
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivating nickel foam with trace titanium oxide for enhanced water oxidation†
Jia-Fang
Xie
 *ab,
Ding
Li
ab,
Hui-Wen
Huo
ab,
Yi-Yin
Huang
c,
Peng
Wu
a,
Quan-Bao
Zhao
ab and
Yu-Ming
Zheng
ab
*ab,
Ding
Li
ab,
Hui-Wen
Huo
ab,
Yi-Yin
Huang
c,
Peng
Wu
a,
Quan-Bao
Zhao
ab and
Yu-Ming
Zheng
ab
aCAS Key Laboratory of Urban Pollutant Conversion, Research Center of Urban Carbon Neutrality, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China. E-mail: jfxie@iue.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cFujian Provincial Key Laboratory of Quantum Manipulation and New Energy Materials, College of Physics and Energy, Fujian Normal University, Fuzhou, Fujian 350117, China
First published on 12th February 2024
Abstract
Nickel-based electrocatalysts for water oxidation suffer from low activity and poor stability. In this work, 0.015 mg cm−2 TiO2 nanosheets anchored on Ni foam addressed these problems after electrochemical activation. In situ investigations, including Raman spectra, corroborated the enhanced generation of highly active Ni(III)–O–O species on Ni foam in the presence of trace TiO2.
Water oxidation is a key half reaction in many renewable energy conversion and storage technologies, but its kinetically sluggish process demands effective electrocatalysts.1,2 Although Ru and Ir-based catalysts have shown excellent performance, their high cost and limited storage pose challenges for broad application.
Ni-based catalysts, characterized by high-valence Ni species, have emerged as a promising alternative, particularly in alkaline water oxidation.3,4 However, catalysts utilizing Ni as the only metal not only necessitate high energy inputs to generate high-valence Ni species but also suffer limited service-life. To address these problems, additional metals or metal oxide semiconductors are incorporated as dopants or substrates, respectively.5–8 TiO2, a low-cost yet interesting semiconductor, is frequently employed as the substrate due to its robust stability under high oxidation conditions.9–12 TiO2 with single and/or nano crystallization and oxygen vacancies has promoted surface reconstruction for the generation of high-valence Ni species.12–14 Despite great achievements, fulfilling the stability of TiO2 with the goal of high activity and stability for Ni-catalyzed water oxidation is still a challenge.
Metal-support interaction, which typically appears in supported metal catalysts, sheds a light on the solution.15,16 This concept suggests that when the metal is supported, its electronic structure is altered by the metal oxide substrate. From the perspective of the substrate, surface atoms on the metal oxide substrate also acquire a modified electronic structure.8,17,18 So, if we transpose the conventional role of Ni as the supported metal and TiO2 as the substrate,19 such as anchoring TiO2 nanomaterials onto a commercial Ni foam (NF) substrate,20 high-valence Ni species would still be generated on NF due to the metal-substrate interaction accompanied by Ti(III) generation on TiO2. Thus, enhanced water oxidation with high activity and stability can be achieved on TiO2 nanomaterial-anchored NF in theory. Further taking the high thermal stability of TiO2 into consideration,21 the utilization of polymer binders can be replaced by heat treatment to realize stably anchored TiO2 on NF, which can also avoid the decomposition risk and low conductivity of binders. However, such a role exchange strategy has not received sufficient attention, which, if realized, would promote the development of cost-effective Ni-based catalysts for highly active and stable water oxidation.
In this work, we take TiO2 nanosheets as a model TiO2 nanomaterial to verify this hypothesis.22 The diffraction peaks of the synthesized TiO2 nanosheets well matched anatase TiO2 (JCPDS No. 21-1272, Fig. S1, ESI†). As shown in Fig. 1a, the trace TiO2 nanosheets with the loading mass of 0.015 mg cm−2 were anchored on NF by mild heat treatment without binders, and the TiO2-loaded material (prepared Ti–Ni) showed the incomplete coverage of TiO2 nanosheets on NF (Fig. S2 and S3, ESI†). Then the prepared Ti–Ni could be electrochemically activated during application as the anode in an alkaline flow cell for CO2 splitting. The resultant activated material is thus denoted as activated Ti–Ni. The electrochemical activation roughened the NF surface but kept the sheet morphology of TiO2 (Fig. S4, ESI†).
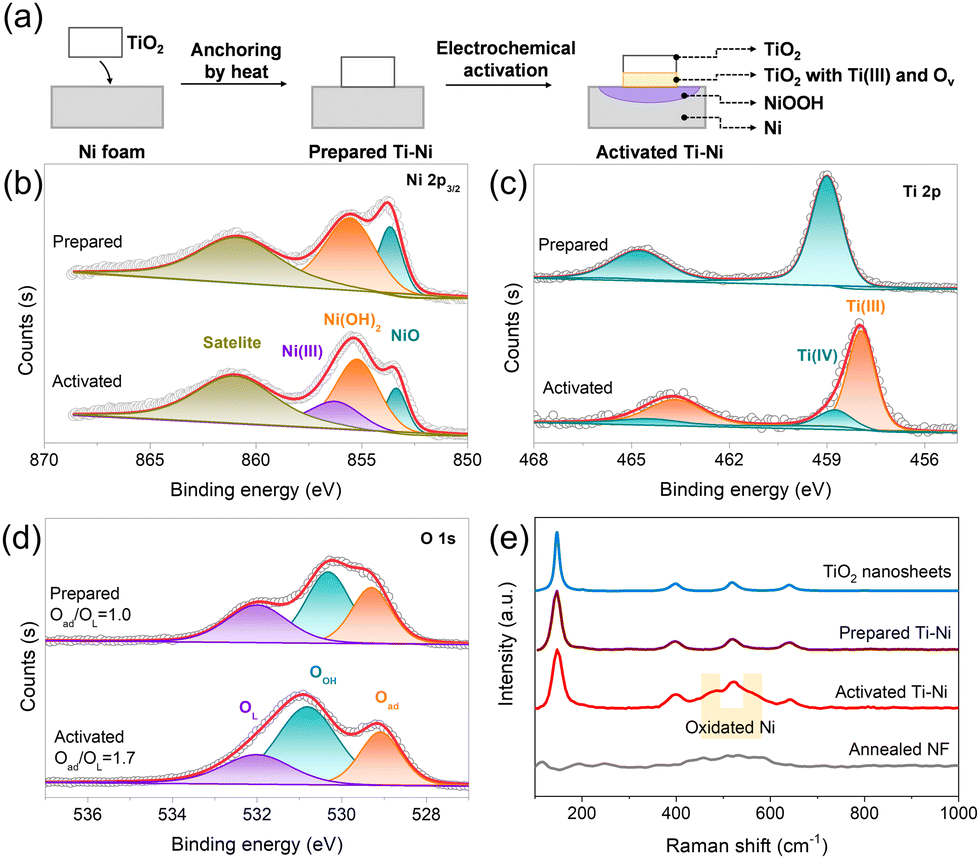 | ||
| Fig. 1 (a) Schematic illustration of the preparation of activated Ti–Ni. XPS spectra corresponding to (b) Ni 2p3/2, (c) Ti 2p, and (d) O 1s of the prepared Ti–Ni and activated Ti–Ni materials. (e) Raman spectra of the prepared Ti–Ni and activated Ti–Ni along with the comparison of annealed NF and TiO2 powder. | ||
The X-ray photoelectron spectroscopy (XPS) revealed more obvious differences on the surface of the samples resulting from electrochemical activation. The prepared Ti–Ni exhibited peaks corresponding to NiO (853.7 eV), Ni(OH)2 (855.5 eV), and Ti(IV) (459.0 eV). As to activated Ti–Ni, a new peak for Ni(III) (856.2 eV) in the high-resolution Ni 2p3/2 spectrum (Fig. 1b) and a new peak for Ti(III) at 458.8 eV in the high-resolution Ti 2p spectrum (Fig. 1c) could be observed, demonstrating the electron transfer from Ni to Ti during electrochemical activation.7,23,24 Bare NF contained a peak of metallic Ni at 852.6 eV (Fig. S5, ESI†), which disappeared in the prepared Ti–Ni and activated Ti–Ni, probably due to the heat treatment. Furthermore, the ratio of adsorbed oxygen to lattice oxygen for activated Ti–Ni was 1.7 (Fig. 1d), which is lower than that of the prepared Ti–Ni (1.0). This increase suggests the occurrence of oxygen vacancies in activated Ti–Ni,7 consistent with Ti(III) generation. Additionally, F was absent in the prepared Ti–Ni and activated Ti–Ni, eliminating the effect of potential F species (Fig. S6, ESI†). The Raman spectra (Fig. 1e) of the prepared Ti–Ni and activated Ti–Ni show the similar four peaks at 146.2 cm−1, 398.1 cm−1, 518.6 cm−1, and 640 cm−1 as TiO2 nanosheets,21,25,26 demonstrating the stable existence of TiO2 nanosheets on NF. Moreover, the new peaks of oxidized Ni located between 450 and 600 cm−1 were observed in activated Ti–Ni compared with the prepared Ti–Ni and the annealed NF without the addition of TiO2,27 also suggesting Ni(III) generation after electrochemical activation. Therefore, activated Ti–Ni with Ni(III) on the NF surface was successfully prepared based on electron transfer from the NF substrate to the TiO2 nanosheets.
The water oxidation on activated Ti–Ni was investigated in 1 M KOH solution, using the prepared Ti–Ni, commercial IrO2, and bare NF as comparison. As shown in the linear sweep voltammetry (LSV) plots (Fig. 2a), activated Ti–Ni had the lowest overpotential of 256 mV to obtain 10 mA cm−2, which was 116 mV lower than that of bare NF (372 mV), proving that water oxidation on the activated Ti–Ni was enhanced. In contrast, the prepared Ti–Ni needed 362 mV overpotential to gain 10 mA cm−2, which is very close to bare NF, suggesting again that highly active Ni(III) was generated during electrochemical activation rather than heat treatment. Furthermore, activated Ti–Ni spent 337 mV overpotential to gain 100 mA cm−2 at 1.56 V, whereas bare NF only showed 3.5 mA cm−2 and IrO2 gained 48.8 mA cm−2. Tafel fitting results (Fig. 2b) obtained from the above-mentioned LSV plots revealed that the reaction kinetics of water oxidation on activated Ti–Ni exhibited a Tafel slope of 59.0 mV dec−1, which is much lower than that of bare NF (84.4 mV dec−1) as well as the prepared Ti–Ni (66.2 mV dec−1). Such an enhanced kinetics suggests that the Ni(III) generated during electrochemical activation accelerates the electron transfer in water oxidation on activated Ti–Ni. Additionally, such a Tafel slope near 60 mV dec−1 indicates that activated Ti–Ni releases the rate-determining first electron transfer compared to bare NF.28
 | ||
| Fig. 2 (a) Water oxidation linear voltammograms of activated Ti–Ni in 1.0 M KOH. (b) The corresponding Tafel plots. (c) Water oxidation linear voltammograms normalized by the electrochemical active surface area. (d) Electrochemical impedance spectroscopy curves at 0.6 V vs. HgO. Prepared Ti–Ni, commercial IrO2 loaded on NF, and bare NF were also shown as a comparison. (e) Electrolysis plots for 24 h at 10 mA cm−2. (f) Comparison of selected electrocatalysts containing TiO2 for alkaline water oxidation. The overpotential at 10 mA cm−2 was shown. | ||
The electrochemical active surface area (ECSA) based on double layer capacitance (Cdl) measurement (Fig. S7, ESI†) was evaluated to exclude the contribution of the quantity of electrochemical active sites. Activated Ti–Ni showed an apparently enhanced Cdl of 1.05 mF cm−2 over bare NF of 0.149 mF cm−2 (Fig. S8, ESI†), suggesting 7 times increased active sites by electrochemical activation with TiO2 nanosheets. Additionally, IrO2 with the same loading mass showed nearly twice higher Cdl than the prepared Ti–Ni, suggesting the limited activity of TiO2 itself.29Fig. 2c shows the LSV plots calculated with area normalized by the resulting ECSA, in which activated Ti–Ni required 282 mV overpotential to obtain 1 mA cm−2 ECSA, slightly lower than IrO2. The minorly higher activity of the prepared Ti–Ni compared to bare NF at applied potentials exceeding 1.53 V might be ascribed to some Ni(III) generation via electron transfer to TiO2 at high potentials.8 The smallest semicircle of activated Ti–Ni in electrochemical impedance spectroscopy (EIS) results (Fig. 2d) suggested again the most enhanced electron transfer ability of the highly active Ni(III) generated during electrochemical activation.
The stability of the activated Ti–Ni for water oxidation was evaluated by continuous electrolysis at 10 mA cm−2 (Fig. 2e). The potentials kept stable for 24 h, proving the good stability of the activated Ti–Ni. As a comparison, IrO2 showed an obvious increase at the beginning and a slight increase of potential in the end of 24 h electrolysis. The overpotential at 10 mA cm−2 obtained from the LSV plot after 24 h electrolysis decreased to 247 for the activated Ti–Ni (Fig. S9, ESI†), demonstrating the intrinsic activity enhancement via electrochemical activation. It is noteworthy that these low overpotentials on activated Ti–Ni are rarely achieved by the reported TiO2-containing electrocatalysts for basic water oxidation, to the best of our knowledge (Fig. 2f and Table S1, ESI†), close to state-of-the-art chemically etched/modified NF, and even surpassing those of IrO2.8,12,13,30–35 These results demonstrated that trace TiO2 nanosheets can enhance the OER activity and stability based on a role exchange strategy of metal-support interaction.
After one year of placement without special protection, activated Ti–Ni still possessed a low overpotential of 255 mV to gain 10 mA cm−2 (Fig. 3a), which was much lower than that of bare NF. Moreover, its potential kept stable at the 24th hour and only increased by 3 mV after 100 h electrolysis at 10 mA cm−2 (Fig. 3b), further demonstrating the excellent stability of the activated Ti–Ni. Turnover frequency (TOF) based on the Ni(II)-to-Ni(III) oxidation peak current helped analyse the intrinsic activity of the active sites more precisely. Based on Ni(III) as a highly active site for water oxidation, CV scans covering the potential region of Ni(II)-to-Ni(III) reaction were conducted to calculate the quantity of Ni(III) sites (Fig. S10, ESI†).7 The slope of the fitting plot for activated Ti–Ni was 0.2221 F, nearly 5-times that of bare NF (Fig. 3c), corresponding to the same ratio of active site quantity. The LSV plots in Fig. 3a were thus calculated to be TOF plots (Fig. 3d), in which activated Ti–Ni possessed a TOF up to 0.677 s−1 at 350 mV overpotential (Fig. S10c, ESI†), nearly 13 times higher than that of bare NF, demonstrating the effectively enhanced intrinsic activity of Ni(III) generated on the activated Ti–Ni. Therefore, electrochemical activation using trace TiO2 nanosheets not only increases the number of Ni(III) sites but also augments their intrinsic activity.
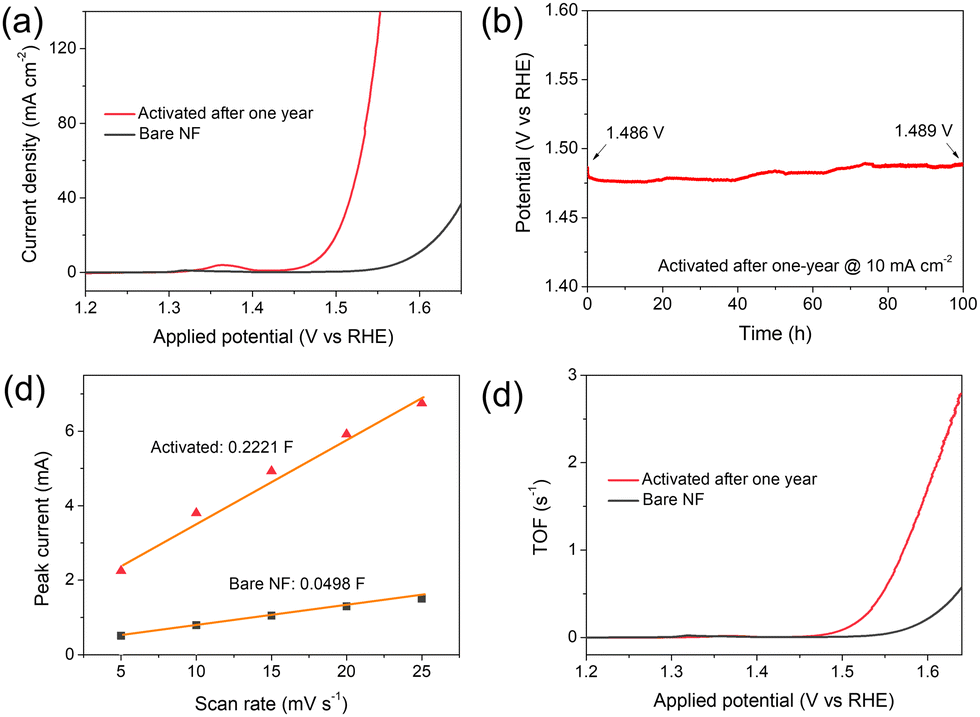 | ||
| Fig. 3 (a) Water oxidation linear voltammograms, (b) stability test at 10 mA cm−2 for 100 h, (c) fitting plots of peak currents in CV scans, and (d) resulting TOF plot for activated Ti–Ni after one-year placement. | ||
To further investigate the role of TiO2 nanosheets on Ni(III) generation in activated Ti–Ni, the in situ Raman spectroscopy on activated Ti–Ni under work, a spectro-electrochemical measurement, was carried out with annealed NF as a comparison (Fig. 4).36 The obvious change of peak intensity for activated Ti–Ni suggested more surface atoms involved in reconstruction at potentials below 1.47 V (Fig. 4a) compared to annealed NF (Fig. 4b), suggesting the importance of TiO2 nanosheets. For activated Ti–Ni, the Eg mode (at about 147 cm−1 and 642 cm−1) and B1g mode (at about 398 cm−1) of the TiO2 nanosheets presented decreasing intensity and broadening of the shape along with the applied potential moving positively (Fig. 4a and b), indicating the formation of oxygen vacancies and Ti(III).23,37 Though Ni(III) species could also be formed on NF, activated Ti–Ni required 100-mV lowered applied potential to increase the Eg bending vibration (δNi) and A1g stretching vibration (νNi) mode than that of annealed NF (Fig. S11, ESI†). Furthermore, the intensities for the Ni(III)–O peaks in activated Ti–Ni were much higher than that of annealed NF, demonstrating the TiO2 nanosheet addition promoted high-valence Ni(III) generation.38 Additionally, the increasing ratio of δNi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :νNi in the activated Ti–Ni may indicate some more complex reconstruction process compared to the stable ratio for that of annealed NF.39–41 Moreover, the peak at ca. 1000 cm−1 belonging to the active oxygen species (NiOO−) on activated Ti–Ni showed a similarly higher intensity and lower applied demand than that of annealed NF (Fig. 4c and d),42,43 further emphasizing the important role of TiO2 nanosheets in generating highly active Ni species of high valence state on activated Ti–Ni for enhanced water oxidation. Finally, we also anchored 0.015 mg cm−2 commercial P25 nanoparticles onto NF and the resulting activated Ti–Ni with P25 also obviously enhanced the OER performance (Fig. S12, ESI†), suggesting that the role-exchange strategy can be expanded to develop high-performance Ni electrodes for water oxidation with other TiO2 nanomaterials.
:νNi in the activated Ti–Ni may indicate some more complex reconstruction process compared to the stable ratio for that of annealed NF.39–41 Moreover, the peak at ca. 1000 cm−1 belonging to the active oxygen species (NiOO−) on activated Ti–Ni showed a similarly higher intensity and lower applied demand than that of annealed NF (Fig. 4c and d),42,43 further emphasizing the important role of TiO2 nanosheets in generating highly active Ni species of high valence state on activated Ti–Ni for enhanced water oxidation. Finally, we also anchored 0.015 mg cm−2 commercial P25 nanoparticles onto NF and the resulting activated Ti–Ni with P25 also obviously enhanced the OER performance (Fig. S12, ESI†), suggesting that the role-exchange strategy can be expanded to develop high-performance Ni electrodes for water oxidation with other TiO2 nanomaterials.
 | ||
| Fig. 4 Raman spectra for activated Ti–Ni and annealed NF under a series of applied potentials (values shown vs. RHE). (a) and (b) Survey spectra ranging from 100 to 1300 cm−1. (c) and (d) Local magnified Raman spectra in the range of 800–1300 cm−1 for the O–O region. OCP: open circuit potential was applied. | ||
In summary, this work demonstrates a role-exchange strategy to fulfill the advantages of TiO2 for water oxidation electrocatalysis. Taking TiO2 nanosheets as an example, the activated Ti–Ni shows enhanced alkaline water oxidation activity with 256 mV overpotential to bring 10 mA cm−2 and TOF up to 0.677 s−1 at 350 mV overpotential. In situ Raman included experimental results reveal that after electrochemical activation derived surface reconstruction, trace TiO2 additive increases both the quantity and the inherent activity of highly active Ni sites with high valence state on NF, leading to reduced overpotentials and accelerated kinetics. Notably, in addition to high activity, activated Ti–Ni showed high stability in continuous 100 h electrolysis after one-year placement. This work provides a new way to prepare cost-effective Ni electrodes for highly active and stable water oxidation.
Jia-Fang Xie: Providing original idea and funding support, conducting experiments, and writing and revising the manuscript. Ding Li, Hui-Wen Huo, and Peng Wu: Helping data collection mainly. Yi-Yin Huang: Helping data analysis and revising the manuscript mainly. Quan-Bao Zhao and Yu-Ming Zheng: Providing funding support and helping data analysis mainly.
This research was conducted as part of the National Key Research & Development Program of China (2021YFA1202700), the National Natural Science Foundation of China (E111C00101, 52027815, and 22175036), the CAS Key Laboratory of Urban Pollutant Conversion Joint Research Fund (KLUPC-2020-3), and the Youth Innovation Promotion Association CAS (2022308) to J.F. Xie, the Science and Technology Planning Project of Fujian Province (E1F2C00201), and the Natural Science Foundation of Fujian Province (2022J01181).
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spori, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Perez-Ramirez, B. R. Cuenya, R. Schlogl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed
.
- N. Y. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed
- H. Ding, H. F. Liu, W. S. Chu, C. Z. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed
- N. K. Shrestha, S. A. Patil, J. H. Seok, A. S. Salunke, S. Cho, A. I. Inamdar, Y. Park, S. U. Lee, H. Kim and H. Im, Mater. Today Phys., 2023, 38, 101252 CrossRef CAS
- R. Lin, L. Mao, Y. Ding and J. Qian, Chem. Commun., 2023, 59, 12875–12878 RSC
- M. Shi, T. Tang, L. Xiao, J. Han, X. Bai, Y. Sun, Y. Ma and J. Guan, Chem. Commun., 2023, 59, 13607–13610 RSC
- Z. Huang, X. Liao, W. Zhang, J. Hu and Q. Gao, ACS Catal., 2022, 12, 13951–13960 CrossRef CAS
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS PubMed
- Q. Guo, Z. B. Ma, C. Y. Zhou, Z. F. Ren and X. M. Yang, Chem. Rev., 2019, 119, 11020–11041 CrossRef CAS PubMed
- C. Zhang, H. Q. Liang, Z. K. Xu and Z. K. Wang, Adv. Sci., 2019, 6, 1900883 CrossRef CAS PubMed
- B. Yang, Z. Zhang, P. Liu, X. Fu, J. Wang, Y. Cao, R. Tang, X. Du, W. Chen, S. Li, H. Yan, Z. Li, X. Zhao, G. Qin, X.-Q. Chen and L. Zuo, Nature, 2023, 622, 499–506 CrossRef CAS PubMed
- C. Liu, J. Qian, Y. Ye, H. Zhou, C.-J. Sun, C. Sheehan, Z. Zhang, G. Wan, Y.-S. Liu, J. Guo, S. Li, H. Shin, S. Hwang, T. B. Gunnoe, W. A. Goddard and S. Zhang, Nat. Catal., 2020, 4, 36–45 CrossRef
- G. Q. Shen, R. R. Zhang, L. Pan, F. Hou, Y. J. Zhao, Z. Y. Shen, W. B. Mi, C. X. Shi, Q. F. Wang, X. W. Zhang and J. J. Zou, Angew. Chem., Int. Ed., 2020, 59, 2313–2317 CrossRef CAS PubMed
- K. Suzuki, X. Li, T. Toda, F. Nagasawa and K. Murakoshi, ACS Energy Lett., 2021, 6, 4374–4382 CrossRef CAS
- T. W. van Deelen, C. H. Mejia and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS
- J. Zhou, F. Wang, H. Wang, S. Hu, W. Zhou and H. Liu, Nano Res., 2023, 16, 2085–2093 CrossRef CAS
- F. D. Lin and S. W. Boettcher, Nat. Mater., 2014, 13, 81–86 CrossRef CAS PubMed
- F. Z. Song, S. Debow, T. Zhang, Y. Q. Qian, Z. C. Huang-Fu, K. Munns, S. Schmidt, H. Fisher, J. B. Brown, Y. Q. Su, Z. Zander, B. G. DeLacy, M. S. Mirotznik, R. L. Opila and Y. Rao, J. Phy. Chem. Lett., 2023, 14, 5692–5700 CrossRef CAS PubMed
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS
- A. Peugeot, C. E. Creissen, D. Karapinar, H. N. Tran, M. Schreiber and M. Pehlivan, Joule, 2021, 5, 1281–1300 CrossRef CAS
- C. Liu, A.-Y. Zhang, D.-N. Pei and H.-Q. Yu, Environ. Sci. Technol., 2016, 50, 5234–5242 CrossRef CAS PubMed
- C. Liu, A.-Y. Zhang, Y. Si, D.-N. Pei and H.-Q. Yu, Environ. Sci. Technol., 2017, 51, 11326–11335 CrossRef CAS PubMed
- E. J. Kim, J. Shin, J. Bak, S. J. Lee, K. H. Kim, D. Song, J. Roh, Y. Lee, H. Kim, K. S. Lee and E. Cho, Appl. Catal., B, 2021, 280, 119433 CrossRef CAS
- L. Yang, T. Wu, R. Zhang, H. Zhou, L. Xia, X. Shi, H. Zheng, Y. Zhang and X. Sun, Nanoscale, 2019, 11, 1555–1562 RSC
- K. Wang, K. Zhao, X. Qin, S. Chen, H. Yu and X. Quan, J. Hazard. Mater., 2022, 424, 127747 CrossRef CAS PubMed
- F. Tian, Y. Zhang, J. Zhang and C. Pan, J. Phys. Chem. C, 2012, 116, 7515–7519 CrossRef CAS
- N. A. Sagui, P. Strom, T. Edvinsson and I. Bayrak Pehlivan, ACS Catal., 2022, 12, 6506–6516 CrossRef CAS PubMed
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed
- D.-N. Pei, L. Gong, A.-Y. Zhang, X. Zhang, J.-J. Chen, Y. Mu and H.-Q. Yu, Nat. Commun., 2015, 6, 8696 CrossRef CAS PubMed
- H. B. Tao, L. W. Fang, J. Z. Chen, H. B. Yang, J. J. Gao, J. W. Miao, S. L. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed
- Y. W. Hu, T. Ding, K. L. Zhang, B. Li, B. C. Zhu and K. B. Tang, ChemNanoMat, 2018, 4, 1133–1139 CrossRef CAS
- M. S. Amer, M. A. Ghanem, P. Arunachalam, A. M. Al-Mayouf and S. M. Hadadi, Catalysts, 2019, 9, 836 CrossRef CAS
- J. Y. Yu, X. D. Wang, X. W. Huang, J. Y. Cao, Q. H. Huo, L. R. Mi, H. P. Yang, Q. Hu and C. X. He, Chem. Eng. J., 2022, 446, 137248 CrossRef CAS
- Y. Liu, Z. Zhang, L. Zhang, Y. Xia, H. Wang, H. Liu, S. Ge and J. Yu, J. Mater. Chem. A, 2022, 10, 22125–22134 RSC
- N. K. Shrestha, S. A. Patil, J. Han, S. Cho, A. I. Inamdar, H. Kim and H. Im, J. Mater. Chem. A, 2022, 10, 8989–9000 RSC
- H.-B. Tao, Z. Tian, Y. Xie, Y. Sun, L. Wang, Z. Kang and Y. Zhang, Chin. J. Appl. Chem., 2022, 39, 528–539 CAS
- R. R. Jia, Y. T. Wang, C. H. Wang, Y. F. Ling, Y. F. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef CAS
- N. K. Shrestha, S. A. Patil, A. S. Salunke, A. I. Inamdar and H. Im, J. Mater. Chem. A, 2023, 11, 14870–14877 RSC
- S. A. Patil, A. C. Khot, V. D. Chavan, I. Rabani, D.-k Kim, J. Jung, H. Im and N. K. Shrestha, Chem. Eng. J, 2024, 480, 146545 CrossRef CAS
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 10295–10299 CrossRef CAS PubMed
- S. Lee, Y.-C. Chu, L. Bai, H. M. Chen and X. Hu, Chem Catal., 2023, 3, 100475 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD patterns, SEM images, CV and LSV plots, and table. See DOI: https://doi.org/10.1039/d3cc05956a |
| This journal is © The Royal Society of Chemistry 2024 |