
An environmental impact statement for molecular anions
Jack
Simons

Henry Eyring Center for Theoretical Chemistry, Department of Chemistry, University of Utah, Salt Lake City, UT 84112, USA. E-mail: jack.simons@utah.edu
First published on 15th December 2023
Abstract
A molecular anion's (MA's) chemical reactivity and physical behavior can be quite different when it is surrounded by other molecules than when it exists in isolation. This sensitivity to the surrounding environment is especially high for anions because their outermost valence electrons are typically loosely bound and exist in rather spatially diffuse orbitals, allowing even weak intermolecular interactions arising from the environment to have strong effects. This Perspective offers illustrations of such sensitivity for a variety of cases including (i) the effect of solvation on electron binding energies, (ii) how some “well known” anions need to have solvent molecules around to even exist as stable species, (iii) how internal Coulomb repulsions within a multiply charged MA can provide temporary stability toward electron loss, (iv) how MAs arrange themselves spatially near liquid/vapor interfaces in manners that can produce unusual reactivity, (v) how nearby cationic sites can facilitate electron attachment to form a MA site elsewhere, (vi) how internal vibrational or rotational energy can make a MA detach an electron.
1. Introduction
In this Perspective article, I am going to discuss how the behavior of molecular anions (MAs) is very sensitive to the surrounding environment (e.g., as isolated species, in strongly solvating solutions, at interfaces between the vapor phase and a liquid or solid surface, in the presence of nearby cations, and more). In all such cases, the sensitivity of the anion's electronic structure arises largely due to the relatively weak electron binding strength characterizing most anions, which causes them to be strongly affected even by moderate-strength intermolecular interactions with their environment. I expect that ion spectroscopists and quantum chemists who have studied MAs will not be surprised by many of the observations I make. However, these communities are not the primary audience I am trying to engage; I am speaking primarily to a broader range of chemists and physicists who study chemical reactions and physical processes involving MAs because I believe they need to be made better aware of the characteristics of MAs that I discuss here. But I also hope those already more informed about MAs will also discover something new in what I have to offer.I am focusing on the surrounding-sensitivity aspect of MA chemistry rather than taking a much broader view because I believe other sources that I have already provided offer the reader an efficient route to such breadth. In particular, in 2008, I published a 110-page review1 treating many experimental and theoretical studies of a wide variety of MAs. In 2023, I provided a Perspective article2 updating the 2008 review by focusing on the research of more recent workers. Also, I maintain a web site (https://hec.utah.edu/simons-group/anions/index.php) that parallels the 2008 review and includes even more detail. In addition to these efforts by my group, I have found John Herbert's article3 on loosely bound electrons to be a wonderful source of information and insight about MAs. For readers primarily interested in molecular electron affinities,4 there exists an extensive review from Fritz Schaefer's and Barney Ellison's groups. In addition, throughout this text, I provide web links to the research pages and Google Scholar sites belonging to several researchers whose work I discuss. I suggest that the sum of these sources offers the broader view that some readers may be looking for.
Before getting to the main emphasis of this Perspective, I’d like to tell you a bit about how I became interested in molecular anions and how my involvement broadened and increased over several decades. In Fig. 1 I show a plot of the number of articles published in ACS journals containing the phrase “ anion” in the title, ranging between 1955 and 2020.
 | ||
| Fig. 1 Plot of the number of articles in all ACS journals having anion in the title over five-year intervals. Reprinted from Fig. 1 in ref. 2 with permission of the American Chemical Society (2023). | ||
I hope these data suggest why this field has attracted my attention for so many years and continues to do so; many chemists seem to be carrying out research involving MAs and I have been amazed at the wide variety of situations in which these studies lie.
1.1. Brief history of my early involvement
My interest in anions dates to the early 1970s and resulted in my first venture into the field producing a paper titled Theory of Electron Affinities of Small Molecules5 in which we used the so-called equation of motion (EOM) mathematical framework to derive working equations for directly calculating intensive electron affinities (EAs). I was aware that other quantum chemists (including Fritz Schaefer and Howard Taylor, two students of my late colleague Frank Harris) had calculated atomic and molecular EAs and had illustrated several difficulties in doing so:i. EAs are relatively small numbers (e.g., ca. 3 eV for halogens but much smaller for many/most atoms and molecules of interest). To extract such numbers by subtracting the total electronic energies of the neutral and anion, both of which are extensive quantities, requires that both energies be determined to high or carefully-balanced precision. In 1973, such calculations were possible only for small molecules or atoms, which is why we included the word “small” in ref. 5's title.
ii. The small EA values give rise to diffuse charge densities for the outermost valence electrons. The atomic orbital basis sets that had been designed for calculations on neutral or positively charged species thus were not sufficient. So considerable effort had to be devoted to creating diffuse basis functions appropriate to anions.
iii. To achieve the level of precision needed to achieve sufficient reliability in calculated EAs, it was necessary to include electron correlation effects in any useful theory. Koopmans’ theorem or subtracting anion and neutral Hartree–Fock energies was not sufficient as such methods often involved errors in excess of 0.5 eV, which is a major fraction of many molecules’ EA.
iv. Some MAs do not have bound electronic states so their study required that one move beyond the conventional bound-state variational or perturbative methods of quantum chemistry. The early work of Howard Taylor provided good guidance on this front.
Based on this experience provided by earlier workers, I decided to pursue using EOM's tools to create a theoretical framework for directly computing EAs (i.e., in a single calculation) focusing on systems for which either the anion or the neutral had a closed-shell electronic structure in its qualitative description. If the anion were closed-shell in such an approximate sense, our EOM equations could be used to determine the EA as the electron detachment energy of the anion. Alternatively, if the neutral were approximately closed-shell, the EA could be obtained as the EOM equations’ electron attachment energy. Within this framework, we approximated the electronic wave function of the closed-shell entity using Rayleigh–Schrödinger perturbation theory (RSPT).
From early test calculations we performed and from others’ experience, I knew that it would not be sufficient to develop EOM equations that would generate EA values of second-order RSPT precision, so we immediately focused on obtaining working equations precise through third-order. Two of my earliest co-workers and life-long friends, Ken Jordan and Poul Jørgensen, played key roles in achieving the third-order EA EOM framework that my group developed and applied to several molecular anions as early as the mid 1970s. John Kenney, Jeanne McHale, Ron Shepard, and Ajit Banerjee deserve much of the credit for these early EA calculations, and were instrumental in our attempts to extend the theory to species whose neutral and or anion required a multi-configuration wave function. Within a few years of our development and early use of this theory we became aware that our final working equations were equivalent to the third-order Greens function equations that Lenz Cederbaum6 and his co-workers derived.7 I had more confidence in following the algebraic and commutator EOM derivation steps used in nuclear physics by Rowe8 than in using the diagrammatic tools of Greens function (GF) theory, so it was only at this later date that we realized that both approaches could yield the same results. In subsequent years, other workers including Vince Ortiz, Rod Bartlett, and Anna Krylov9–12 have greatly improved the EOM and GF methods and applied them to numerous species and incorporated them into widely used computer codes.
In the late 1970s, I became aware of and interested in anions that are electronically metastable. Bob Donnelly introduced the complex coordinate technique into our EOM framework13 and Zlatko Bacic used such methods14 as well as stabilization techniques,15–17 which Gina Frey also used,18 for studying metastable states not of anions but as they arise in atom–molecule and molecule–molecule collisions.
In this same timeframe, Ken Jordan19 introduced me to the fact that polar molecules could bind an electron not in a valence orbital but to a so-called dipole-bound orbital20 if the molecule's dipole moment were large enough. In these early days, Ken and my group studied electron binding to closed-shell polar molecules,21 but only for species with quite large dipole moments (e.g., alkali halides) because our computational tools were not yet advanced enough to allow us to study dipole-bound anions having much smaller EAs. Ken's group has continued the study20 of these MAs over many years. My interest in metastable and dipole-bound MAs persists to this day and is described in some detail in ref. 1 and 2 where the methodological advances needed as well as the interesting behaviors of these species are explained.
1.2. My ongoing love affair with MAs
From the early 1980s through the present, there have been a variety of methodological developments and MA studies carried out within my group including:i. Providing straightforward ways22,23 to estimate lifetimes of metastable states using data from a stabilization graph15 including improvement24 by collaboration with Ken Jordan;
ii. Providing propensity rules25,26 for electron ejection from MAs induced by vibration/rotation energy transfer;
iii. Contributing27 to the characterization of double-Rydberg MAs;
iv. Teaming up with Alex Boldyrev28 and Maciej Gutowski29 to study the electronic and geometrical stabilities of multiply charged30 MAs;
v. Proposing31 and then experimentally studying32,33 in collaboration with Lai-Sheng Wang34 MAs containing a planar tetra-coordinate carbon atom through collaboration with Alex Boldyev;
vi. Continuing studies of dipole-bound anions, now including Maciej Gutowski and Piotr Skurski,35 designing basis sets36 appropriate for their treatment and illustrating that electron correlation is important to include;37
vii. Beginning in 200238 and persisting for several years, a series of studies involving Piotr Skurski and his group illustrating the mechanism39 by which a very low-energy electron attaches to a DNA base and subsequently generates a base-sugar C–O bond cleavage;
viii. Starting in 200340 and lasting through 2014,41 development and testing the so-called Utah-Washington mechanism by which disulfide and backbone N–Cα bond cleavage occurs in electron-capture and electron-transfer mass spectrometry, again by collaboration with Piotr Skurski and his group.
I should also note that I derived tremendous benefit by building and maintaining close relationships with several experimental groups that study MAs. These include groups lead by John Brauman, Carl Lineberger, Kit Bowen, Lai-Sheng Wang, Dan Neumark, Mark Johnson, Frank Turecek, Scott McLuckey, and Jan Verlet. I am a strong believer that it is important for theoretical chemists to understand the experiments they attempt to help interpret.
2. Molecular anions are very sensitive to their surroundings – the focus of this perspective
2.1. Interesting but not surprising cases
In Fig. 2 I show the photoelectron spectra42 associated with detaching an electron from an I−1 ion solvated by various numbers of N2O or CO2 molecules. These experiments use a laser of fixed energy hv and then measure the kinetic energy (KE) distributions of the ejected electrons from which the electron binding energy (EBE) can be determined as EBE = hv − KE. These binding energies can determine the vertical electron detachment (VDE) energy and the adiabatic electron detachment (ADE) energy from the maximum and onset of the KE distribution's peak, respectively. For cases where two peaks are shown in Fig. 2, the peak at the highest KE relates to producing the neutral I atom in its ground state; the peak at lower KE involves generating I in its spin–orbit excited state. | ||
| Fig. 2 Photoelectron spectra of I-(N2O)n (a) and I-(CO2)n (b). Reprinted from Fig. 2 and 4 from ref. 42 with permission of the AIP (1995). | ||
In all cases, the peaks shift to lower and lower KE values as the number of solvent molecules increases, meaning the solvated anion is more and more stabilized. Of course, the degree of differential solvation is not the same for N2O and CO2 because the strengths of their interactions with I− are different.
These findings are not surprising but they do introduce one issue I wish to discuss. These data clearly suggest that the behavior of I−, when related to its willingness to donate an electron to a potential reactive partner (or to share an electron pair) varies quite substantially depending on how strongly the anion is solvated. Obviously, this willingness will be small when the anion is fully surrounded by strongly solvating species such as H2O. But, for the purposes of this Perspective, the emphasis I wish to place is on situations when the anion finds itself not surrounded by one or more full solvation shells but, for example, when existing on or near the surface of a small atmospheric water microdroplet, at an air–liquid surface interface, or on the surface of an ice crystal. In these cases, the anion is likely to have a much smaller EBE than when fully solvated as Fig. 2 suggests.
2.2. Interesting cases with surprises
The differences between solvated and surface-bound anions discussed above characterizes what happens with many atomic and molecular anions and is not surprising because solvation differentially stabilizes the anion relative to its neutral. But now let me show you what happens when one examines the solvation of certain multiply charged anions. In Fig. 3 I show the photoelectron spectra43 of a series of SO42−(H2O)n clusters. | ||
| Fig. 3 Photoelectron spectra of SO42−(H2O)n (a) and vertical (upper data) and adiabatic (lower data) electron binding energies (b). Reprinted from Fig. 1 and 2 of ref. 43 with permission of the AIP (2000). | ||
The peaks on the left in Fig. 3(a) having the lowest EBE values relate to ejecting an electron from the closed-shell SO42− moiety to generate SO41− in its lowest-energy state. Not surprisingly, this peak shifts to higher and higher EBE as n increases, which is analogous to what we saw earlier for I−.
However, you should notice that no data are shown for clusters containing three or fewer water molecules in Fig. 3(a). In Fig. 3(b), I show the EBE values (VDEs and ADEs) as functions of the number of water molecules for the SO42−(H2O)n clusters. Clearly, the data trend suggests that for n = 0, 1, and 2, the EBEs fall below zero; this suggests that such SO42−(H2O)n clusters are not electronically stable. This is why Alex Boldyrev and I used44 the title “Isolated SO42− and PO43− Anions Do Not Exist” when we examined the electronic stability of the sulfate dianion and phosphate trianion. It turns out that SO42− and PO43− do exist but have such short lifetimes as to be difficult if not impossible to detect experimentally. Shortly, I will explain why they have short lifetimes, but first I want to illustrate that the evolution from stable to metastable MAs does not just occur for multiply charged anions.
In Fig. 4 I show the VDEs for several singly charged molecular cluster anions45 as functions of the number of molecules in the cluster. For some of the species, the clusters exist having more than a single geometrical shape; in such cases, the different geometries are distinguished using I, II, and III as labels. The VDEs are plotted with the horizontal axis being n−1/3, which would be proportional to the radius of a spherical cluster containing n molecules.
 | ||
| Fig. 4 Electron binding energies of several families of Mn− cluster anions. Reprinted from Fig. 4 in ref. 45 with permission of the American Chemical Society (2012). The notation I, II, III labels clusters of different geometric structure. | ||
The first thing to note is that the VDEs for a given molecule and a given geometry when extrapolated to n → ∞ give estimates of the bulk VDEs that correlate reasonably with the expected solvation strength of the corresponding solvated electron in that solvent (e.g., electrons in water, THF, and formamide have large VDEs while in benzene and toluene smaller VDEs arise). The other thing to notice is that many VDE plots appear to approach zero for small cluster sizes, but this is not true in all cases. Recall that for the I−(N2O)n and I−(CO2)n clusters, the EBE (related to VDE) remained positive for all n whereas for SO42−(H2O)n the EBE fell below zero for n < 3. This difference occurs because the bare I− is electronically stable whereas bare SO42− is not. Likewise, H2O−, NH3−, benzene−, and toluene− are not electronically stable; in fact, all of these molecules need several “partners” to form a cluster large enough to bind an electron.
Another thing to notice is that some of the clusters appear to have positive VDEs even when the data is extrapolated to small n much like was the case for I−. Such is the case for H3C–CN clusters. It is thought that these clusters involve not intact H3C–CN molecules but at least one dimer such as shown in Fig. 5.
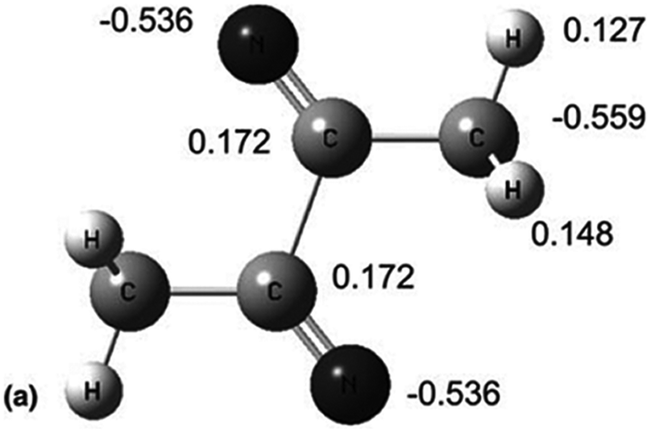 | ||
| Fig. 5 Presumed structure and Mulliken partial charges of the (H3C–CN)2− anion. Reprinted from Fig. 17 in ref. 45 with permission of the American Chemical Society (2012). | ||
This covalently linked dimer actually has a positive VDE and its anion is assumed to form the starting point for subsequently larger (H3C–CN)n− clusters whose VDEs are plotted in Fig. 4. An analogous dimerization is thought to also occur for formamide, again yielding VDE data that do not approach zero at very small cluster sizes. In this sense, these data are similar to that of solvated I−.
This kind of study has been pursued in great depth for the (H2O)n− clusters which can also be viewed as involving solvation of an electron. In Fig. 6 I show (H2O)n− data46 analogous to that displayed in Fig. 4.
 | ||
| Fig. 6 Plots of vertical electron binding energies of (H2O)n− cluster anions having various isomeric structures. Reprinted from Fig. 3 in ref. 46 with permission of the American Association for the Advancement of Science (2005). | ||
In this case various experimental ion-generation source conditions assisted by electronic structure calculations to identify geometries allowed data to be collected and interpreted for three geometrical isomer classes of (H2O)n− clusters and these are labeled I, II, and III in Fig. 6. The vertical binding energy (VBE) data for isomer I extrapolate at large n to a value close to that of the fully-solvated electron in water. The other two isomers appear to have geometries that do not evolve into that representative of bulk-solvated electron. For all three isomers, the VBE values approach zero at small n.
2.3. Some MAs that are not stable without substantial solvent stabilization still exist
The good news is that sometimes these MAs don’t simply “disappear” when there is insufficient solvation to render them electronically stable; some of them become metastable meaning they exist with finite lifetimes. The origins of metastability are different for multiply charged MAs like SO42− and singly charged MAs such as benzene− and toluene−.For the multiply charged MAs it is the Coulomb repulsions among the two or more charged sites that give rise to the metastability. I illustrate this in Fig. 7(a) for the case of a doubly deptrotonated fluorescein (Fl) molecule. Consider the effective potential an electron feels as it is brought from being infinitely distant from a singly charged Fl− anion to the site it occupies in Fl2−. I offer a qualitative depiction of such a potential in Fig. 7(b). The energy of the F− plus electron system starts at r = ∞ at D0, the energy of the doublet F− anion. As r decreases the electron experiences the long-range Coulomb repulsion provided by the remaining charged site of F−. Of course this repulsive potential is highly anisotropic as it depends on the direction with which the second electron is approaching; in Fig. 7(b) I just illustrate its overall repulsive character ignoring the angular dependence. As the approaching electron comes closer, it eventually experiences the attractive valence-range potential of the oxygen–atom site it eventually occupies in the singlet F2− dianion whose energy is denoted S0. The competition between the Coulomb repulsion and the valence-range attraction gives rise to the barrier-and-well shape for the overall potential. The barrier along this potential is called the repulsive Coulomb barrier (RCB).47 When two negatively charged sites are highly localized in a dianion, the height of the RCB can be estimated (in eV) as 14.4 (eV)/R(Å) where R is the distance between the two charged sites in Å.
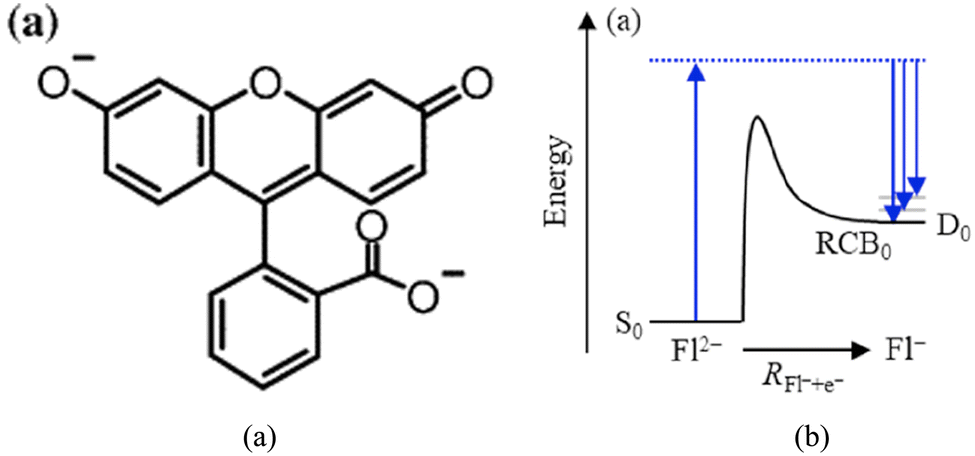 | ||
| Fig. 7 Structure of doubly deptrotonated fluorescein Fl2− (a) and depiction of repulsive Coulomb barrier connecting Fl2− to singly deprotonated Fl1− (b). Reprinted from Fig. 7 of ref. 2 (a) and Fig. 8 of ref. 2 (b) with permission of the American Chemical Society (2023). | ||
In the F2− case, it turns out that the distance between the two charged oxygen sites is large enough to allow the intrinsic valence-range attraction potential to overcome it and thus produce an electronically stable F2− dianion as suggested in Fig. 7(b). However, for SO42− the situation is qualitatively different.
When applied to the SO42− case discussed earlier, the RCB can be estimated in terms of the oxygen-to-oxygen distance R = 2.45 Å; this turns out to be 14.4/2.45 = 5.88 eV. The strength of the valence-range attraction can be estimated by calculating the EBE of the bisulfate anion HO–SO3−, which is 5.13 eV. This suggests that the SO42− dianion's energy lies 0.75 eV above that of SO4−. Thus, it would be more appropriate to describe the electron-anion interaction potential as illustrated in Fig. 8, which shows the energy of the dianion higher than that of the singly charged anion.
 | ||
| Fig. 8 Depiction of the repulsive Coulomb barrier appropriate for SO42−. | ||
In this case, one of the two excess electrons of SO42− can undergo autodetachment by tunneling through the RCB. In 2000 we estimated48 the tunneling rate for SO42− to be of the order of 108 s−1. Later, using stabilization methods49 rather than estimating the height of the RCB as above, we predicted SO42− to be unstable by 1.1 eV and to have a tunneling rate of ca. 1010 s−1. In either case, this shows that sulfate is metastable and does not live for very long before undergoing electron loss.
The bottom line is that SO42− is metastable and short-lived. This is also the case for PO43− and for CO32−, and is the fate of many small multiply charged MAs because their charged sites are too close to one another for their valence attractions to overcome their internal Coulomb repulsions. Of course there are multiply charged MAs that are electronically stable; Fl2− discussed above offers one example. They tend to have their charged sites far from one another and/or to have their charges distributed over multiple sites as in TeF82− where the two charges are shared among eight equivalent sites which are also far from one another because of the large size of the central Te atom.
The internal Coulomb repulsion story just described does not explain what happens in cases like benzene− and toluene−. In these cases a barrier along the electron–molecule interaction energy profile also occurs but such barriers arise from the angular momentum L carried by the excess electron. Within the Schrödinger equation's kinetic energy operator, the term  appears, and it is this factor that gives a repulsive contribution that, when combined with the valence-range attractions, generates a barrier such as I show in Fig. 9(a).
appears, and it is this factor that gives a repulsive contribution that, when combined with the valence-range attractions, generates a barrier such as I show in Fig. 9(a).
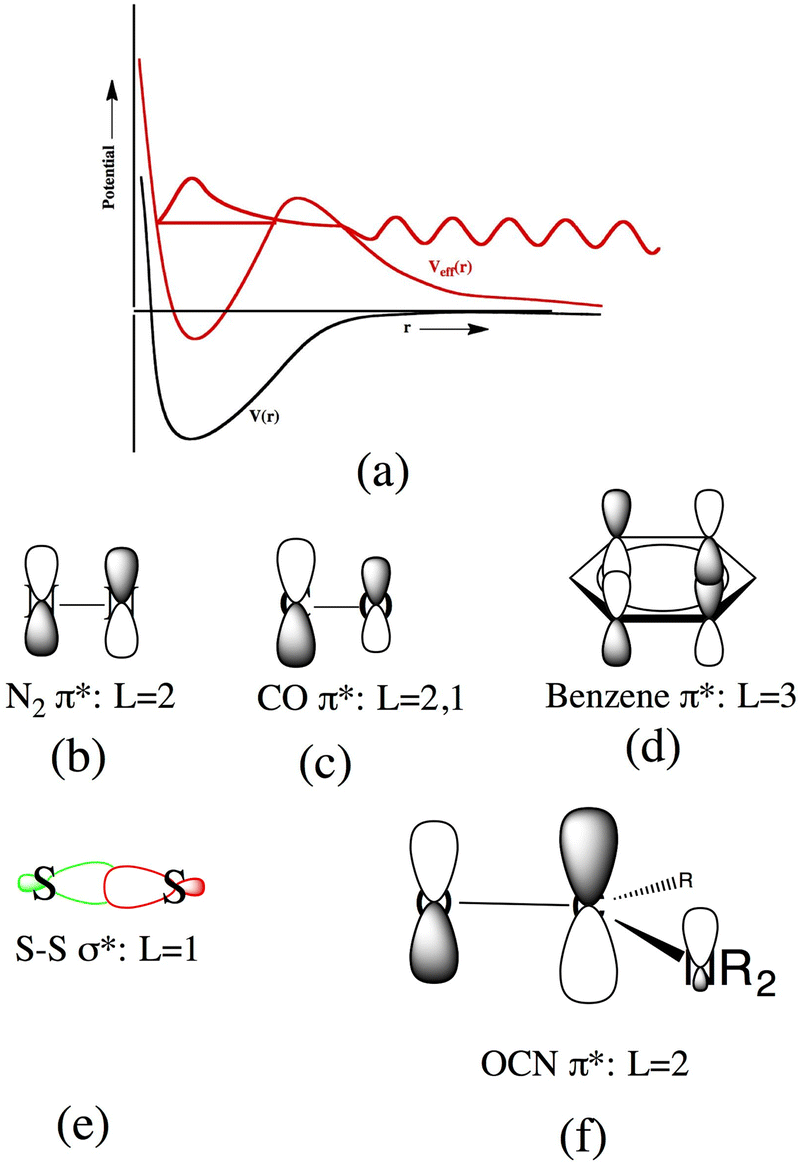 | ||
| Fig. 9 Qualitative depiction of valence-range potential in the absence of centrifugal effects (black) and including centrifugal potential (red). In (b)–(f), approximate L values of several antibonding orbitals are illustrated. | ||
A brief diversion into the role of electronic angular momentum is now useful. When studying the metastable electronic states of atomic anions such as Mg− (2P), it is clear that the “extra” electron occupies an orbital (3p in this case) of p-symmetry. The orbital in this case can be written as a product of a radial function R(r) and an angular function Yl,m(θ,ϕ) with the latter being of pure L = 1 character. The Schrödinger equation governing this orbital can then be reduced to one governing the radial function in which the potential V(r) generated by the electron's interaction with the neutral Mg atom is augmented by a so-called centrifugal potential ħ2L(L + 1)/2mer2 (L = 1 in this case) to produce an effective potential Veff(r). This centrifugal potential has a magnitude given by 3.81L(L + 1)/r2 in eV where r is given in Å. I illustrate V(r) and Veff(r) in Fig. 9(a). The attractive V(r) potential spans a region over which the orbitals of the neutral Mg atom are bound. The Veff(r) potential traps part of the metastable state's wave function behind its barrier, which arises due to its repulsive centrifugal term. The latter wave function's tunneling through this barrier is what determines the lifetime of the metastable state as suggested in Fig. 9(a).
For MAs, the separation into radial and angular orbital contributions and potentials is not as straightforward since these species do not possess spherical symmetry. The orbital holding the excess electron could, in principle, be written as a linear combination of radial functions Rn,l,m(r) multiplied by Yl,m(θ,ϕ) angular functions all located at a common origin (e.g., at the center of the aromatic ring in benzene or at the midpoint of the N–N bond in N2). However, in most quantum chemistry studies of metastable molecular anions, this orbital is expressed as a linear combination of the atom-centered basis functions, which can be of s, p, d, etc. symmetry. The metastable state's active orbital is delocalized over two or more atomic centers in many cases and has angular nodes arising from its bonding or antibonding character. In Fig. 9(b)–(f) I illustrate qualitatively such nodal properties for the orbitals holding the excess electron in N2−, CO−, benzene−, a disulfide-bond anion, and an amide OCN-bond anion.
In these MS cases, the metastable state's orbital spans two or more atomic centers as well as the centrifugal barrier region and beyond. For this reason, it is convention to focus on the angular nodal characteristics of the active orbital not at any one atomic site but when viewed over the wider region where the orbital has its valence and tunneling amplitudes. For example, in N2− the orbital's important angular nodes occur at the plane perpendicular to the bond axis at the bond midpoint and the plane containing the bond; it is because there are two nodal planes that we speak of this orbital having L = 2 (i.e., d orbital) character. For CO−, the active orbital is quite similar to that in N2− but it does not “quite” have a nodal plane perpendicular to the C–O bond axis, although it does have a nodal plane containing the bond. Because of this orbital's antibonding character, it does undergo a sign change between its values at the C and O centers; it is for this reason and its similarity to the N2− orbital that we still label this state as L = 2 and L = 1 even though its L = 2 character is only approximate.
The active orbital in benzene has three nodal planes (the plane of the six C atoms, and two perpendicular to that plane), so we label this orbital as L = 3. For the disulfide orbital, its antibonding nature produces a sign change (regardless of what substituents are bound to these atoms), so this orbital has dominant L = 1 character. In the amide antibonding orbital case in Fig. 9(f), the situation even more ambiguous; its dominant components reside on the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C portion and have one plane perpendicular to the O–C bond axis as well as a sign-change between the O and C atoms. So, we label this with L = 2.
C portion and have one plane perpendicular to the O–C bond axis as well as a sign-change between the O and C atoms. So, we label this with L = 2.
Even though the use of angular momentum L-values is not as rigorous and clear for MAs as it is for atomic anions, the fact remains that the angular derivative terms in the Schrödinger equation's kinetic energy operator produce undulations in the effective potentials that govern the radial motion of the excess electron. These undulations generate the barriers that tend to trap the excess electron and render the state metastable. I hope this explains how we make use of angular momentum nomenclature to label the metastable states arising in MAs.
The metastable MAs arising from centrifugal barriers are called shape-resonance species. They often decay by tunneling at rates that exceed even those mentioned above for SO42−; typical rates are in the 1013 to 1015 s−1 range. The fact that the repulsive Coulomb potential operative for multiply charged MAs decays as r−1 tends to make RCBs very wide, which generates low tunneling rates and thus longer lifetimes than for shape resonance whose long-range potential varies as r−2 and thus does not produce as wide a barrier through which tunneling must occur.
So, both for multiply charged MAs and MAs in which the active electron occupies an orbital of non-zero angular momentum, metastable species should be anticipated. I hope to have shown that
i. Even MAs that are electronically stable as isolated species have EBEs that vary substantially from one environment to another; such variation can alter the MA's chemical behavior;
ii. Many multiply charged MAs that we chemists think we know are actually not stable as isolated species but can exist as short-lived metastable species;
iii. Such metastable MAs can become electronically stable when solvated, and their subsequent EBEs will depend significantly on the degree and strength of their solvation;
iv. Metastability can arise from internal Coulomb repulsions among negative sites in multiply charged MAs or from centrifugal barriers existing in shape resonances involving MAs having their excess electron in an orbital of non-zero angular momentum.
3. Examples of when surroundings play key roles in MA behavior
There are two species that I will primarily be discussing when I talk about a MA's surroundings: solvent molecules and counter cations. Because of the ubiquitous importance of water as a solvent, I will focus on aqueous solutions in explaining the concepts I want to address. Moreover, I will discuss several situations that involve the behavior of ions at or near the liquid–vapor interface because the degree of solvation a MA experiences decreases as one approaches this interface from the bulk solution. This should be of special interest to chemists interested in atmospheric aerosols, clouds, and related earth-science and space-science topics. Many research groups are currently active in the area of aerosols and liquid–vapor interfaces including, for example, (i) a large number of research groups at Univ. of Calif. San Diego operating under the Center for Aerosol Impacts on Chemistry of the Environment50 (CAICE) umbrella; (ii) a team including Dick Zare51 (Stanford), Graham Cooks52 (Purdue), Mark Johnson53 (Yale) Teresa Head-Gordon54 (Berleley), Wei Min55 (Columbia) and Jahan Dawlaty56 (USC); (iii) Veronica Vaida57 (Colorado), (iv) Joseph Francisco58 (Penn), and (v) Gill Nathanson59 (Wisconsin). These workers are interested in many aspects of the behavior of the water and various ions and other solutes, and study both charged and neutral aerosol droplets. In this Perspective, I don’t intend to and could not overview such a wide range of topics undertaken by these groups. Instead I will use a small subset of their findings to illustrate how MA behave differently in different surroundings, as this is the focus of this Perspective. Doing so, I hope will convince the reader that this field of study is interesting, challenging, and important. I think it will become even more widely studied in years to come.3.1. How anions and cations are distributed spatially at solution–vapor interfaces
The first thing I want to show relates to how anions and cations are spatially distributed in aqueous solutions at and near vapor–liquid interfaces. I choose to focus on water solutions because they are so prevalent and important in chemistry and atmospheric/earth science. In Fig. 10 I show results from a molecular dynamics simulation60 of 1.2 M aqueous solutions of HCl, HBr, NaOH, and NaCl with the spatial distribution of the cations and anions involved plotted as functions of distance z from the vapor/liquid interface into the bulk. | ||
| Fig. 10 Snapshot of simulations of 1.2 M solutions (left); ions’ spatial distributions near air–water interface (near z = 15 Å) into the bulk (z = 0 and below) with OH− oxygen shown as pink; Cl− as yellow, Br− as orange. The Na+ and H+ counter cations are colored green and red, respectively. Adapted from Fig. 1 in ref. 60 with permission of the American Chemical Society (2005). | ||
I ask you to notice several features of these distribution profiles:
i. The concentration of anions and cations are not at all constant as one moves from the interface into the bulk solution; there exist oscillations (e.g., due to solvent shells surrounding the ions) in the profiles that persist over at least 15 Å;
ii. In some cases (e.g., HCl, HBr, and NaCl) the anion concentration near the interface is enhanced relative to the bulk concentration; however, in others (e.g., NaOH), the anion concentration is lower than in the bulk; so OH− appears to behave differently from halide anions;
iii. In some cases (e.g., HCl and HBr) when the anion concentration near the interface is enhanced, the H3O+ cation concentration near the interface is also enhanced but not necessarily to the same degree and not necessarily at the same distance from the interface; in other cases (e.g., NaOH and NaCl) the Na+ cation concentration appears to be pushed away from the interface;
iv. In HCl although the H3O+ cation and Cl− anion profiles appear to favor the interface and at nearly identical distances from the edge of the interface, this does not mean that they exist as near-contact ion pairs; the snapshot photo on the left shows that the ions reside in different locations in directions perpendicular to the z axis.
I don’t mean to suggest that the findings of these simulations provide a full or final description of how anions and cations tend to distribute in aqueous solutions near the vapor–liquid interface, as there are ongoing studies of this issue that might enhance or alter our understanding. However, I do think they suggest that one should be aware of the high probability that the concentration of ions near such interfaces can differ substantially from the bulk concentrations.
I introduced these spatial distribution data because one should keep in mind the observation made earlier that the electronic stability of MAs can depend significantly on the degree to which the MA is solvated. Solvation within the bulk is substantially different from that near the interface. So remember (i) that MAs might/likely have different concentrations near the surface than in the bulk and (ii) that those near the interface can have chemical reactivity quite unlike that of the fully solvated species.
Before progressing to offer some illustrations of (perhaps) unexpected behavior by MAs, I think it useful to discuss the situation of the OH− and H3O+ ions in pure H2O at the liquid–vapor interface because these two ions will be present in all aqueous aerosols. This is a very complicated situation that is still very much under active study, but I think it safe to discuss what at present seems to be going on when it comes to the equilibrium spatial distribution of these ions. There are at least three issues that arise that are relevant to the focus of this discussion:
i. Is the degree to which H2O dissociates into H3O+ and OH− near an interface much different, if at all, from that of bulk water?
ii. Do either H3O+ or OH− (or both) favor being located near the liquid–vapor interface? The data in Figure might suggest that OH− is pushed away from the interface, but perhaps this is only when Na+ is the cation; this might not be the case when H3O+ is the cation.
iii. Do the OH− ions nearest the interface (even if they are pushed somewhat away from the interface) have different chemical behavior than those in the bulk? We already know the answer to this question is “yes”.
In 2015 the Voth group61 examined the spatial distributions of H3O+ and OH− ions within horizontal simulation slabs (x, and y being coordinates in the horizontal direction and z being a coordinate connecting the vapor and liquid) representing an air–water interface making use of reactive multistate empirical valence bond potentials for the HO− and H3O+ ions they had developed and tested. In Fig. 11 I show their potentials of mean force (PMF) experienced by these two ions as functions of the distance z from (positive z) and into (negative z) the air–water interface. These potentials determine where the ions are most likely to be found.
 | ||
| Fig. 11 Potentials of mean force associated with hydroxide (blue) and hydronium (red and black) ions. Reprinted from Fig. 1 of ref. 61 with permission of the American Chemical Society (2015). | ||
There are two PMFs shown for H3O+ because the authors tested how results differed when using two of their most recent ion–water interaction potentials.
Although the results obtained for the two potentials differ substantially, they seem to suggest that H3O+ experiences a PMF that is more attractive near the interface than in the bulk and that OH− experiences a PMF that is repulsive near the interface. The latter result is in line with what was reported earlier in Fig. 10 for NaOH where the OH− ions appear to be pushed away from the interface.
In summary, this study appears to offer evidence that air–water interfaces likely have enhanced (relative to bulk) H3O+ and diminished OH− concentrations. Note that this result does not mean that the dissociation constant for H2O producing H+ and OH− is different near the interface; it just means that the OH− ions tend to move inward away from the interface while the H3O+ ions behave in an opposite fashion. Importantly for future discussion, notice that although the concentration of OH− ions immediately at the interface might be less than in the bulk, there remains a bulk-level OH− concentration within the ca. 15 Å distance that was shown in Fig. 10 to characterize the width or thickness of the interface region.
In anticipation of results on air–water interfaces not in slab geometries as studied by the Voth team but in aerosol microdroplets (e.g., presumed nearly spherical droplets of ca. 20 μm diameter), I think it useful to mention work62 aimed at measuring the pH within such microdroplets. In these experiments, droplets containing gold nanoparticles to which a pH-indicator molecule is attached were employed. Using surface-enhanced Raman spectra (i.e., the enhancement arising from the gold nanoparticles) of various vibrational modes of the indicator molecule, and studying these spectra at various locations within the bulk and in the interface region of the microdroplet, these workers were able to determine the pH at such a range of locations. They interpreted their findings as showing that the centers of the microdroplets had pH values near 11 while the interface regions had pH values ca. 4 units smaller, again suggesting that OH− appears less likely than H3O+ to concentrate near the air–water interface. I should note that the bulk solution used in these studies was phosphate buffered which is why the bulk pH was 11 rather than 7 as it would be for pure H2O.
Another thing to keep in mind when analyzing data on (presumed approximately spherical) microdroplets relates to the volume that can reasonably be assigned to the interface region. We saw earlier that the spatial distributions of cations and anions near interfaces displayed considerable variation over a range of ca. 15 Å. Using this as an estimate of the width W of the interfacial layer, one can estimate the volume fraction F of this layer for a droplet of radius R as  which is valid for the droplets discussed here. For microdroplets of diameter 20 μm, R = 10 μm = 100
which is valid for the droplets discussed here. For microdroplets of diameter 20 μm, R = 10 μm = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å; assuming W = 15 Å, this gives a value for F of 0.00045. The point I wish to make is that for microdroplets in the size range discussed here, the interfacial layer is expected to contain only a small fraction of the droplet's total volume, so any chemical reactivity that can be attributed largely to the interface and not to the bulk must be due to something qualitatively “special” about the reactants’ characteristics within the interface.
000 Å; assuming W = 15 Å, this gives a value for F of 0.00045. The point I wish to make is that for microdroplets in the size range discussed here, the interfacial layer is expected to contain only a small fraction of the droplet's total volume, so any chemical reactivity that can be attributed largely to the interface and not to the bulk must be due to something qualitatively “special” about the reactants’ characteristics within the interface.
3.2. An example of “special” chemical reactivity at an aqueous interface
I am going to discuss one study that appears to demonstrate exceptional chemical reactivity involving MAs within a water/vapor interface of microdroplets, but I will also point out that there are several other such studies. I choose to focus on what one might assume to be the most simple case: a pure water microdroplet. Certainly, if the situation is as complex and not-fully-understood in this case as it appears, it must be even more so in droplets also containing other substances. I want to emphasize that the understanding of the molecular-level mechanisms giving rise to the unusual reactivity I will discuss likely has not been finalized by the scientific community, although I find what has been postulated to be reasonable even if not yet fully accepted. I will offer some of my own thoughts about possible alternatives, but doing so does not alter the fact that there does indeed appear to be enhanced chemical reactivity of species within the liquid–vapor interface region.In 201963 and again in 2022,64 Dick Zare's group reported that H2O2 was formed from microdroplets of water having diameters in the 1–50 μm range (i.e., R = 5000 to 250000 Å) with higher local concentrations of H2O2 being generated in smaller droplets than in larger as shown in Fig. 12, clearly suggesting that the reaction generating H2O2 occurs near the surface of the droplet.
 | ||
| Fig. 12 Fluorescense intensity measures of H2O2 concentrations for various microdroplet sizes. Reprinted from Fig. 2 of ref. 63 (2019). | ||
Those workers did their best to exclude the possibility that the flow gas used to generate the microdroplets or the capillary through which the water and flow gas were passed could be the source of reactants capable of producing H2O2 and concluded that it was likely OH− ions residing near the surface of the droplets that were operative and suggested that OH radicals produced from OH− were reacting to generate the H2O2.
Another aspect of their study involved addressing why OH− MAs near the surface were more efficient than OH− anions in the interior of the droplet. Two possibilities come to mind: the concentration of OH− near the surface could be much higher than in the bulk or the OH− ions near the surface might be more susceptible to losing an electron to generate OH radicals that then combine to form the H2O2. The possibility that the concentration of OH− ions near the surface is much higher than in the bulk can likely be ruled out based on what we discussed earlier (OH− appears to be pushed away). So, the Zare group proposed that OH− MAs near the surface are more susceptible than such anions in the bulk to losing an electron to generate OH radicals. In my opinion, it is reasonable to make such a proposal as we know that the EBE of OH− is higher when this MA is more strongly solvated, and near the surface it should be less solvated and thus have a lower EBE.
In 2019, the Zare group also reported65 that, in microdroplets of diameter 1–50 μm formed from a 0.1 μM aqueous solution of various organic molecules, reduction of the organic species was observed even though reduction did not occur in bulk solutions of the same materials. Moreover, the extent of reduction appeared to be highest in the smallest droplets, again suggesting that the reaction takes place at or near the interface. In that paper and in ref. 64 the authors suggested that strong local electric fields at or near the interface, as predicted by Teresa Head-Gordon66 and by Chris Mundy,67 might facilitate detachment of an electron from OH− to generate an OH radical and a free electron. Thus the mechanism suggested by Zare involves OH− MAs existing near the interface (n.b., their concentration might be lower than in the bulk as discussed earlier but there remains a sufficient concentration) having their excess electron detached by strong local electric fields to generate OH radicals and electrons that lead to reduction. In this proposed mechanism it is not an increase in OH− concentration near the interface that is giving rise to unusual reactivity, it is the unusual reactivity (i.e., lowered EBE) of the OH− ions that is the source.
However, I note that in ref. 64 where the authors studied the formation of H2O2 the authors state “Where the electron released from OH− goes remains an unsettled question at present”. I would like to suggest two possibilities that offer slightly different perspectives on what causes the electron to be ejected from the OH− MA and where the electron goes.
First, it is known that it takes about 1.8 eV to remove an electron from an isolated OH−, and that in bulk water, the EBE increases to ca. 3.5 eV (see Fig. 6). In 2016 John Herbert examined68 binding of an excess electron at the water vapor–liquid interface and within the bulk liquid focusing primarily on how the EBEs for electrons bound near the surface compare to the EBEs of electrons more fully solvated and how an electron initially bound to the surface dynamically evolves into full solvation. In Fig. 13 I show a depiction that Herbert offers to illustrate how such an electron can evolve into the bulk.
 | ||
| Fig. 13 Dynamic evolution of electron binding energies (VEBE is the same as my VDE) and orbital occupied by the excess electron. Reprinted from Fig. 1 of ref. 68 with permission of the Americal Chemical Society (2016). | ||
For the purposes of this Perspective, the two things I want to emphasize from Fig. 13 are that (i) even in the absence of any solvent relaxation (i.e., at t = 0), surface sites exist to which the electron can bind with an EBE of 0.42 eV, but this would not be sufficient to overcome the ca. 1.8 eV EBE of an unsolvated (or very weakly solvated OH−) but (ii) it takes only 0.1 ps for the EBE at this site to grow to 2.07 eV, which exceeds the 1.8 eV EBE of isolated OH−. Thus one is tempted to suggest that surface sites similar to those associated with the t = 0.10 ps situation might exist and be active in detaching the electron from OH−, thus answering the question “where does the electron go?”
However, there is one weakness in following this line of reasoning. An electron residing in an OH− anion near the interface might have a good possibility of residing near a surface site similar to that shown in Fig. 13(a). However, for a surface site similar to that shown in Fig. 13(b) to exist, the interfacial solvent molecules would have to have undergone a substantial spontaneous reorganization (i.e., a large fluctuation) prior to the electron attachment. The reorganization associated with Fig. 13(b) was caused by the presence of the excess electron, but in the case under discussion, the electron still resides on a nearby OH− ion. In other words, in Fig. 13(b) the reorganization did not occur as a spontaneous fluctuation.
But, perhaps there are other sites on the surface that, without any further reorganization, can bind an electron by more than 1.8 eV. We know from studies of electrons bound to small to medium size water clusters that surface-bound electrons can have EBEs in this range especially near surface water molecules whose two O–H bonds are directed outward into the vapor region (this geometry is often labeled AA). See the plot of VDEs for isomer I of H2O in Fig. 6 that shows values exceeding 1.8 eV for clusters containing ca. 50 molecules. Therefore it is reasonable to suggest that an OH− MA at or near the microdroplet's surface could transfer its electron to a nearby site having this H2O AA geometry. In such a mechanism it would be the availability of H2O sites of sufficient EBE strength on the microdroplet's surface that induce the electron detachment from near-surface OH− MAs. In my opinion, this mechanism is quite similar to the strong local field picture suggested by Zare but at least it also predicts where the detached electron goes- to a surface site of sufficient EBE.
A second possibility that I believe is worth considering involves Rydberg-like states in which an electron is attached to an H3O+ cation. We know/expect that there are ample H3O+ ions near the interface, and it is known that such cations can bind an electron (by ca. 4.5 eV for the lowest-energy Rydberg state of the bare H3O+). Rydberg orbitals are quite diffuse with substantial amplitudes ranging 5–10 Å from the Oxygen nucleus. An electron transfer  could occur at an inter-ion separation at which the ionic and neutral energy surfaces intersect. Assuming H3O to lie 4.5 eV below H3O+ and OH− to lie 1.8 eV below OH, this distance would be given by14.4/R = 4.5–1.8 which yields R = 5.3 Å. There are also higher-energy Rydberg states of H3O that could have sufficient EBEs to overcome the 1.8 eV EBE of OH−, and they could also be involved.
could occur at an inter-ion separation at which the ionic and neutral energy surfaces intersect. Assuming H3O to lie 4.5 eV below H3O+ and OH− to lie 1.8 eV below OH, this distance would be given by14.4/R = 4.5–1.8 which yields R = 5.3 Å. There are also higher-energy Rydberg states of H3O that could have sufficient EBEs to overcome the 1.8 eV EBE of OH−, and they could also be involved.
Thus for R ≈ 5.3 Å, the electron could depart from the OH−, thus generating an OH radical and form a neutral H3O. This Rydberg radical is known to be quite unstable and to dissociate into H2O and H. I note that the Rydberg orbital of H3O extends to and beyond 5.3 Å, so this proposition may be reasonable. This mechanism suggests that neutral H atoms would be generated from dissociation of the H3O Rydberg species, and it seems this aspect could be amenable to experimental testing.
Notice that I did not include any solvent dielectric screening between the OH− and H3O+ ions in the above estimate. This is because these two ions are assumed to exist at or near the liquid–vapor interface where their EBEs and electrostatic interactions will be more like those of isolated ions than of strongly solvated ions. I think there is at least one weakness in this proposed mechanism. It requires that an H3O+ion reside in close enough proximity to the OH− ion to effect the electron transfer. At least in bulk H2O, the average distance between these ions is much too large, but perhaps near the interface, things might be different.
I hope this one example aimed at understanding what species are present and active at aqueous vapor–liquid interfaces and how those species’ chemical reactivity may be different from what is expected shows how excitingly complicated this field of study is.
3.3. Two examples of how “nearby” counter cations have substantial effects on MAs
In 2007 Evan Williams’ group studied69 what happens when a low-energy free electron is attached to a positively charged Ca(H2O)n2+ cluster. They found two competing pathwaysi. the boiling off of k solvent molecules induced by the exothermic electron capture event:  ; and
; and
ii. the creation of a solvated CaOH1+core in combination with boiling off of fewer solvent molecules:

The degree of competition between these two routes is illustrated in Fig. 14 for clusters initially containing 4 to 41 solvent molecules.
 | ||
| Fig. 14 Fraction of ions undergoing reactions (i) (triangles) and (ii) (squares). Reprinted from Fig. 4 of ref. 69 with permssion of the American Chemical Society (2007). | ||
It seems clear that for cluster sizes having more than 30 water molecules, the first pathway (exothermic electron capture boiling off solvent molecules) is dominant whereas for clusters with fewer than 20 molecules the second path dominates.
In 2008, we studied this kind of competition for clusters containing Mg2+ cation cores70 instead of Ca2+ cores and put forth an explanation for the sudden shift from one pathway to the other illustrated in Fig. 14. The key idea underlying our suggestion is illustrated in Fig. 15 where we show how the energy of an H2O molecule varies as one stretches one of its bonds (i) in the absence of a Mg2+ cation and having no excess electron (filled circles), (ii) with an excess electron attached to the water molecule's O–H antibonding σ* orbital but with no Mg2+ cation present (squares), (iii) and with an excess electron attached to the water molecule's O–H antibonding σ* orbital with a Mg2+ cation located at distances approximating those of the second (triangles) and third (diamonds) solvation shells, respectively. In arriving at this interpretation, we used visualization of the orbital holding the attached electron to conclude that this electron resides in an O–H antibonding σ* orbital. However, we note that to date we are unaware of any experimental findings that show an excess electron can attach to such an orbital of an isolated H2O molecule.
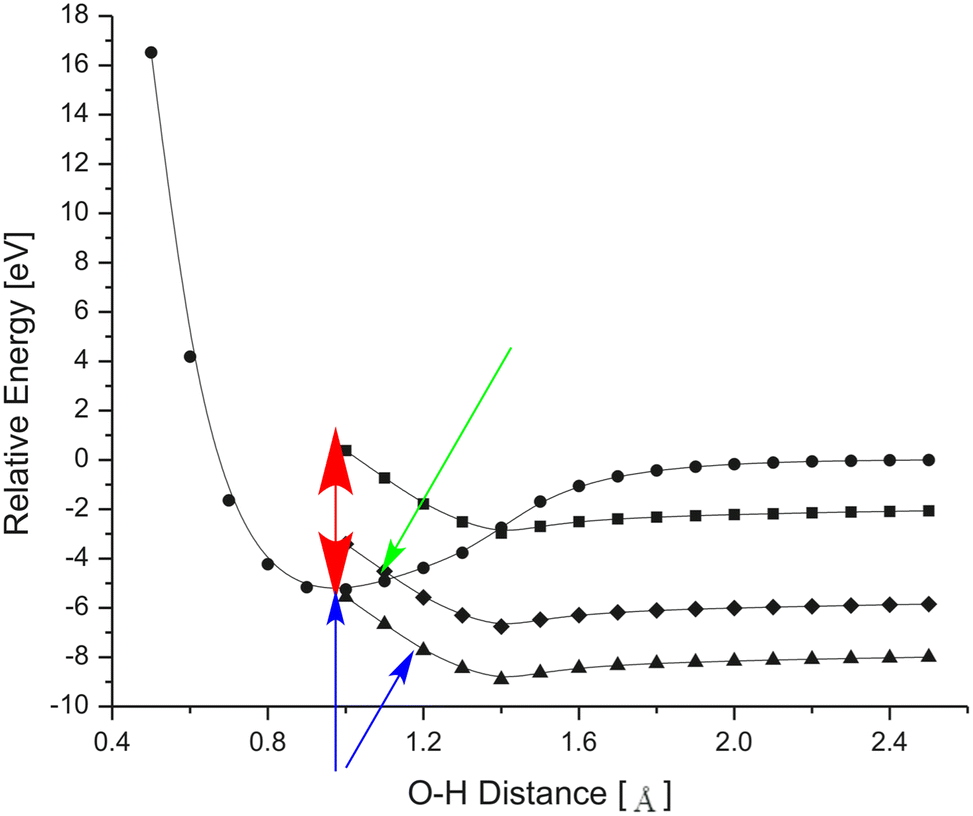 | ||
| Fig. 15 Energy of bare neutral H2O as one O–H bond is lengthened (circles), of H2O with an excess electron in an O–H σ* orbital (squares), and with an excess electron in an O–H σ* orbital and one Mg2+ ion at a second hydration distance (triangles) or third hydration distance (diamonds). Adapted from Fig. 4 in ref. 70 with permission of Elsevier (2008). | ||
The main points derived from this analysis are that
i. a bare H2O molecule at its equilibrium geometry (i.e. O–H distance near 0.8 Å) cannot attach an excess electron (i.e., its vertical electron affinity is negative as illustrated by the red arrow);
ii. with an Mg2+ cation located near the location of the second hydration shell of Mg(H2O)n2+, electron attachment to the dication into an OH O–H σ* orbital can occur exothermically as illustrated by the two blue arrows;
iii. with an Mg2+ cation located near the location of the third hydration shell of Mg(H2O)n2+, electron attachment to the dication into an OH O–H σ* orbital is slightly endothermic as illustrated by the green arrow.
Our results suggested that if a free electron can access the H2O molecules residing in the first or second solvation shell, electron attachment to an O–H antibonding σ* orbital at the equilibrium geometry can occur in an exothermic manner, and this is how the second pathway operates generating an OH− MA that attaches to the Mg2+ cation as well as an ejected H atom and using its exothermicity to eject some solvent molecules. However, if the second solvation shell is full, electron attachment to a third-shell (or higher-shell) O–H antibonding σ* orbital at the equilibrium geometry cannot occur. In this case, the electron can attach instead to a surface-localized orbital such as I show in Fig. 16, using the exothermicity of this attachment process to eject solvent molecules.
 | ||
| Fig. 16 Surface-localized orbital electron attaches to. Reprinted from Fig. 5 of ref. 70 with permission of Elsevier (2008). | ||
I am using this example to illustrate the effects that a surrounding cation can have on a MA. You might say that this system is not really an anion as its overall charge remains positive. I prefer to think of it as an anion in disguise. The MA is that formed by attachment of an electron to the σ* orbital of an H2O molecule; the electron does not attach to the Mg2+ ion. Without the presence of the Mg2+ cation, the electron attachment cannot occur exothermically; with an assist from the Coulomb potential of the cation, the attachment can be rendered exothermic. The degree of Coulomb stabilization C generated by the cation can be estimated as  where ZCation is the charge of the cation.
where ZCation is the charge of the cation.
I wanted to use this example of Coulomb stabilization as an introduction to another case in which the presence of positive charges within a molecule can qualitatively alter its chemical behavior by allowing anion sites to be rendered stable and chemically active. Now, let me show you an example of such a process that I expect you might find compelling and that also involves a MA in disguise.
In electron capture dissociation (ECD) mass spectroscopy experiments positively charged (often via protonation) parent ions are generated in the gas phase (e.g., by electrospray) and subjected to collisions with very low-energy (often thermal) free electrons. The exothermic capture of a free electron by the positively charged parent ion causes fragmentation into various product ions, but by no means is the exothermicity randomly distributed. Instead, only certain bonds in the polypeptide are cleaved. For this reason (i.e., that only a few types of bonds are cleaved), the ECD tool has proven especially useful in proteomics studies in which sequencing a polypeptide is the goal. ECD's is quite selective in generating fragmentation primarily at disulfide bonds and at N–Ca bonds along the peptide's backbone. It is thought that the latter fragmentations occur through capture of a free electron into a backbone amide OCN π* orbital as illustrated in Fig. 17.
Upon capture of the electron, a carbon-centered radical site (red dot in upper right) is generated. The existence of this radical site, in turn, reduces the barrier to cleavage of the neighboring N–Ca bond because it allows for formation of a new C–N π bond, which is energetically stabilizing. This series of events ultimately generates fragment ions that are labeled c and z.
In 200371 and 200572 my group put forth a proposal for how the ECD electron attachment might occur. We knew that vertical attachment of an electron to an amide π* orbital is ca. 2 eV endothermic (attachment to a disulfide S–S σ* orbital is 1 eV endothermic), so one had to wonder how a mechanism such as suggested in Fig. 17 could take place. The key concept in answering this involved that of Coulomb stabilization by nearby positively charged groups. In the polypeptide case, these groups are often protonated side chain amine units. So in the resultant so-called Utah-Washington mechanism73 (so named due to close collaboration with Frank Turecek74) we postulated that the total Coulomb stabilization (i.e., summed over all charged units in the system) at a given N–Ca site's amide π* orbital must exceed 2 eV for direct exothermic electron attachment to occur and induce that site's bond cleavage. Likewise the total Coulomb stabilization at any disulfide linkage must exceed 1 eV to induce cleavage at that site. This model is especially useful since it provides for predictions of which bonds will cleave and which won’t; only those close enough to positive sites to experience sufficient Coulomb stabilization will be broken.
 | ||
| Fig. 17 Description of Utah-Washington mechanism for electron attachment to amide π* orbital leading to N–Ca bond cleavage. Reprinted from Scheme 1 of ref. 73 with permission of the American Chemical Society (2014). | ||
To demonstrate the kind of study we carried out to arrive at this mechanistic story, I show in Fig. 18 a doubly protonated (at the two terminal Lys side chains) polypeptide (H–Lys–Ala20–S)22+ that we used to examine both cleavage of backbone N–Ca bonds and the central disulfide linkage.
To explain and emphasize the role of the Coulomb-stabilization model embodied in the UW mechanism, I note the following:
 | ||
| Fig. 18 Structure of doubly charged (H–Lys–Ala20–S)22+ (a); plots (b) of the energy profiles (eV vs. r in Å) for cleaving the central S–S bond in the absence of protonation at the terminal Lys sites (black) and with an electron attached to the S–S σ* orbital in the presence of the two protonated Lys sites (red); reprinted from Fig. 3 of ref. 73 with permission of the American Chemical Society (2014). | ||
i. near the equilibrium S–S bond length of ca. 2.1 Å, attachment of an electron into the S–S σ* orbital is ca. 1 eV endothermic, so its formation would lead to a metastable shape resonance state;
ii. the Coulomb stabilization exerted at the S-S site from each protonated Lys site (each of which is ca. 32 Å distant) can be estimated by 14.4 eV Å/(32 Å) = 0.45 eV, so the total Coulomb stabilization at the S–S σ* orbital site would be 0.9 eV, perhaps barely enough to render electron attachment to the S–S σ* orbital energetically feasible;
iii. but, in addition, each of the α-helices formed by the two Lys20 units has a very large dipole moment, and these moments, both with their positive ends directed toward the S–S site, combine to produce an additional stabilization of 4 eV;
iv. so, it is a combination of Coulomb stabilization and dipole stabilization from the two (Ala)20Lys+ units that lowers the energy of the electron-attached state to 4 eV below the energy of the neutral (and 5 eV below the energy of the metastable S–S σ* orbital).
Analogous studies of electron attachment the backbone amide π* orbital have also been carried out and form the basis for our proposing the mechanism shown in Fig. 17 for these cleavages.
So, much like the presence of the central Mg2+ ion in the Mg2+(H2O)n clusters allowed attachment to a σ* orbital of a surrounding H2O molecule to generate OH− and H, in ECD it is the positively charged groups in the polypeptide that allow attachment to the amide π* or disulfide σ* orbital causing subsequent bond rupture. I think this should cause one, when dealing with materials containing one or more positively charged site, to consider chemical reactivity generated by electron attachment to functional groups that might not have positive EAs in the absence of the positive groups.
3.4. An example where time scales influence what the environment is
A lot of experimental and theoretical work75 has been done studying how low-energy electrons might cause strand-break damage in DNA. In real life, the concern relates to free electrons formed when ultraviolet light detaches electrons from H2O molecules or proteins in living organisms. In such processes, the ejected electrons initially have kinetic energies (KE) in the 10–20 eV range, and they can cause damage by ionizing or electronically exciting various DNA functional units. These electrons can be cooled down by undergoing collisions with other molecules, but their danger to DNA does not disappear even when their KEs fall below ionization thresholds and bond strengths in the DNA molecule. In fact, we suggested75 that damage could occur even from electrons having KEs in the 1 to 2 eV range and this prediction was subsequently verified76 by experiments.Once a free electron reaches a KE of 1 eV, its velocity is approximately 6 × 1015 Å s−1, so in 10−14 s (the lifetime of many metastable shape resonances), the electron can traverse ca. 60 Å. I want to keep this order-of-magnitude estimate of the free electron's speed in mind as it plays an important role in what I am now going to discuss. In considering how a free electron might attach to and induce bond cleavage in DNA, several questions need to be addressed:
i. How and into what orbital does the electron attach?
ii. How does the presence of the excess electron cause a bond to break?
iii. How fast does all this happen and do the geometries of the DNA and of the surrounding molecules have time to “adjust” to the presence of the excess electron?
It is known77 that some of the nucleotides (i.e., base, sugar, phosphate units) of DNA have small (<0.17 eV) vertical EAs (some having dipole-bound ground states). Upon geometry relaxation, the resultant ground-state anions achieve modest VDEs (0.3 to 0.7 eV) after which the excess electron resides primarily in a base π* orbital. It would then seem to make sense to suggest that a free electron might attach to a nucleotide in its initial geometry (although one would have to account for how it undergoes a free-to-bound transition) after which the stable nucleotide MA could undergo geometry (and surrounding) relaxation to generate an even more electronically stable MA. The nascent MA's vibrational motions then might allow various barriers to bond ruptures to be overcome as suggested78 in Fig. 19.
 | ||
| Fig. 19 Adapted from Fig. 6 of ref. 78 with permission from Oxford Univ. Press (2010). | ||
Here three possible bond cleavages are considered: the base-sugar glycosidic bond, the sugar-phosphate C5′–O5′ bond, and the sugar-phosphate C3′–O3′ bond. As Fig. 19 shows, the latter bond cleavage is predicted to have the lowest barrier (ca. 6 kcal mol−1 as in Fig. 19(a)). In ref. 78 the authors also examine the energy profiles for these three bond cleavage options using a polarized continuum model to simulate the relaxation of the surrounding solvent (taking a dielectric constant of 78, about which I will remark later). The energy profiles in Fig. 19(a) contain no solvent effects but they do assume the electron-attached species has undergone its own geometry relaxation.
In my opinion, much of what I just outlined from ref. 78 makes sense. However, there are details that I think need to be clarified and, in some cases, modified. First, I want to discuss how the initial electron attachment might take place. If the electron is to enter a base π* orbital, it is important to realize that it will encounter a long-range centrifugal potential that will produce a barrier to attachment. For example, at a distance of 5 Å from the approximate “center” of the base, this potential would be  , for an orbital of angular momentum L. The base π* orbital has at least one nodal plane, so it is reasonable to assign a value of at least 1 to L, so KE would have to be at least 0.3 eV to surmount the barrier. An electron having KE = 0.3 eV would transit the ca. 3.5 Å distance from one base to another stacked above or below it in about 10−15 s. I don’t think this is enough time for the electron to lose energy and become bound before it moves past the base being discussed. For electrons having even higher KEs, the time available for losing energy to become bound is even shorter.
, for an orbital of angular momentum L. The base π* orbital has at least one nodal plane, so it is reasonable to assign a value of at least 1 to L, so KE would have to be at least 0.3 eV to surmount the barrier. An electron having KE = 0.3 eV would transit the ca. 3.5 Å distance from one base to another stacked above or below it in about 10−15 s. I don’t think this is enough time for the electron to lose energy and become bound before it moves past the base being discussed. For electrons having even higher KEs, the time available for losing energy to become bound is even shorter.
However, if the electron's KE happened to be in a range to allow it to tunnel through and be trapped within the barrier caused by the centrifugal potential and the valence-range attractive potential of the π* orbital, it could form a metastable shape-resonance MA. The four bases of DNA are known79 to possess shape resonance MAs at KE values ranging from <0.5 eV to ca. 5 eV as shown in Fig. 20.
 | ||
| Fig. 20 Electron transmission spectra for electrons entering base π* orbitals. Reproduced from Fig. 2 in ref. 75 with permission from the American Chemical Society (2006). | ||
Notice that these spectral peaks are quite broad largely due to the short lifetimes (e.g., a lifetime of 10−14 s corresponds to a lifetime-determined width of 0.06 eV) and Franck–Condon profiles of the shape resonances. So free electrons with KEs between zero and 5 eV would be expected to have ample opportunity to attach to DNA bases forming shape resonance MAs.
Such metastable species tend to exist (before undergoing electron loss) for of the order of 10−14 to 10−13 s. During that timeframe, full geometrical and solvent relaxation would not have time to occur, but it would be possible for the electronic structure (i.e., orbitals) of the MA and its environment to adjust/relax absorbing some of the electronic energy and thus stabilizing the MA. For this reason, I think it would be more appropriate to model the effects of surroundings on the nascent electron-attached species using a dielectric constant in the range of the high-frequency value (ca. 2) rather than 78 as was done in ref. 78. This would only qualitatively alter the story told in Fig. 20, but it would involve less differential stabilization of the anion energy surfaces relative to that of the neutral than shown there.
In any event, I believe it reasonable to assume the electron initially resides in a base π* orbital, but then what happens? As suggested in Fig. 21 and as I detailed in ref. 75, a through-bond electron transfer takes place.
 | ||
| Fig. 21 Depiction of electron attached to base π* orbital (left) and transferred to sugar-phosphate σ* orbital (right). Adapted from Fig. 8 in ref. 75 with permission from the American Chemical Society (2006). | ||
This electron transfer event requires surmounting a modest barrier, in which the electron migrates into a sugar-phosphate C–O σ* orbital. Once in that orbital, the energy landscape evolves downhill breaking the C–O bond and leaving the excess electron on the phosphate moiety. The very high EBE of the phosphate MA provides the main thermodynamic driving force that favors this reaction path. Indeed, subsequent experiments80 showed that DNA strand breaks do occur for free electrons in this KE range as shown in Fig. 22.
 | ||
| Fig. 22 Yield of single strand breaks (squares) as a function of electron kinetic energy. Reproduced from Fig. 11 in ref. 75 with permission from the American Chemical Society (2006). | ||
Moreover, additional experiments81,82 carried out to analyze which bonds were cleaved showed that substantial sugar–phosphate C–O bonds are broken.
I hope this example illustrates how it is important to consider the timescales involved in forming the MA by electron attachment as well as timescales needed to effect chemical bond changes that might then occur. In these considerations, it is important to realize that full geometrical relaxation and full solvent reorganization cannot occur in 10−15 or 10−14 s.
3.5. MAs can be susceptible to vibration- or rotation-induced electron ejection; here the internal excess energy plays the role of an “environment”
For an MA (individual molecule or cluster) that has a very low but positive EBE, it is possible for vibrational or rotational energy to be transferred to the electronic degrees of freedom to cause electron detachment. Vibrational detachment is expected when the EBE is in the range of the vibrational energies of modes that are active in geometrical regions where the orbital holding the excess electron resides. Rotational detachment is most prevalent for dipole-bound anions because they have EBEs similar in magnitude to rotational energies.In the case of vibration-induced electron ejection, the rate depends on the degree to which a vibration alters the orbital from which the electron is ejected. For example, in OH−, the vibrational motion has little influence on the oxygen-centered pπ orbital containing the electron so the detachment rate will be low. In contrast, for an olefin anion such as Cl2CCCl2−, the C–C stretching vibration substantially alters the π* orbital, so the detachment rate can be high. In 2020, I published an article83 over viewing the propensity rules and rates of such non-adiabatic electron ejection processes.
To illustrate the first of such processes, I will use the case of NH−(2Π) which, when excited from its v = 0 vibrational level to v = 1 can eject an electron84 as was observed in Carl Lineberger's group in 1985. In Fig. 23 I offer a simplified (ignoring the λ-doubling and not including rotational-level labels) description of this situation's electronic energy curves. In 1981 I explained the selection rules85 that govern the vibration-induced electron ejection and in 1986 we illustrated86 how the theory of such processes can be applied to assist in interpreting the experimental data of ref. 84.
 | ||
| Fig. 23 Neutral NH (blue) energy as a function of bond length with two lowest vibrational levels shown; NH− anion energy (red) with lowest vibrational level shown. | ||
The basic idea is as follows. The electron affinity of NH is 0.37 eV, which is less than the v = 0 → v = 1 vibrational level splitting in NH−. Tuning a laser to excite NH− from v = 0 to v = 1 (also selecting specific rotational and λ-doublet levels if desired), one can induce electron ejection, but the ejection has two mechanisms that can be operative. First, the direct electric-dipole bound-to-continuum transition (NH−v = 0 + hv → NH v = 0 + e−) can be operative. Secondly, the photon can populate the v = 1 level of NH− after which vibration-to-electronic energy transfer (a non-adiabatic process) can occur to then populate the v = 0 level of the neutral NH and eject an electron. In both cases, the electron is ejected with the same kinetic energy, but interference between the wave function's amplitudes belonging to the two pathways can give rise to so-called Fano line shapes. Although this line shape aspect was not addressed in the NH− study of ref. 84, it was demonstrated nicely in a study87 of electron detachment from (H2O)n− clusters carried out in Mark Johnson's and John Tully's labs. In that study, it was excitation of the O–H stretching vibrations of the cluster MA that induced the electron ejection and these modes’ energies exceeded the EBE of the MA. The point of these two examples is to alert the reader to be prepared to consider vibration-induced electron detachment whenever the EBE is similar in magnitude to one or more of the MA's vibrational modes’ energies.
Now, let me turn to the case of rotation-induced electron ejection. In 1987, Carl Lineberger's group studied88 the H2C–CN− ion, which has a closed-shell 1A1 ground state with the excess electron occupying a valence orbital. Those workers determined this anion state to lie ca. 12500 cm−1 below the 2B1 ground state of the neutral H2C–CN. Pertinent to the present discussion, they also studied the dipole-bound excited state of the H2C–CN− ion, which lies only 60 cm−1 below the 2B1 neutral, in particular focusing on the rate at which electrons were detached when various rotational states of the dipole-bound anion (DBA) were populated by laser excitation from the ground state. Once the degree of rotational excitation exceeded 60 cm−1, detachment could occur.
The rotational energy levels of H2C–CN− can be labeled by J, M, and K quantum numbers with K describing spinning of the H2C-unit around the anion's C2 axis. In ref. 88 it was found that the line widths associated with populating various J, K levels of the DBA were less strongly dependent on K than on J. This is because the J quantum number describes to a large extent the overall tumbling motion of the H2C–CN− molecular framework, which has a stronger impact on the dipole-bound electron than does spinning of the H2C-unit. On this basis it was suggested that rotational energy of the DBA was being converted into electronic energy to induce detachment of the electron.
David Clary came up with a very useful way89 to understand how such detachment takes place as I will now illustrate by considering what happens when the J = 34; K = 0 level of the DBA is accessed by laser excitation from the 1A1 ground state. I should note that the Lineberger-lab experiments were able to access such high J levels of the DBA because the temperature of their sample allowed high-J levels of the ground state to be populated prior to the laser excitation.
In Fig. 24, I show a series of diabatic and one adiabatic energy curves associated with the distance R between the to-be-ejected electron and the H2C–CN molecular framework.
 | ||
| Fig. 24 Diabatic and adiabatic potentials of the Clary model associated with J = 34. Reproduced from Fig. 1 of ref. 90 with permission of the AIP (1989). | ||
The energy of the J = 34; K = 0 neutral H2C–CN together with an excess electron having L = 0 angular momentum and zero KE is shown as the horizontal line at an energy of 412 cm−1 (the J = 0; K = 0 energy of the neutral is chosen to define E = 0). The energies of the J = 33 to 31; K = 0 rotational levels of neutral H2C–CN are also shown on the right side of Fig. 23; to each of them is added the centrifugal potential  for values of L ranging from L = 1 to L = 3 (to retain the total angular momentum of 34), and it is these functions of R that are depicted as the diabatic curves.
for values of L ranging from L = 1 to L = 3 (to retain the total angular momentum of 34), and it is these functions of R that are depicted as the diabatic curves.
In Fig. 25 I show a Table of the energies of many of the rotational levels of the anion and neutral to illustrate three points.
 | ||
| Fig. 25 Energies of several rotational levels of the neutral and anionic system. Reproduced from Table 1 of ref. 90 with permission of the AIP (1989). | ||
First, it is only for J ≥ 14 that the rotational energy of the DBA exceeds 60 cm−1. Second, at J = 34 (the example illustrated in Fig. 23), the energy of the anion (346 cm−1) is below the energy of the J = 32 neutral but above the J = 31 neutral, so this level of the anion can only produce neutrals having J ≤ 31, which means at least 3 units of angular momentum must be transferred in the detachment process for this anion level. Third, as one moves to lower J levels of the anion, more and more units of angular momentum must be transferred to access the first energetically accessible level of the neutral; in contrast, as one moves to higher J levels, fewer units of angular momentum are needed. These trends will be important to keep in mind for what I speculate about later.
In Clary's model such diabatic energy surfaces (i.e., the curves for J = 34 downward and L = 0 upward with the sum remaining 34) couple when the excess electron's interaction (i.e., short-range repulsion and longer-range charge-dipole term) with the molecule is included to produce a lowest-energy adiabatic curve that is also shown in Fig. 24. This adiabatic potential is then said to support the J = 34; K = 0 level of the DBA with an energy shown by the horizontal line 60 cm−1 below the J = 34 energy of the neutral. In the original Clary work, it was assumed that the electron remained on the lowest adiabatic surface, thus ultimately leading to the neutral in J = 0 with the ejected electron carrying away all of the excess energy. In a follow up to Clary's original work, I was able to show90 that by allowing for flux to populate all energetically accessible levels of the neutral (i.e., J ≤ 31), the model's predictions of the observed line widths’ dependence on J were improved.
The lifetime of the J = 34; K = 0 level of the MA is determined largely by the rate at which the electron can tunnel though the barrier on the adiabatic curve. For the J = 34; K = 0 level, the line width was observed to be 2.2 × 10−3 cm−1, which corresponds to a lifetime of 1.5 × 10−8 s; this is how long it takes for this metastable state to undergo electron loss. Higher J levels tunnel more quickly and lower J levels tunnel more slowly as explained earlier. Using 1014–1015 s−1 as an estimate of the frequency with which the DBA's excess electron strikes the barrier through which it must tunnel to escape, a lifetime of ca. 10−8 s suggests that the tunneling probability is of the order of 10−7 to 10−6.
As you can see in Fig. 24, for the J = 34 anion, the excess electron must tunnel out quite far before it crosses the energy of the J = 31; L = 3 diabatic curve and can escape to produce the neutral in J = 31 with an L = 3 electron; to generate lower J-levels, the tunneling will span even larger distances. The reason I am pointing this out is that one might think an electron so distant from a molecule is experiencing no interaction with the molecule, but that is not the case especially for these DBAs which bind the excess electron very “loosely” and in a very diffuse orbital. To emphasize this point, for L = 3 at R = 215 Å, the centrifugal potential  is equal to 0.001 eV = 1 meV = 8 cm−1, which is the KE of the electron ejected when the DBA detaches it to generate J = 31 neutral.
is equal to 0.001 eV = 1 meV = 8 cm−1, which is the KE of the electron ejected when the DBA detaches it to generate J = 31 neutral.
Alternatively, if the J = 34 DBA were to eject an electron and generate a J = 0 neutral, the ejected electron would have a KE of 346 cm−1. This electron would exit the L = 34 centrifugal barrier at R = 325 Å. This illustrates how the first open channel (to produce J = 31 neutral) has the shortest tunneling distance while processes leading to lower neutral L-values have considerably longer tunneling distances. Because the tunneling probabilities depend exponentially on distance, the ejection rate can be dominated by the channel having the smallest L-value change.
To summarize what is thought to take place for the J = 34 case according to the so-called rotationally adiabatic Clary model:
i. The anion in its J = 34 state has a rotational energy (b × J × (J + 1) = 0.341 × 34 × 35) of 406 cm−1, which means the anion is rotating at a frequency of 1.2 × 1013 s−1;
ii. Therefore, during the ca. 10−8 s it takes for the tunneling to produce electron detachment, the anion can rotate approximately 105 times, and several paths can be followed by the wave function:
a. Electron tunneling outward to ca. 215 Å to generate a J = 31 neutral and a free electron having L = 3 and KE = 8 cm−1 (during the 105 rotations of the anion, three units of angular momentum are transferred from the rotational to the electronic degrees of freedom via couplings of the J = 31, 32, 33, and 34 diabatic states);
b. During this same ca. 10−8 s, more than three units of angular momentum might be lost by the rotation and transferred to the excess electron thus allowing the population of lower-J diabatic curves which can then lead to producing lower-J neutrals and ejected electrons having higher-L and higher-KE. The probabilities of transferring more than three units of angular momentum decay as ΔL increases but is non-zero, and it is important to keep in mind that there are ca. 105 rotational periods for this to occur.
c. It is even possible for a J = 0 neutral to be formed by ejecting an electron having KE = 346 cm−1 and L = 34, albeit this outcome is less probable than for producing higher-J neutrals. Using  to relate the electron's KE and L values to the “impact parameter” B with which the ejected electron would depart gives B = 320 Å.
to relate the electron's KE and L values to the “impact parameter” B with which the ejected electron would depart gives B = 320 Å.
Allow me to now speculate about how an analogous process might be operative not in detaching electrons from DBAs but in forming DBAs that have been suggested to be present in certain inter-stellar environments acting as doorway states leading the formation of more stable MA states. I do not have the knowledge to provide a broad overview of the status of anions in such environments, but I want to offer one anion-formation speculation for your consideration. For much more detail about the field of anions in astronomical situations, I offer three sources: Ryan Fortenberry91 has written a nice article highlighting the role quantum chemistry can play in imagining new MAs that might occur; Millar et al.92 provide a very thorough overview of the experimental and theoretical efforts that have produced evidence for the existence of MAs in space; and at the University of Innsbruck,93 Roland Wester, Paul Scheier, and Franco Gianturco have produced many studies of MAs including those in space (here is one94 that I think relates closely to the subject I am discussing as I will explain later).
Given that anions have been found to occur in “outer space”, one wonders “how are such MAs formed?” When it comes to carbon-rich linear radicals such as C4H, C6H, and C3N, it has been speculated that DBAs might be involved in the formation process, and this is discussed in ref. 94. Briefly, the idea is that a free electron collides with one of these neutral radicals to initially form a DBA, which then undergoes relaxation (intramolecular vibrational or radiative) to form a valence-bound ground state anion (the three radicals mentioned are known94 to form such anions with quite substantial (>3 eV) EBEs).
The question that remains to be resolved is how are such DBAs formed from the neutral radical and a free electron. One possibility comes to my mind after thinking about the Clary model discussed above. Reflecting back on Fig. 24, one might ask whether it would be possible to form the metastable J = 34 H2C–CN− DBA by colliding a free electron (having KE = 346 cm−1 and L = 34 (i.e., with an impact parameter B = 320 Å)) with a neutral H2C–CN in a low rotational level (because the temperatures in these environs are only few °K (1 °K ≈ 0.7 cm−1)). After all, we know that J = 34 H2C–CN− can decay into low-J levels of H2C–CN allowing the ejected electron to carry away this specified excess energy and angular momentum. In ref. 94 it is suggested that attachment of a low-energy electron to form a DB state of C4H, C6H, or C3N could provide a doorway to generating the more stable ground states of the resulting anions. In that same reference, state of the art electron–molecule scattering calculations were carried out focused on the collision of such low-energy electrons with these radicals. Although those studies provided clear support for the proposal that forming the DB MAs, they did not explain how the free electron undergoes the free-to-bound transition as they were carried out within a frozen nuclear framework model. It is in this final electron-attachment step that I suggest the Clary model can be of use.
Such an electron capture process might not be likely for a species such as H2C–CN− that has an EBE of 60 cm−1 which requires major angular momentum transfer to reach even the first (J = 14) metastable level of the DBA to be accessed from a low-J level of the neutral. But for a species whose DBA has a lower EBE, it might make sense as I will now illustrate.
Linear molecules such as C4H, C6H, C3N have moments of inertia that produce rotation b-values similar to that (0.341 cm−1) of H2C–CN, so I will use b = 0.341 cm−1 in this illustrative hypothetical example. I will assume an EBE of 20 cm−1 rather than the 60 cm−1 EBE of H2C–CN− to allow smaller changes in angular momentum to be possible.
At a temperature of 5°k, the most populated level of such a rotor (given by  ) is J = 2, and this level has an energy given by b × J × (J + 1) = 0.3 × 2 × 3 = 2 cm−1. However, the J = 5 level has a Boltzmann population ((2J + 1)e−bJ(J+1)kT) 21% as high as the J = 2 level, so it is to J = 5 I will consider attaching an electron (again, to keep the angular momentum change small).
) is J = 2, and this level has an energy given by b × J × (J + 1) = 0.3 × 2 × 3 = 2 cm−1. However, the J = 5 level has a Boltzmann population ((2J + 1)e−bJ(J+1)kT) 21% as high as the J = 2 level, so it is to J = 5 I will consider attaching an electron (again, to keep the angular momentum change small).
If the DBA had an EBE of 20 cm−1 (2.5 meV which is in the low range of DBA EBE values), to be unstable with respect to electron loss induced by rotation, 0.341 J(J + 1) cm−1 would have to exceed 20 cm−1, which would require J to be 8 or higher. I show in Fig. 26 a Clary type potential plot for the case of J = 9 with EBE = 20 cm−1, choosing E = 0 to define the neutral molecule in its J = 0 level.
 | ||
| Fig. 26 Clary-model diabatic and adiabatic potentials for J = 9 of the hypothetical linear anion. | ||
I want to consider whether a free electron could strike the neutral molecule in J = 5 to generate a metastable J = 9 DBA. For the case at hand, an electron having KE = 11–10 = 1 cm−1 could strike this neutral in J = 5 and, if it had an impact parameter to generate a collisional angular momentum of L = 4, it could tunnel inward to produce a DBA in J = 9. Alternatively an electron having KE = 11–2 = 9 cm−1 and L = 7 could strike a neutral in J = 2 and tunnel inward to form the J = 9 DBA.
Three issues still remain to be addressed: (i) can an electron of such low KE generate such L values? (ii) what happens after the metastable J = 9 DBA is formed? and (iii) how long is the tunneling distance in these cases?
To estimate the tunneling distance, consider the electron with KE = 1 cm−1 and L = 4. For this case, the tunneling inward begins approximately at where the J = 5; L =4 diabatic curve intersects the J = 6: l = 3 curve, and this turns out to be at R = 300 Å.
Of course, once such a DB MA is formed, it can still eject the electron via the same tunneling mechanism, but it can have substantial time to do so. We saw that the J = 34 level of H2CCN− had ca. 10−8 s before undergoing electron loss. As noted earlier, for C4H, C6H, and C3N, there exists a lower-energy valence-bound state of the MA to which the metastable DBA could relax via an IVR process during this time interval.
4. Summary
You likely will note that there are many classes of MAs that I have not discussed. That is because the focus of this Perspective has been on how a MA's environment can substantially alter its physical and chemical behavior. To achieve that goal, I decided to discuss only enough types of MAs sufficient to make my points. I believe the environmental-impact aspect of MA's behavior will become even more widely appreciated because many atmospheric, space, and nano-science phenomena involve processes taking place at interfaces where the reactive species are neither isolated nor strongly solvated. I continue to be amazed by the novel behavior of these species, and I hope to have succeeded in convincing you of this.Conflicts of interest
There are no conflicts to declare.References
- J. Simons, Molecular Anions, J. Phys. Chem. A, 2008, 112, 6401–6511 CrossRef CAS PubMed.
- J. Simons, Molecular Anions Perspective, J. Phys. Chem. A, 2023, 127, 3940–3957 CrossRef CAS PubMed.
- J. M. Herbert, in Reviews of Computational Chemistry, ed. A. L. Parrill and K. B. Lipkowitz, 2015, vol. 28, pp. 391–517 Search PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Atomic and Molecular Electron Affinities: Photoelectron experiments and Theoretical Computations, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed.
- J. Simons and W. D. Smith, Theory of Electron Affinities of Small Molecules, J. Chem. Phys., 1973, 58, 4899–4907 CrossRef CAS.
- https://scholar.google.com/citations?user=jYhIeQcAAAAJ&hl=en. Lenz has a long track record of excellent work on MAs.
- L. S. Cederbaum, G. Hohlneicher and W. von Niessen, Improved calculations of ionization potentials of closed−shell molecules, Mol. Phys., 1973, 26, 1405 CrossRef CAS.
- D. J. Rowe, Equations-of-motion method and the extended shell model, Rev. Mod. Phys., 1968, 40, 153–166 CrossRef.
- J. V. Ortiz, Dyson orbital concepts for description of electrons in molecules, J. Chem. Phys., 2020, 153, 070902 CrossRef CAS PubMed.
- J. V. Ortiz, Toward an exact one electron picture of chemical bonding, Adv. Quantum Chem., 1999, 35, 33–52 CrossRef CAS.
- M. Nooijen and R. J. Bartlett, Equation of motion coupled cluster method for electron attachment, J. Chem. Phys., 1995, 102, 3629–3647 CrossRef CAS.
- A. I. Krylov, Eqations-of-Motion Coupled-Cluster Methods for Open-Shell and Electronically Excited Species: The Hitchhiker's Guide to Fock Space, Ann. Rev. Phys. Chem., 2008, 59, 433–462 CrossRef CAS PubMed.
- R. A. Donnelly and J. Simons, Complex Coordinate Rotation of the Electron Propagator, J. Chem. Phys., 1980, 73, 2858–2866 CrossRef CAS.
- Z. Bacic and J. Simons, Application of the Coordinate Rotation Method to Metastable Atom-Diatom Scattering Resonances, Int. J. Quantum Chem. Symp., 1980, 14, 467–475 CAS.
- A. U. Hazi and H. S. Taylor, Stabilization Method of Calculating Resonance Energies: Model Problem, Phys. Rev. A: At., Mol., Opt. Phys., 1970, 1, 1109–1120 CrossRef.
- J. Simons, Analysis of Stabilization and Extrapolation Methods for Determining Energies and Lifetimes of Metastable Electronic States, J. Phys. Chem. A, 2021, 125, 7735–7749 CrossRef CAS PubMed.
- Z. Bacic and J. Simons, Resonance Energies and Lifetimes from Stabilization-Based Methods, J. Phys. Chem., 1982, 86, 1192–1200 CrossRef CAS.
- R. F. Frey and J. Simons, Resonance State Energies and Lifetimes via Analytic Continuation of Stabilization Graphs, J. Chem. Phys., 1986, 84, 4462–4469 CrossRef CAS.
- https://scholar.google.com/citations?hl=en&user=5cllXmIAAAAJ. Ken also has a long track record of excellent research on MAs.
- K. D. Jordan and F. Wang, Theory of Dipole-Bound Anions, Annu. Rev. Phys. Chem., 2003, 54, 367–396 CrossRef CAS PubMed.
- You can see several such papers on my personal publication list at https://simons.hec.utah.edu/publications.html and here is my Google Scholar page https://scholar.google.com/citations?hl=en&user=5LIV3DwAAAAJ.
- J. Simons, Resonance State Lifetimes from Stabilization Graphs, J. Chem. Phys., 1981, 75, 2465–2467 CrossRef CAS.
- R. F. Frey and J. Simons, Resonance State Energies and Lifetimes via Analytic Continuation of Stabilization Graphs, J. Chem. Phys., 1986, 84, 4462–4469 CrossRef CAS.
- J. S.-Y. Chao, M. F. Falcetta and K. D. Jordan, Application of the stabilization method to the N2(12Pg) and Mg (12P) temporary anion states, J. Chem. Phys., 1990, 93, 1125–1135 CrossRef CAS.
- J. Simons, Propensity Rules for Vibration-Induced Electron Detachment of Anions, J. Am. Chem. Soc., 1981, 103, 3971–3976 CrossRef CAS.
- J. Simons, Ejecting Electrons from Molecular Anions via Shine, Shake/Rattle, and Roll, J. Phys. Chem. A, 2020, 124, 8778–8797 CrossRef CAS PubMed.
- M. Gutowski and J. Simons, Double-Rydberg Anions: Ground-State Electronic and Geometric Stabilities, J. Chem. Phys., 1990, 93, 3874–3880 CrossRef CAS.
- https://scholar.google.com/citations?hl=en&user=ZB8zKToAAAAJ. Alex discovered new classes of MAs (e.g., superhalogen MAs) and has published much in this area.
- https://scholar.google.com/citations?hl=en&user=erZ4_hMAAAAJ, Maciej has been very active in studying dipole bound and other MAs.
- A. I. Boldyrev, M. Gutowski and J. Simons, Small Multiply Charged Anions as Building Blocks in Chemistry, Acc. Chem. Res., 1996, 29, 497–502 CrossRef CAS.
- A. I. Boldyrev and J. Simons, Tetracoordinated Planar Carbon in Pentaatomic Molecules, J. Am. Chem. Soc., 1998, 120, 7967–7972 CrossRef CAS.
- X. Li, L.-S. Wang, A. I. Boldyrev and J. Simons, Tetracoordinated Planar Carbon in the Al4Cl− Anion. A Combined Photoelectron Spectroscopy and Ab Initio Study, J. Am. Chem. Soc., 1999, 121, 6033–6038 CrossRef CAS.
- L.-S. Wang, A. I. Boldyrev, X. Li and J. Simons, Experimental Observation of Pentaatomic Tetracoordinated Planar Carbon-Containing Molecules, J. Am. Chem. Soc., 2000, 122, 7681–7687 CrossRef CAS.
- https://scholar.google.com/citations?hl=en&user=eQFN1ewAAAAJ. Lai-Sheng has many experimental papers dealing with a wide variety of MAs.
- https://scholar.google.com/citations?hl=en&user=jfdD7E8AAAAJ. Piotr and his group have studied many superhalogens, electron damage to DNA, and electron-induced fragmentation in mass spectrometry.
- P. Skurski, M. Gutowski and J. Simons, How to Choose a One-Electron Basis Set to Reliably Describe a Dipole-Bound Anion, Int. J. Quantum Chem., 2000, 80, 1024–1038 CrossRef CAS.
- M. Gutowski, P. Skurski, A. I. Boldyrev, J. Simons and K. Jordan, The Contribution of Electron Correlation to the Stability of Dipole-Bound Anionic States, Phys. Rev., 1996, 54, 1906–1909 CAS.
- R. Barrios, P. Skurski and J. Simons, Mechanism for Damage to DNA by Low-Energy Electrons, J. Phys. Chem. B, 2002, 106, 7991–7994 CrossRef CAS.
- J. Simons, How Do Low-energy (0.1–2 eV) Electrons Cause DNA Strand Breaks?, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed.
- A. Sawicka, P. Skurski, R. R. Hudgins and J. Simons, Model Calculations Relevant to Disulfide Bond Cleavage via Electron Capture Influenced by Positively Charged Groups, J. Phys. Chem., 2003, B107, 13505–13511 CrossRef.
- I. Anusiewicz, P. Skurski and J. Simons, Refinements to the Utah-Washington Mechanism of Electron Capture Dissociation, J. Phys. Chem. B, 2014, 118, 7892–7901 CrossRef CAS PubMed.
- D. W. Arnold, S. E. Bradforth, E. H. Kim and D. M. Neumark, Study of I−(CO2)n and I−(N2O)n clusters by anion photoelectron spectroscopy, J. Chem. Phys., 1995, 102, 3510–3518 CrossRef CAS.
- X.-B. Wang, J. B. Nicholas and L.-S. Wang, Electronic instability of isolated SO42− and its solvation stabilization, J. Chem. Phys., 2000, 113, 10837–10840 CrossRef CAS.
- A. I. Boldyrev and J. Simons, Isolated SO42− and PO43− Anions Do Not Exist, J. Phys. Chem., 1994, 98, 2298–2300 CrossRef CAS.
- R. Young and D. M. Neumark, Dynamics of Solvated Electrons in Clusters, Chem. Rev., 2012, 112, 5553–5577 CrossRef CAS PubMed.
- J. R. R. Verlet, A. E. Bragg, A. Kammrath, O. Cheshnovsky and D. M. Neumark, Observation of Large Water-Cluster Anions with Surface-Bound Excess Electrons, Science, 2005, 307, 93–96 CrossRef CAS PubMed.
- A. Dreuw and L. S. Cederbaum, Nature of the Repulsive Coulomb barrier in multiply charged negative ions, Phys. Rev. A: At., Mol., Opt. Phys., 2000, 63, 012501 CrossRef.
- J. Simons, P. Skurski and R. Barrios, Repulsive Coulomb Barriers in Compact Stable and Metastable Multiply Charged Anions, J. Am. Chem. Soc., 2000, 122, 11893–11899 CrossRef CAS.
- A. Whitehead, R. Barrios and J. Simons, Stabilization Calculation of the Energy and Lifetime of Metastable SO42−, J. Chem. Phys., 2002, 116, 2848–2851 CrossRef CAS.
- https://caice.ucsd.edu/ .
- https://chemistry.stanford.edu/people/richard-zare-0l, https://scholar.google.com/citations?hl=en&user=NWIMAqIAAAAJ.
- https://www.chem.purdue.edu/people/profile/cooks; https://scholar.google.com/citations?hl=en&user=_Jxju_sAAAAJ.
- https://chem.yale.edu/faculty/mark-johnson; https://scholar.google.com/citations?hl=en&user=Q9Ys_NUAAAAJ.
- https://chemistry.berkeley.edu/faculty/chem/teresa-head-gordon; https://scholar.google.com/citations?hl=en&user=LP_1xT4AAAAJ.
- https://www.chem.columbia.edu/content/wei-min .
- https://dornsife.usc.edu/chemistry/dawlaty/ .
- https://www.colorado.edu/chemistry/veronica-vaida; https://scholar.google.com/citations?hl=en&user=-OvYYRcAAAAJ.
- https://www.researchgate.net/scientific-contributions/Joseph-S-Francisco-39101962 .
- https://chem.wisc.edu/staff/nathanson-gilbert/; https://www.researchgate.net/scientific-contributions/Gilbert-M-Nathanson-38178492.
- M. Mucha, T. Frigato, L. M. Levering, H. C. Allen, D. J. Tobias, L. X. Dang and P. Jungwirth, Unified Molecular Picture of the Surfaces of Aqueous Acid, Base, and Salt Solutions, J. Phys. Chem. B, 2005, 109, 7617–7623 CrossRef CAS PubMed . These workers are among the leaders in such studies.
- Y.-L. S. Tse, C. Chen, G. E. Lindberg, R. Kumar and G. A. Voth, Propensity of Hydrated Excess Protons and Hydroxide Anions for the Air-Water Interface, J. Am. Chem. Soc., 2015, 137, 12610–12616 CrossRef CAS PubMed.
- H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. Willner, L. C. Marr and P. J. Vikesland, Aerosol microdroplets exhibit a stable pH gradient, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7272–7277 CrossRef CAS PubMed.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Spontaneous generation of hydrogen peroxide from aqueous microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298, DOI:10.1073/pnas.1911883116.
- M. A. Mehrgardi, M. Mofidfar and R. N. Zare, Sprayed Water Microdroplets Are Able to Generate Hydrogen Peroxide Spontaneously, J. Am. Chem. Soc., 2022, 144, 7606–7609 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Micrometer-Sized Water Droplets Induce Spontaneous Reduction, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- H. Hao, I. Leven and T. Head-Gordon, Can Electric Fields Drive Chemistry for an Aqueous Microdroplet?, Nat. Commun., 2022, 13, 280 CrossRef CAS PubMed.
- S. M. Kathmann, L. F. W. Kuo and C. J. Mundy, Electronic Effects on the Surface Potential at the Vapor–Liquid Interface of Water, J. Am. Chem. Soc., 2008, 130, 16556–16561 CrossRef CAS PubMed.
- M. P. Coons, Z.-Q. You and J. M. Herbert, The Hydrated Electron at the Surface of Neat Liquid Water Appears To Be Indistinguishable from the Bulk Species, J. Am. Chem. Soc., 2016, 138, 10879–10886 CrossRef CAS PubMed.
- R. D. Leib, W. A. Donald, M. F. Bush, J. T. O’Brien and E. R. Williams, Nonergodicity in Electron Capture Dissociation Investigated Using Hydrated Ion Calorimetry, J. Am. Soc. Mass Spectrom., 2007, 18, 1217–1231 CrossRef CAS PubMed.
- D. Neff and J. Simons, Theoretical study of electron capture dissociation of [Mg(H2O)n]2+ clusters, Int. J. Mass. Spectrosc., 2008, 277, 166–174 CrossRef CAS.
- A. Sawicka, P. Skurski, R. R. Hudgins and J. Simons, Model Calculations Relevant to Disulfide Bond Cleavage via Electron Capture Dissociation Influenced by Positively Charged Groups, J. Phys. Chem. B, 2003, 107, 13505–13511 CrossRef CAS.
- I. Anusiewicz, J. Berdys-Kochanska and J. Simons, Electron Attachment Step in Electron Capture Dissociation (ECD) and Electron Transfer Dissociation (ETD), J. Phys. Chem. A, 2005, 109, 5801–5813 CrossRef CAS PubMed.
- I. Anusiewicz, P. Skursi and J. Simons, Refinements to the Utah-Washington Mechanism of Electron Capture Dissociation, J. Phys. Chem. B, 2014, 118, 7892–7901 CrossRef CAS PubMed.
- https://chem.washington.edu/people/frantisek-turecek; https://scholar.google.com/citations?hl=en&user=QAMyaBMAAAAJ.
- I summarize much of this here: J. Simons, How Do Low-energy (0.1–2 eV) Electrons Cause DNA Strand Breaks?, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed.
- F. Martin, P. D. Burrow, Z. Cai, P. Cloutier, D. Hunting and L. Sanche, DNA Strand Breaks Induced by 0-4 eV Electrons: The Role of Shape Resonances, Phys. Rev. Lett., 2004, 93, 068101–1-068101-4 CrossRef PubMed.
- J. Gu, Y. Xie and H. F. Schaefer, III, Electron attachment to DN single strands: gas phase and aqueous solution, Nucleic Acids Res., 2007, 35, 5165–5172 CrossRef CAS PubMed.
- J. Gu, J. Wang and J. Leszczynski, Electron attachment-induced DNA single-strand breaks at the pyrimidine sites, Nucleic Acids Res., 2010, 38, 5280–5290 CrossRef CAS PubMed.
- K. Aflatooni, G. A. Gallup and P. D. Burrow, Electron Attachment Energies of the DNA Bases, J. Phys. Chem. A, 1998, 102, 6205–6207 CrossRef CAS.
- F. Martin, P. D. Burrow, Z. Cai, P. Cloutier, D. Hunting and L. Sanche, DNA Strand Breaks Induced by 0-4 eV Electrons: The Role of Shape Resonances, Phys. Rev. Lett., 2004, 93, 068101-1–068101-4 Search PubMed.
- Y. Zheng, P. Cloutier, D. Hunting, L. Sanche and J. R. Wagner, Chemical Basis of DNA Sugar-Phosphate Cleavage by Low- Energy Electrons, J. Am. Chem. Soc., 2005, 127, 16592–16598 CrossRef CAS PubMed.
- Y. Zheng, P. Cloutier, D. Hunting, J. R. Wagner and L. Sanche, Phosphodiester and N-glycosidic bond cleavage in DNA induced by 4-15 eV electrons, J. Chem. Phys., 2006, 124, 064710-1–064710-9 Search PubMed.
- J. Simons, Ejecting Electrons from Molecular Anions via Shine, Shake/Rattle, and Roll, J. Phys. Chem. A, 2020, 124, 8778–8797 CrossRef CAS PubMed.
- D. M. Neumark, K. R. Lykke, T. Andersen and W. C. Lineberger, Infrared spectrum and autodetachment dynamics of NH−, J. Chem. Phys., 1985, 83, 4364–4373 CrossRef CAS.
- J. Simons, Propensity Rules for Vibration-Induced Electron Detachment of Anions, J. Am. Chem. Soc., 1981, 103, 3971–3976 CrossRef CAS.
- G. Chalasinski, R. A. Kendall, H. Taylor and J. Simons, Propensity Rules for Vibration-Rotation Induced Electron Detachment of Diatomic Anions: Application to NH− → NH+ e−, J. Phys. Chem., 1988, 92, 3086–3091 CrossRef CAS.
- S. T. Edwards, M. A. Johnson and J. C. Tully, Vibrational Fano Resonances in Dipole-Bound Anions, J. Chem. Phys., 2012, 136, 154305 CrossRef PubMed.
- K. R. Lykke, D. M. Neumark, T. Andersen, V. J. Trapa and W. C. Lineberger, Autodetachment Spectroscopy and Dynamics of CH2CN− and CD2CN−, J. Chem. Phys., 1987, 87, 6842–6853 CrossRef CAS.
- D. C. Clary, Photodetachment of Electrons from Dipolar Anions, J. Phys. Chem., 1988, 92, 3173–3181 CrossRef CAS.
- J. Simons, Modified Rotationally Adiabatic Model for Rotational Autoionization of Dipole-Bound Molecular Anions, J. Chem. Phys., 1989, 91, 6858–6865 CrossRef CAS.
- R. C. Fortenberry, Interstellar Anions: The Role of Quantum Chemistry, J. Phys. Chem. A, 2015, 119, 9941–9953 CrossRef CAS PubMed.
- T. J. Millar, C. Walsh and T. A. Field, Negative Ions in Space, Chem. Rev., 2017, 117, 1765–1795 CrossRef CAS PubMed.
- https://www.uibk.ac.at/ionen-angewandte-physik/index.html.de .
- F. Carelli, M. Satta, T. Grash and F. A. Gianturco, Carbon-Rich Molecular Chains in Protoplanetary and Planetary Atmospheres: Quantum Mechanisms and Electron Attachment Rates for Anion Formation, Astrophys. J., 2013, 774, 97 CrossRef.
| This journal is © the Owner Societies 2024 |