the reaction by 4.6-fold (see Fig. S8, ESI†). Furthermore, the DFT computational studies were performed to determine the relative enantiotopic C–H energy within CMD, which showed a 2.48 kcal mol−1 lower energy for the favorable TS than for the unfavorable TS (see Fig. S10, ESI†). Based on control experiments, we have proposed a plausible catalytic cycle (Scheme 5). Initially, amine 1a, Pd(OAc)2, and ligand L2 with a Cs+ cation form a Pd-aminate, I, as suggested by 19F NMR experiments (see Fig. S9, ESI†) and a previous report.10 Furthermore, the Pd-aminate I undergoes enantioselective C–H activation to generate a chiral palladacycle, II. The chiral palladacycle II interacts with the olefin through π-bond interactions, leading to the formation of a Pd-alkyl intermediate, III. Consequently, intermediate III would undergo β-H elimination to afford chiral 2-alkenylated ferrocenylamine. The 2-alkenylated ferrocenylamine was then oxidized by Cu(OAc)2 through one-electron oxidation of triflamide to form copper-amidate IV, followed by hydrolysis, yielding chiral 2-alkenylated ferrocenyl formaldehydes.9
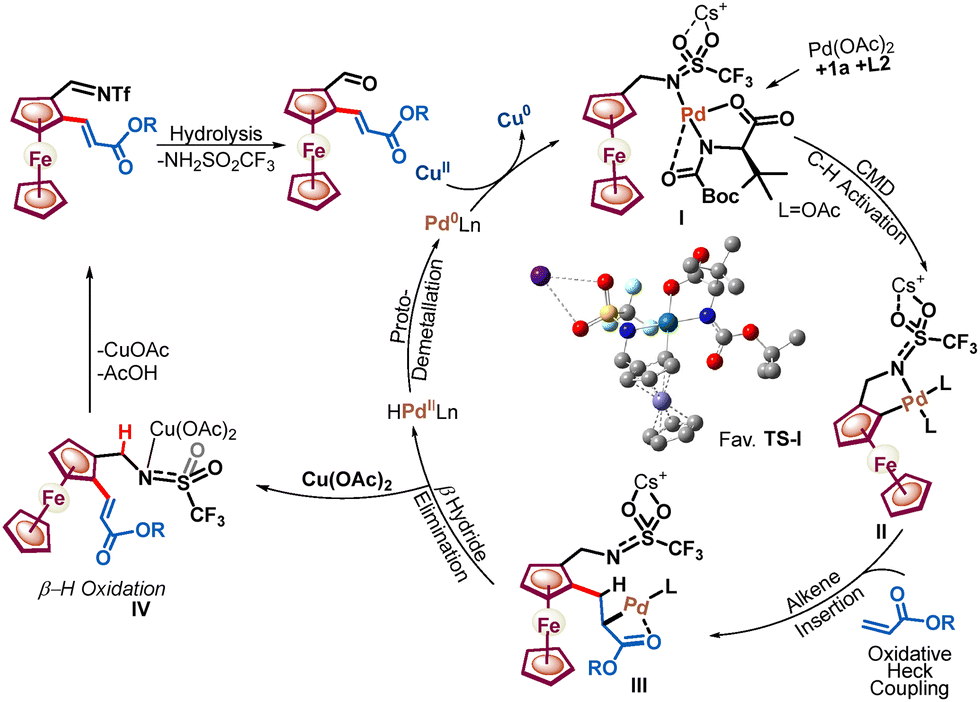 | ||
| Scheme 5 Plausible catalytic cycle. The favourable TS shown was obtained by DFT calculations. | ||
In summary, we have developed a mild in situ oxidizable directing group methodology for the highly enantioselective and monoselective synthesis of chiral platform ferrocene formaldehydes. The developed methodology effectively rationalizes the difficult enantioselective C–H activation step first and the facile β-H oxidation step later, which represents an efficient alternative to the transient directed strategy using a dual PdII/CuII cycle in stacked molecules.
SK acknowledges DST-ANRF (CRG/2023/002473), New Delhi, and IISER Bhopal for financial support. DP and SJ acknowledge the UGC, New Delhi, for fellowships [NTA ref. no. 191620241186] and [NTA ref. no. 201610131472]. AD acknowledges DST INSPIRE (IF210534) for the fellowship.
Conflicts of interest
There are no conflicts to declare.Data availability
Detailed synthetic procedures, reaction condition optimization, data characterization, controlled experiments, and geometries and energies of theoretically investigated structures are provided in the ESI.†Notes and references
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (b) C. Sambiagio, D. Schonbauer, R. Blieck, T.-D. Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J.-W. Delord, T. Besset, B. U. W. Maes and A. Schnurch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (c) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed; (d) R. L. Carvalho, R. G. Almeida, K. Murali, L. A. Machado, L. F. Pedrosa, P. Dolui, D. Maiti and E. N. D. S. Júnior, Org. Biomol. Chem., 2021, 19, 525–547 RSC; (e) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed; (f) R. S. Thombal, P. Yuosef, M. Rubio, D. Lee, D. Maiti and Y. R. Lee, ACS Catal., 2022, 12, 5217–5230 CrossRef CAS; (g) B. B. Zhan, L. Jin and B.-F. Shi, Trends Chem., 2022, 4, 220–235 CrossRef CAS; (h) Q. Zhang, L.-S. Wu and B.-F. Shi, Chem, 2022, 8, 384–413 CrossRef CAS; (i) K. Wu, N. Lam, D. A. Strassfeld, Z. Fan, J. X. Qiao, T. Liu, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2024, 63, e202400509 CrossRef CAS PubMed.
- (a) D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351–365 CrossRef CAS PubMed; (b) Z.-J. Cai, C.-X. Liu, Q. Wang and S.-L. You, Nat. Commun., 2019, 10, 4168 CrossRef PubMed; (c) C.-X. Liu, Q. Gu and S.-L. You, Trends Chem., 2020, 2, 737–749 CrossRef CAS; (d) Y. Huang, C. Pi, X. Cui and Y. Wu, Adv. Synth. Catal., 2020, 362, 1385–1390 CrossRef CAS; (e) Q. Wang, Y. H. Nie, C. X. Liu, W. W. Zhang, Z. J. Wu, Q. Gu, C. Zheng and S.-L. You, ACS Catal., 2022, 12, 3083–3093 CrossRef CAS; (f) L. Zhou, H. G. Cheng, L. Li, K. Wu, J. Hou, C. Jiao, S. Deng, Z. Liu, J. Q. Yu and Q. Zhou, Nat. Chem., 2023, 15, 815–823 CrossRef CAS PubMed; (g) X. Kuang, J.-J. Li, T. Liu, C.-H. Ding, K. Wu, P. Wu and J.-Q. Yu, Nat. Commun., 2023, 14, 7698 CrossRef CAS PubMed; (h) Q.-J. Yao, F.-R. Huang, J.-H. Chen and B.-F. Shi, Nat. Commun., 2024, 15, 7135 CrossRef CAS PubMed; (i) J.-Y. Ma, Q.-J. Yao, L.-C. Jiang, F.-R. Huang, Q. Yue and B.-F. Shi, J. Am. Chem. Soc., 2025, 147, 7061–7069 CrossRef CAS PubMed.
- (a) R. G. Array, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674–7715 CrossRef PubMed; (b) K. Yoshida and R. Yasue, Chem. – Eur. J., 2018, 24, 18575–18586 CrossRef CAS PubMed.
- (a) O. Riant, O. Samuel, T. Flessner, S. Taudien and H. B. Kagan, J. Org. Chem., 1997, 62, 6733–6745 CrossRef CAS; (b) A. Urbano, G. Hernández-Torres, A. M. del Hoyo, A. Martínez-Carrión and M. C. Carreño, Chem. Commun., 2016, 52, 6419–6422 RSC; (c) C.-X. Liu, F. Zhao, Q. Gu and S.-L. You, ACS Cent. Sci., 2023, 9, 2036–2043 CrossRef CAS PubMed.
- Z. Yu, Q. Liu, Q. Li, Z. Huang, Y. Yang and J. You, Angew. Chem., Int. Ed., 2022, 61, e202212079 CrossRef CAS PubMed.
- (a) R. A. Thorat, Y. Mandhar, D. Parganiha, K. B. Patil, V. Singh, B. Shakir, S. Raju and S. Kumar, ChemistrySelect, 2023, 8, e202203945 CrossRef CAS; (b) R. A. Thorat, D. Parganiha, S. Jain, V. Choudhary, B. Shakir, K. Rohilla, R. K. Jha and S. Kumar, Org. Lett., 2025, 27, 552–558 CrossRef PubMed; (c) D. Parganiha, R. A. Thorat, A. D. Dhumale, Y. D. Upadhyay, R. K. Jha, S. Raju and S. Kumar, Chem. Sci., 2025, 16, 700–708 RSC.
- (a) J. M. González, X. Vidal, M. A. Ortuño, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2022, 144, 21437–21442 CrossRef PubMed; (b) P. Losada, L. Goicoechea, J. L. Mascarenas and M. Gulías, ACS Catal., 2023, 13, 13994–13999 CrossRef CAS PubMed.
- S. Mallick, T. Mandal, N. Kumari, L. Roy and S.-D. Sarkar, Chem. – Eur. J., 2024, 30, e202304002 CrossRef CAS PubMed.
- J. Kim, G. Golime, H. Y. Kim and K. Oh, Asian J. Org. Chem., 2019, 8, 1674–1679 CrossRef CAS.
- L. Chu, X. C. Wang, C. E. Moore, A. L. Rheingold and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344–16347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02611c |
| This journal is © The Royal Society of Chemistry 2025 |