
Improved nanoformulation and bio-functionalization of linear-dendritic block copolymers with biocompatible ionic liquids†
Christine M.
Hamadani
 a,
Indika
Chandrasiri
a,
Mahesh Loku
Yaddehige
a,
Gaya S.
Dasanayake
a,
Iyanuoluwani
Owolabi
b,
Alex
Flynt
b,
Mehjabeen
Hossain
c,
Lucy
Liberman
de,
Timothy P.
Lodge
de,
Thomas A.
Werfel
fgc,
Davita L.
Watkins
a and
Eden E. L.
Tanner
*a
a,
Indika
Chandrasiri
a,
Mahesh Loku
Yaddehige
a,
Gaya S.
Dasanayake
a,
Iyanuoluwani
Owolabi
b,
Alex
Flynt
b,
Mehjabeen
Hossain
c,
Lucy
Liberman
de,
Timothy P.
Lodge
de,
Thomas A.
Werfel
fgc,
Davita L.
Watkins
a and
Eden E. L.
Tanner
*a
aDepartment of Chemistry & Biochemistry, The University of Mississippi, University, MS 38677, USA. E-mail: eetanner@olemiss.edu; Tel: +662-915-1165
bDepartment of Biological Sciences, The University of Southern Mississippi, Hattiesburg, Mississippi 39406, USA
cDepartment of BioMolecular Sciences, The University of Mississippi, University, MS 38677, USA
dDepartment of Chemical Engineering & Materials Science, University of Minnesota, Minneapolis, Minnesota 55455, USA
eDepartment of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA
fDepartment of Biomedical Engineering, The University of Mississippi, University, MS 38677, USA
gDepartment of Chemical Engineering, The University of Mississippi, University, MS 38677, USA
First published on 1st April 2022
Abstract
Linear-dendritic block copolymers (LDBCs) have emerged as promising materials for drug delivery applications, with their hybrid structure exploiting advantageous properties of both linear and dendritic polymers. LDBCs have promising encapsulation efficiencies that can be used to encapsulate both hydrophobic and hydrophilic dyes for bioimaging, cancer therapeutics, and small biomolecules. Additionally, LDBCS can be readily functionalized with varying terminal groups for more efficient targeted delivery. However, depending on structural composition and surface properties, LDBCs also exhibit high dispersities (Đ), poor shelf-life, and potentially high cytotoxicity to non-target interfacing blood cells during intravenous drug delivery. Here, we show that choline carboxylic acid-based ionic liquids (ILs) electrostatically solvate LDBCs by direct dissolution and form stable and biocompatible IL-integrated LDBC nano-assemblies. These nano-assemblies are endowed with red blood cell-hitchhiking capabilities and show altered cellular uptake behavior ex vivo. When modified with choline and trans-2-hexenoic acid, IL-LDBC dispersity dropped by half compared to bare LDBCs, and showed a significant shift of the cationic surface charge towards neutrality. Proton nuclear magnetic resonance spectroscopy evidenced twice the total amount of IL on the LDBCs relative to an established IL-linear PLGA platform. Transmission electron microscopy suggested the formation of a nanoparticle surface coating, which acted as a protective agent against RBC hemolysis, reducing hemolysis from 73% (LDBC) to 25% (IL-LDBC). However, dramatically different uptake behavior of IL-LDBCs vs. IL-PLGA NPs in RAW 264.7 macrophage cells suggests a different conformational IL-NP surface assembly on the linear versus the linear-dendritic nanoparticles. These results suggest that by controlling the physical chemistry of polymer-IL interactions and assembly on the nanoscale, biological function can be tailored toward the development of more effective and more precisely targeted therapies.
Introduction
Linear-dendritic block copolymers (LDBCs) are unique materials that form architecture-dependent nanostructures with properties that change according to the weight percentage of hydrophobic to hydrophilic components.1–5 Their amphiphilic nature means that they can self-assemble when placed into particular solvent environments, such as water. LDBCs are promising materials relative to traditional linear block copolymers because the resulting nanoaggregates are known to have high mechanical and colloidal stability, and the hydrophilic dendrimer structure affords tunability to the surface, yielding multifunctional surface properties. In contrast, non-linear block co-polymer-based nanoassemblies, such as star-like and bottlebrush-like block-co-polymers, can be architecturally manipulated through various synthetic schemes and show high stability for stimuli-responsive release.6,7 However, importantly, LDBCs also maintain a high level of synthetic versatility, meaning that different shapes, sizes, and surface charges can be obtained by manipulating the chemical composition of the dendritic or linear polymer units prior to assembly.1,3,4 The driving forces for this assembly are noncovalent interactions—such as the hydrophobic effect—between the building blocks that induce segregation between polymer types. However, LDBC aggregation behavior does not always align with standard principles of polymer assembly. Aspects such as the carbon framework, molecular mobility, and environment can affect the assembly.8–12 In addition, differences in the molecular shape influence the packing parameter and correlate to the overall properties of the resulting structure, including surface viscosity, water permeability, and cellular uptake. Prior research has shown that the LDBCs can load hydrophobic and hydrophilic drugs and successfully deliver this cargo into cells.13 However, the clinical applications of LDBCs are significantly underexplored, relative to their more conventional dendritic, linear, and nonlinear block co-polymer (BCP) counterparts, because of the often-unpredictable nature of their molecular self-assembly into stable nanostructures.Ionic liquids (ILs) are composed of asymmetric, bulky cations and anions and are liquid below 100 °C.14 A host of favorable properties, including negligible vapor pressure, high thermal stability, and tunability, have resulted in the use of ILs in a wide variety of applications, including synthesis,15 catalysis,16 and electrochemical sensors.17 Even very minor structural changes of the IL constituents impact the interactions both between the charged components and any other materials present, including water. This results in measurably different bulk properties such as viscosity and conductivity as the structures are altered and water is added.18,19 In the presence of other materials, ILs can show complex long range ordering with their charged components that balance electrostatics, dispersion forces, and often hydrogen bonding.20 The formation of nano-environments can be precisely controlled by altering the structure of the IL components, which makes them attractive materials for use in systems where assembly occurs on the nanoscale.21
Because the asymmetric nature of the IL components leads them to form long-range nanoscopic structures in the presence of other materials, ILs can successfully stabilize and increase the monodispersity of a range of nanomaterials22 by ordering around the starting materials, including metals such as gold23 and platinum.17 Traditional amphiphilic linear block copolymers, composed of solvophilic poly(ethylene oxide) with either poly(ethyl glycidyl ether) or poly(glycidyl propyl ether) as the solvophobic component, were assembled in water and pure protic alkyl ammonium nitrate ILs.24 In water, the polymers form spheres, whereas in the IL, the polymers form disks with the curvature of the disks being dictated by the volume of the cation. Lodge and colleagues25 investigated the assembly of poly((1,2-butadiene)-block-ethylene oxide) (PB-PEO) polymers in neat [Bmim][PF6]. They found that the block copolymers readily assembled into morphologies dictated by the length of the PEO segment that were stable in temperatures of 25–100 °C.
When ILs are crafted from materials with negligible impact on mammalian cell and tissue physiology, they create so-called biocompatible ionic liquids (BILs),26–28 which show promise for clinical use or other related applications in biotechnology. Already, BILs have demonstrated great promise as drug delivery agents through skin,29,30 the buccal cavity,31 and the intestines,32 and were recently employed to coat linear block-copolymeric particles as a means of preventing protein adsorption after intravenous drug delivery.33
Here, we show that choline-carboxylic-acid-based ILs electrostatically solvate LDBCs by direct dissolution and form stable and biocompatible IL-integrated LDBC nanoassemblies. The IL functionalization resulted in nanoaggregates spontaneously hitchhiking onto RBCs and altered cellular uptake behavior ex vivo compared to their non-functionalized counterparts. By controlling the physical chemistry of polymer-IL interactions and assembly on the nanoscale, we can imbue polymeric materials with altered interactions with biological systems for more effective and targeted intravenous nanotherapies for drug delivery.
Experimental
Materials
Carboxylic acid-terminated poly(D,L-lactide-co-glycolide) (PLGA) [50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50] polymer was purchased from Sigma Aldrich (Resomer 504H, #719900-5G). Polycaprolactone polyaminoamide amine (PCL-PAMAM-NH3+) 50 kDa polymer was synthesized in-house as previously reported.13 Choline bicarbonate (80% in H2O, #C7519-500ML) and 98% trans-2-hexenoic acid (#W316903-1KG-K) were obtained from Sigma Aldrich. Ultrapure MilliQ water was obtained from a Millipore MilliQ purifier (#Milli-Q IQ 7000), and deuterium oxide was purchased from Sigma Aldrich (#435767-1KG). HPLC-Grade acetonitrile was obtained from Sigma Aldrich (#34851-4L). Tetramethylsilane (TMS) internal standard for NMR quantification was obtained from Sigma Aldrich (#87920-25ML). Amicon 4 mL 30 kDa MWCO ultracentrifugation filter tubes were purchased from Sigma Aldrich (Ultra-4, #UFC803096). A Thermo Scientific Sorvall Ultracentrifuge (Model #ST8R) was used to filter ionic liquid-polymer nanoparticle samples post-synthesis. Sub-450 nm filters (Titan-3) and sub-100 nm filters (Millipore, Millex-VV, #SLVVR33RS) were purchased from Sigma Aldrich. Pooled-gender BALB/c mouse blood was acquired from Bio-IVT (NY, USA). 96-well clear plates (Corning, #353075) and 96-well opaque black-bottom COSTAR plates were obtained from Corning. DiD far-red dye was obtained from Sigma Aldrich (Life Technologies, DiD’ solid, #D7757), and HPLC-grade rhodamine B dye was obtained from Sigma Aldrich (#R6626-100G).
:50] polymer was purchased from Sigma Aldrich (Resomer 504H, #719900-5G). Polycaprolactone polyaminoamide amine (PCL-PAMAM-NH3+) 50 kDa polymer was synthesized in-house as previously reported.13 Choline bicarbonate (80% in H2O, #C7519-500ML) and 98% trans-2-hexenoic acid (#W316903-1KG-K) were obtained from Sigma Aldrich. Ultrapure MilliQ water was obtained from a Millipore MilliQ purifier (#Milli-Q IQ 7000), and deuterium oxide was purchased from Sigma Aldrich (#435767-1KG). HPLC-Grade acetonitrile was obtained from Sigma Aldrich (#34851-4L). Tetramethylsilane (TMS) internal standard for NMR quantification was obtained from Sigma Aldrich (#87920-25ML). Amicon 4 mL 30 kDa MWCO ultracentrifugation filter tubes were purchased from Sigma Aldrich (Ultra-4, #UFC803096). A Thermo Scientific Sorvall Ultracentrifuge (Model #ST8R) was used to filter ionic liquid-polymer nanoparticle samples post-synthesis. Sub-450 nm filters (Titan-3) and sub-100 nm filters (Millipore, Millex-VV, #SLVVR33RS) were purchased from Sigma Aldrich. Pooled-gender BALB/c mouse blood was acquired from Bio-IVT (NY, USA). 96-well clear plates (Corning, #353075) and 96-well opaque black-bottom COSTAR plates were obtained from Corning. DiD far-red dye was obtained from Sigma Aldrich (Life Technologies, DiD’ solid, #D7757), and HPLC-grade rhodamine B dye was obtained from Sigma Aldrich (#R6626-100G).
Equipment
Dynamic light scattering characterization was done on a Zetasizer Pro (Model #MAL1252117, Malvern, UK). Flow cytometry studies were performed on an acoustic instrument with special gating for red blood cell population scatter vs. far-red fluorescence (APC) (Model #AFC2). Fluorescence and absorbance endpoint studies as well as cytotoxicity assessments were performed on a UV-Vis/fluorescent plate reader (Biotek H1 Synergy Hybrid Multi-mode). Live cell imaging was carried out with a white-laser equipped STED-stellaris microscope with a temperature stage.Methods
:1 molar ionic ratio was synthesized as previously published.34 Briefly, choline bicarbonate was combined with trans-2-hexenoic acid in a 500 mL round bottom flask in an oil bath at 40 °C and allowed to stir overnight. The next day, the ionic liquid was placed onto a rotary evaporator for 2 h at 60 °C and 15 mbar, and dried in a vacuum oven for 48 h at 60 °C to remove residual water and carbon dioxide. The final ionic liquid product was a viscous, light-yellow liquid. (94.9% yield, 2.9% (w/w) water content via Karl Fischer titration). 1H NMR spectroscopy (400 MHz, DMSO) δ 6.27 (dt, J = 15.4, 7.0 Hz, 1H), 5.62 (dd, J = 15.4, 1.6 Hz, 1H), 3.85 (dq, J = 8.0, 2.7 Hz, 2H), 3.49–3.42 (m, 2H), 3.15 (s, 9H), 1.99 (qd, J = 7.1, 1.5 Hz, 2H), 1.36 (p, J = 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H).
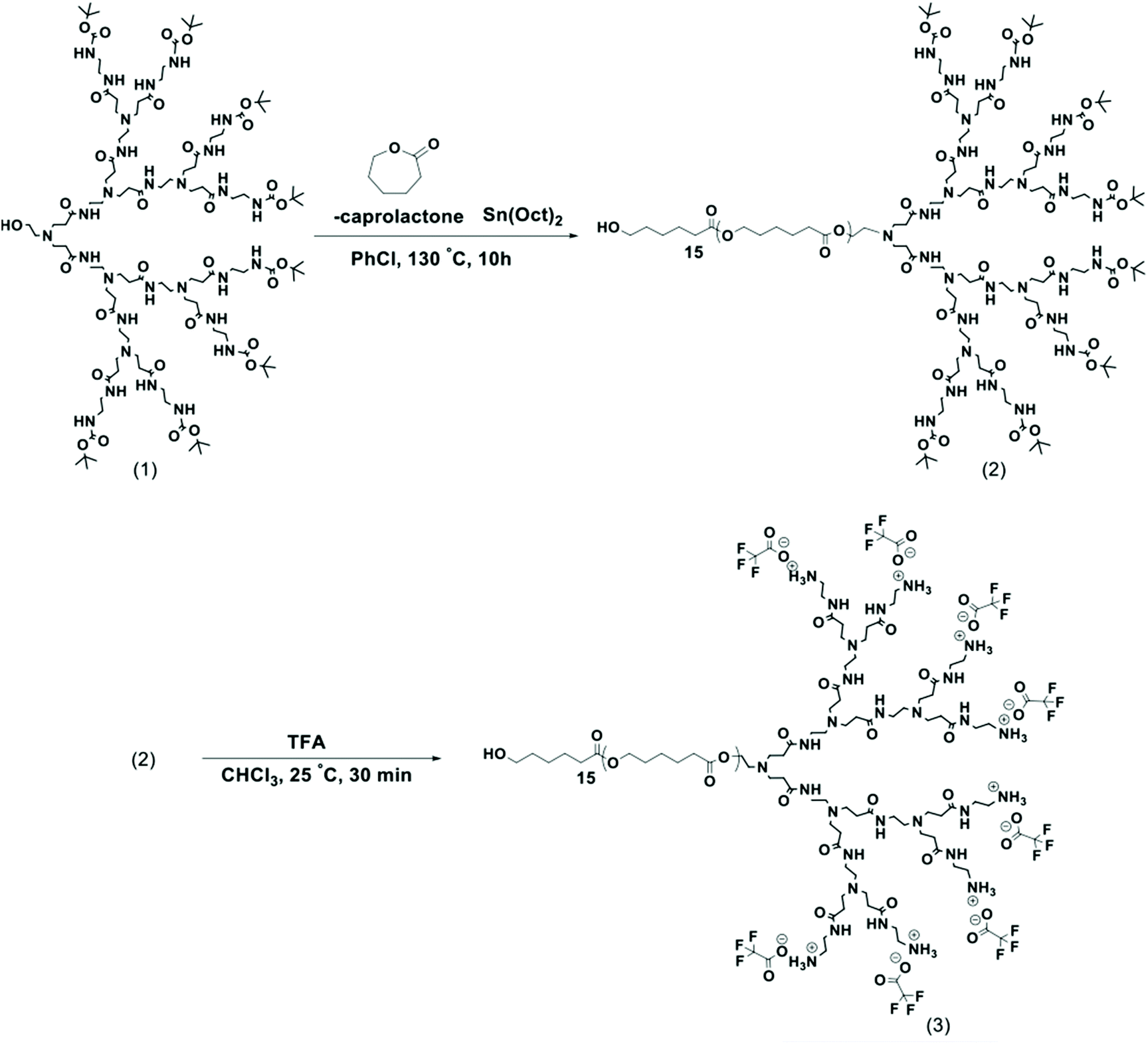 | ||
| Scheme 1 General synthetic route to LDBC by ring-opening polymerization of ε-caprolactone monomer initiated by PAMAM-G3-Boc macroinitiator (1) to yield 50-PCL-PAMAM-Boc (2), which is then deprotected to afford the final amphiphilic LDBC 50-PCL-PAMAM-NH3 + (3) with the 15 units of PCL (number of repeating units of CL = 15).13 | ||
:1 IL mg−1 PLGA was added to the center of the stirring vortex in the form of a single drop (1 liquid drop from a 1000 uL pipette tip set at 1000 uL), and 1 h later, the stir rate was brought up to 900 rpm. The nanoparticles were stirred at 900 rpm for 1 more hour in the dark (for 2 total hours stirring post IL-addition) at 25 °C to allow electrostatic-driven molecular assembly of the cation and anion with the PLGA NP COO– functionalized surface in an aqueous environment.
After 2 h of magnetic stirring post IL-addition, 30 kDa MWCO ultracentrifugation was performed at 2500 rpm for 50 min at 4 °C to ensure any unmodified ionic liquid and dye dialyzed out of solution, and then the filtered nanoparticles were brought back up to a final concentration of 1 mg mL−1 with either MilliQ water, D2O, or 1× PBS (pH 7.4) depending on application, and stored in the dark at 4 °C until further use. Approximate shelf life for both PLGA and IL-PLGA nanoparticles was determined to have an optimal biological use up to 2 weeks at 4 °C, and a total shelf life up to 3.5 weeks at 4 °C. 1H NMR spectroscopy (400 MHz, D2O) δ 6.79–6.67 (m, 2H), 5.88 (dd, J = 15.5, 2.0 Hz, 2H), 4.79 (s, 38H), 4.11 (dq, J = 5.5, 2.8 Hz, 4H), 3.83 (dt, J = 6.9, 4.6 Hz, 1H), 3.76–3.69 (m, 2H), 3.65–3.56 (m, 6H), 3.26 (s, 22H), 2.14 (d, J = 1.9 Hz, 388H), 1.53 (q, J = 7.4 Hz, 5H), 0.97 (t, J = 7.4 Hz, 7H), 0.16–0.00 (TMS, 12H).
:50], synthesized as described above, was massed by analytical balance in a timely manner to avoid absorption of water, and combined with 50 μL of IL, which was added drop-by-drop until all the ionic liquid was in the vial and submerging the polymer. To scale-up the concentration of LDBCs from 1 mg mL−1 to 10 mg mL−1, IL was added at 50 μL IL mg−1 polymer. To the same glass scintillation vial containing IL and LDBC, 2% far-red DiD dye (bio-tracking in blood) or 4% rhodamine dye (for confocal cell work) was added by mass of LDBCs (% wt/wt).
For IL-LDBC formulations, dye aliquots were taken from 1 mg mL−1 freshly prepared stock solutions of DiD or Rhodamine in HPLC-grade ACN. Importantly, these specific formulations were experimentally found to be optimal as they allowed the dye to be solvated or ‘protected’ by the IL while the dye underwent direct dissolution with the LDBCs and allowed for ready evaporation of the ACN during sonication and vortex/sonication-hybrid nanoprecipitation. The specific dye percentage (wt/wt) was selected to be 2% (DiD), which was found to allow high encapsulation of the dye while maintaining core size under 100 nm, allowing for additional surface modification of the IL and for the particles to remain under 200 nm for IV-formulation. Rhodamine is approximately half of the molecular weight of DiD, thus formulation at 4% allowed for equivalent NP sizes.
The scintillation vial containing IL, LDBC, and dye was then vortexed on the highest setting for 6 minutes total, in 2-minute intervals. The solution was collected after each interval in order to resubmerge the polymer. Then, the solution was sonicated to solvation for 5 h, kept between a temperature range of 35–40° C (to prevent further temperature escalation and potential dye degradation, ice cubes were periodically added to the water bath over the 5-hour period). Direct dissolution was evidenced by a bright blue initial phase to dark emerald green colored product after filtration.
After sonication was complete to form a homogenous phase of IL, dye, and LDBCs, 2 mL of Milli-Q water (or D2O) was added dropwise (1 drop per second) using a 1 mL glass Pasteur pipette while vortexing the scintillation vial at the highest setting. Without capping the vial, vortexing continued for an additional 5 min at the highest setting. After vortexing, the scintillation vial was capped and wrapped with parafilm and then sonicated for an additional 30 min without heat before being transferred into a 4 mL Amicon 10 kDa MWCO centrifuge filter (Millipore, 4 mL, #UFC801024) and centrifuged at 2500 rpm for 50 min at 4 °C. After filtering, the deep-emerald-colored filtrate was gently reconstituted from the filter and transferred into a 1.5 mL centrifuge tube and brought up to 1 mL (final concentration 1 mg mL−1) by adding Milli-Q, D2O, 1× DPBS (cell-grade, no magnesium or calcium), or 1× PBS pH 7.4 depending on application. IL-LDBCs were then stored in the dark at 4 °C for up to 3 weeks, where optimal biological use was determined to be within 2 weeks at 4 °C by Dynamic Light Scattering (DLS), with 3 weeks as the shelf-life maximum in buffer [ESI Fig. 1†].
Once the organic phase was added, solvent evaporation took place over 2 h via sonication at 25 °C in the dark. After sonication, the scintillation vial containing LDBCs was capped, wrapped with parafilm and aluminum foil, and stored under ambient conditions overnight in the dark at 25 °C. LDBCs were then gently filtered using sub-450 nm filters with a 1 mL-syringe adaptor and brought to 1 mg mL−1 final concentration with application buffers as previously. For final scale-up concentrations of 10 mg mL−1, 10 kDa MWCO centrifuge filtration took place at the same settings above (2500 rpm for 50 min), but at 25 °C. Unlike the PLGA, IL-PLGA, or IL-LDBC NPs, bare LDBCs were stored in a parafilm-wrapped glass scintillation vial at 25 °C in the dark in a dry place up to 7 days, as storage at 4 °C destroyed native LDBC composition. To maintain consistency across the different shelf life of all four treatments, PLGA, IL-PLGA, LDBC, and IL-LDBCs were synthesized at the same time and used in tandem within the first 72 hours of synthesis for biological experiments. 1H NMR spectroscopy (400 MHz, D2O) δ 6.77–6.66 (m, 5H), 5.86 (d, J = 15.6 Hz, 6H), 4.11 (s, 10H), 3.82 (s, 4H), 3.73–3.66 (m, 8H), 3.64–3.56 (m, 14H), 3.25 (s, 41H), 2.36 (s, 1H), 2.20 (d, J = 7.4 Hz, 10H), 1.94 (s, 1H), 1.68 (s, 2H), 1.51 (q, J = 7.5 Hz, 10H), 0.95 (t, J = 7.7 Hz, 14H), 0.00 (TMS, 12H).
:50 dilution from originally concentrated fraction from 250 μL of K2-EDTA treated whole blood) at a 1:10 (v/v) ratio in quadruplicate in a 96 clear-well COSTAR plate (final volume 200 μL per well). The samples were then incubated for 1 h at 37 °C and then centrifuged at 4 °C for 10 min at 500g. 100 μL of supernatant was collected to measure peak hemolytic absorbance in quadruplicate at 405 nm with a UV-Vis/fluorescent plate-reader. 20% Triton X-100 and 1× PBS pH 7.4 were used as positive and negative internal controls at the same treatment dilution. The 1× PBS negative control was subtracted as a minimum baseline (0%) from all samples, and the Triton-X-100 positive control was used as a maximum baseline (100%) to calculate normalized hemolytic percentages with standard error of mean (n = 4). A two-tail t-test of means was used to determine significance between any two samples.33,35
:10 (v/v) ratio, roughly equivalent to [100:1] NP:RBC. All samples were then simultaneously removed from ice and mechanically inverted by hand up to 1 min at room temperature before being transferred to an incubator to shake at 500 rpm for 20 min at 37 °C. Afterward, all samples were centrifuged for 10 min at 1000g and 4 °C to isolate and separate serum and red blood cell fractions. Serum was washed once at 1000g for 10 min at 4 °C to remove any residual white blood cell fraction that did not pellet. Then, each red blood cell fraction was centrifuged at 200g at 4 °C for 5 min, and the supernatant was removed and replaced to original volume with 1× PBS pH 7.4. After three such washes, each isolated fraction was brought up to 1 mL with 1× PBS, and a 200 μL aliquot of each sample was removed and brought to 2 mL with 1× PBS for fluorescence-activated cell sorting (FACS). For FACS measurements, desired cell populations were gated via forward vs side scattering to carefully isolate out red blood singlets vs. doublets/debris, which were then plotted against far-red fluorescent encapsulated dye (SSC-A vs. RL1-A) to understand the relative amounts of NPs that hitchhiked for IL- vs. non-IL-functionalized NPs. The lowest flow rate (12.5 μL min−1) was used. FACS was completed within 2 h of the conclusion of the washing process. For fluorescent quantification on a plate reader, each final pellet/serum fraction instead was brought up to 200 μL final volume with 1× PBS and the entire fraction was read by fluorescent plate reader relative to NP controls and a corresponding calibration curve (excitation 640 nm, emission 670 nm). A two-tail t-test of means was again used to determine significance between any two samples, while a one-way ANOVA was used to determine significance across samples of 3 or more.
Results and discussion
In order to establish the impact of the presence of choline 2-hexenoate IL on the assembly of both linear and LDBC nanoaggregates, the aggregates were prepared in the presence and absence of IL as described in detail in the Experimental section.The assembly of the commercially available linear PLGA (50:50) was investigated first. The nanoparticles were synthesized by nanoprecipitation from acetonitrile, with IL addition to the already-formed nanoparticles. As shown in Fig. 1 dynamic light scattering (DLS) (n = 5) suggested that modification of the PLGA NP surface with IL increased its average size (from 65 ± 2 nm to 149 ± 2 nm) and narrowed the distribution significantly (dispersity 0.21 to 0.13). In addition, the change in surface charge post-modification (zeta potential −20 ± 3 mV to −60 ± 4 mV) indicates that the IL undergoes electrostatic self-assembly on the surface such that the outermost layer that would interface with a biological environment is the anionic carboxylic acid, consistent with previously published results.33
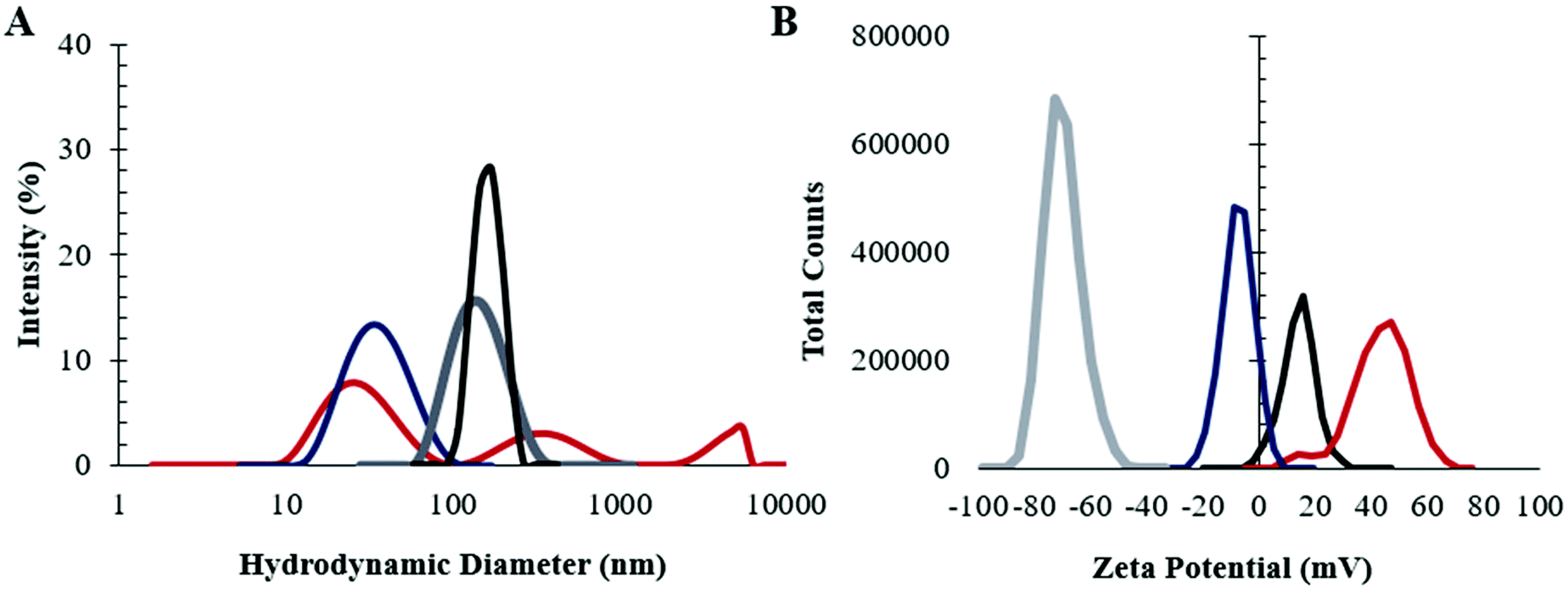 | ||
| Fig. 1 Both the size and surface charge of the nano-assemblies change with IL modification. Dynamic light scattering (DLS) measurements of the (A) nanoparticle diameter (nm) and (B) surface charge (mV) of red: LDBCs; black: IL-LDBCs, blue: PLGA NPs, grey: IL-PLGA NPs. All NPs contain DiD far-red fluorescent dye. The size distribution narrows upon IL modification of LDBCs and is comparable to IL-modified PLGA NPs. | ||
Next, LDBCs synthesized in-house with a PCL/PAMAM ratio of 50% and a terminal amine were prepared by nanoemulsion. In the absence of IL, LDBC NPs were observed to be highly disperse (reduced second cumulant of 0.47), with a large population under 100 nm (hydrodynamic diameter 69 ± 7 nm), and were found to have a positive surface charge (33 ± 4 mV), consistent with its amine-terminated functionality. When modified with choline 2-hexenoate IL, the LDBCs were larger, with a diameter of 171 ± 7 nm and a reduced dispersity (0.23), roughly half that of the unmodified LDBCs. The surface charge also showed a shift of two-fold towards neutrality (15 ± 4 mV). Additionally, formulation of the LDBCs with IL extended its shelf-life at 4 °C from 1 week to 3 weeks (Fig. S1†). Results from DLS thus suggest three different aspects to the nanoformulation. First, the IL is able to tune and control physicochemical properties, such as stability, dispersity, size, and surface charge of the NPs. Second, the larger size increase in the case of the LDBC formulation indicates that there may be a larger quantity of ionic liquid nanoformulation compared to IL-PLGA NPs. Third, it appears, based on the less negative surface charge of the IL-LDBCs compared to their linear counterparts, that the proportions of cation and anion throughout the outermost interfacing layer of the IL coating differ on the linear PLGA compared to the hybrid LDBCs.
As the DLS data suggested two distinct IL-integrated nanoassemblies, we explored the chemical nature of these modifications by proton NMR spectroscopy (1H NMR). LDBCs, IL-LDBCs, PLGA, and IL-PLGA NPs were separately formulated in D2O, with TMS as an internal quantification standard (Fig. 2). Note that in the case of the assembled LDBCs, only the peaks corresponding to the hydrophilic PAMAM are visible, because the hydrophobic PCL is inside the core of the nanoassembly (Fig. 2c). We observed that relative to PLGA NPs, IL-PLGA NP retained all IL proton peaks (Fig. 2b and d), however they are present at a 0.8:1 cation/anion ratio (trans-2-hexenoic 1.53 (q, J = 7.4 Hz, 5H), vs. choline 4.11 (dq, J = 5.5, 2.8 Hz, 4H)). This aligns with the strong anionic presence on the external interfacing layer of the IL-PLGA coating. However, interestingly, the protons closest to the C2 trans-double bond are masked by the PLGA or not present (peak C in Fig. 2c), which may point to a unique conformational assembly of the anionic alkyl chain in the outer layer of the IL-PLGA NP. In direct contrast, IL-LDBCs assemble with an exact 1:1 cation/anion ratio (choline: 4.11 (s, 10H), trans-2-hexenoic acid: 1.51 (q, J = 7.5 Hz, 10H)). This is consistent with the notion that both anion and cation participate equally in the IL-LDBC coating. Additionally, TMS quantification suggests that relative to IL-PLGA nanoassemblies, IL-LDBCs form with approximately double the amount of IL, which is consistent with the relative increase in IL-LDBC size. However, both IL-PLGA and IL-LDBC formulations revealed new small multiplet peaks around the 2H choline peak 4 (Fig. 2b and d), potentially suggesting altered solvation of the polymer at the IL/polymer interface.
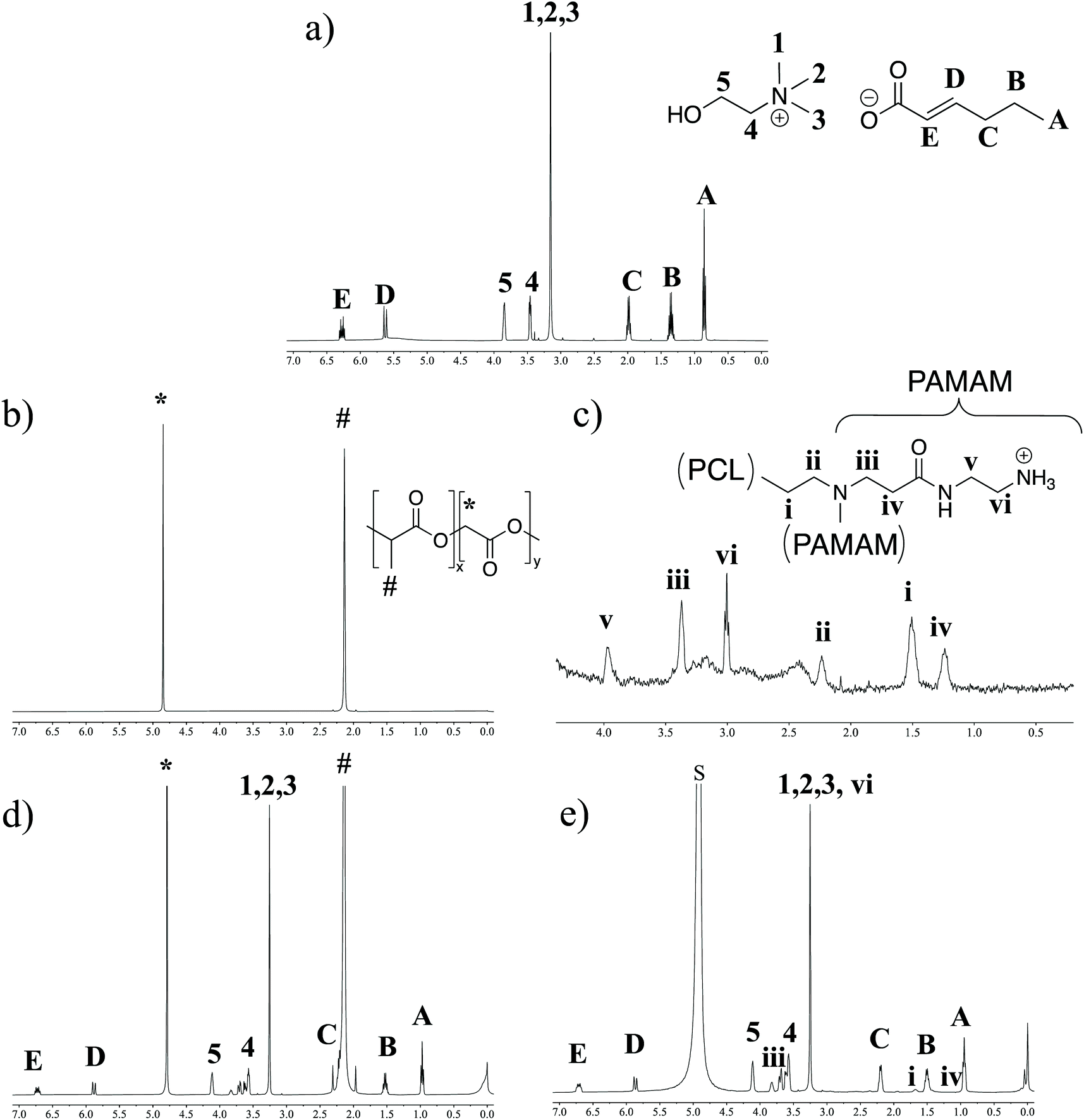 | ||
| Fig. 2
1H-NMR shows different chemical composition of the IL layering in PLGA NPs compared to LDBCs. (a) 1H NMR of choline and trans-2-hexenoic acid [1:1]. (b) PLGA NPs, (c) LDBCs, (d) IL-PLGA NPs, and (e) IL-LDBCs. (d) and (e) include 25 μL [TMS]/700 μL [NPs in D2O] for IL quantification. | ||
In order to understand the morphology of the IL modification at the interface of the polymer, we performed two different sets of Transmission Electron Microscopy (TEM) imaging: the first involving room temperature imaging to distinguish the IL-LDBC core and coating composition, as well as cryo-TEM to analyze and compare the nature of IL-PLGA NP morphology and composition. Because the IL-PLGA samples were destroyed by the harsh conditions and electron beam of traditional TEM, cryo-TEM sample preparation and imaging allowed for undistorted analysis of compositional morphology, although with smaller size restrictions, and as such, only a series of small nanoparticles were imaged. As seen in Fig. 3A and B, cryo-TEM revealed two distinct compositional differences in morphology between PLGA and IL-PLGA NPs. The IL-PLGA NPs showed a distinct core–shell composition, in which the PLGA cores show a slight distortion from their original spherical morphology, possibly due to the physical impact from the anion and cation rearrangement in aqueous solution and electrostatically-driven self-assembly onto the PLGA NP surfaces. This may have created a new hybrid material at the interface observed by the new IL peaks around Peak 4 in Fig. 2B and D, where it is possible the first IL cationic layer interfaced with terminal PLGA carboxylic acid termination.
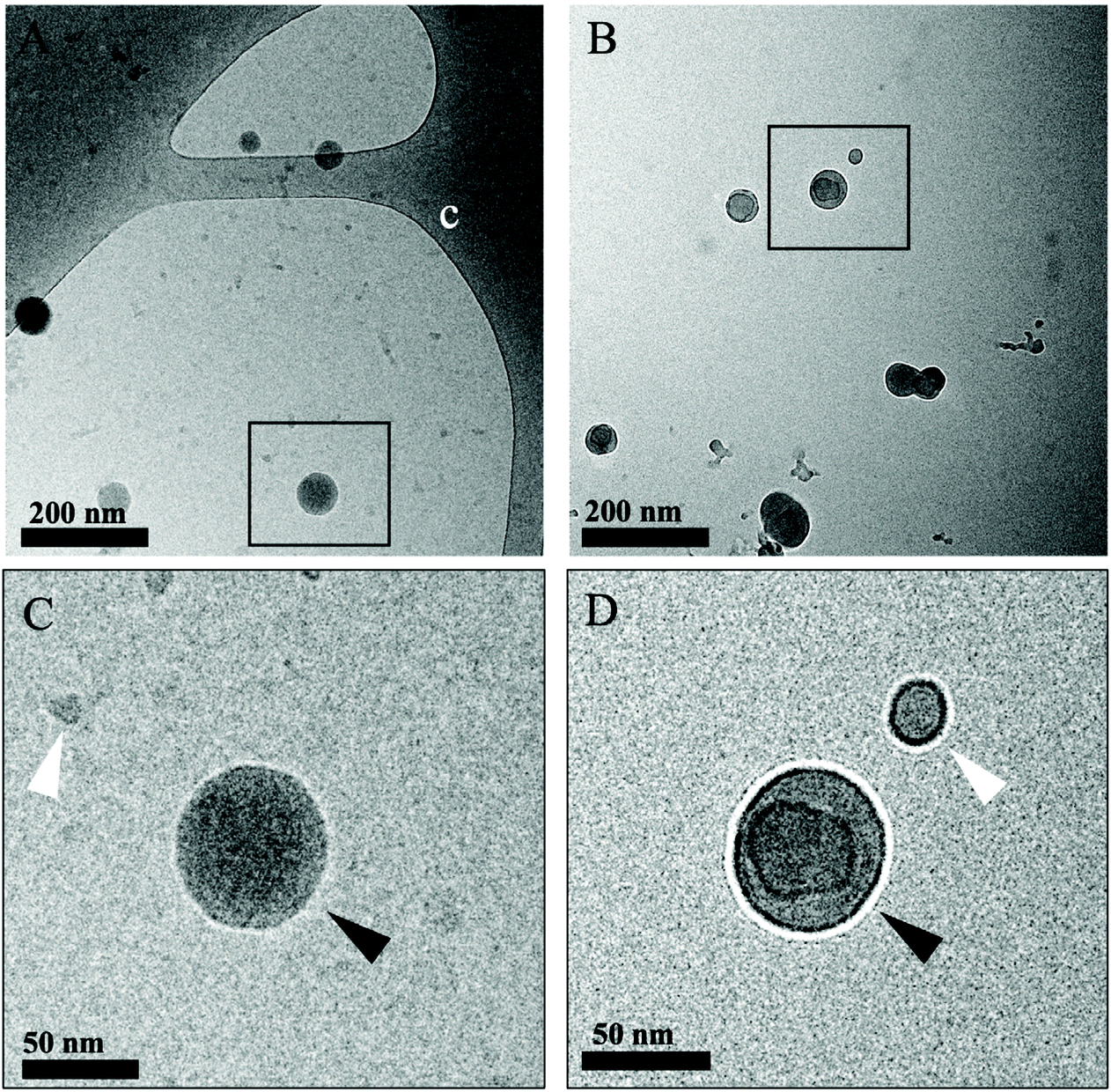 | ||
| Fig. 3 Cryo-TEM of PLGA (A and C) and IL-PLGA nanoparticles (B and D) show core shell morphology. C and D show zoomed-in regions marked by black rectangles in A and B, respectively. c in A denotes holey carbon support film. White arrowhead in C points to a ∼10 nm PLGA nanoparticle and black arrowhead points to a ∼60 nm particle, representative of a smaller and larger PLGA nanoparticle population. Black arrowhead in D shows a 50 nm PLGA particle surrounded by an ionic liquid shell, observed by a distinct contrast between the PLGA core and the ionic liquid shell. White arrowhead points to a 20 nm ionic liquid nanodroplet. | ||
Complementing Fig. 3, Fig. 4 below shows distinct morphological differences between LDBC and IL-LDBCs captured by room temperature TEM. Note that these samples were stained with uranyl acetate prior to imaging unlike the cryo-TEM samples, which were not stained, altering the relative contrast of each image. Because of the larger size of the LDBC aggregates compared with PLGA, cryo-TEM was not feasible. Upon assembly with IL, the LDBC NP hydrodynamic diameter was observed to increase to just under 200 nm as confirmed by DLS, as well as increasing in spatial dispersion, likely due to the electrostatic coating repelling each IL-LDBC nanoparticle away from each other (Fig. 4B). In contrast, unmodified LDBCs are observed to be much smaller in size and disperse in their size and assembly as they demonstrate a tendency to aggregate (Fig. 4A). When zoomed in on their structural differences, the LDBC assembly has two features: an initiator core (blue arrow) and an outer assembly (purple arrow) (Fig. 4C), while IL-LDBC NPs have a similar core structure (yellow arrow) with a dense coating of ionic liquid integrated into the LDBC structure all around it (red arrow), shown by the dark grey halo of contrast surrounding the lighter core. However, there is no visible difference in core morphology, further suggesting IL-PLGA and IL-LDBCs may assemble with different conformations but still confer similar biofunctionalization.
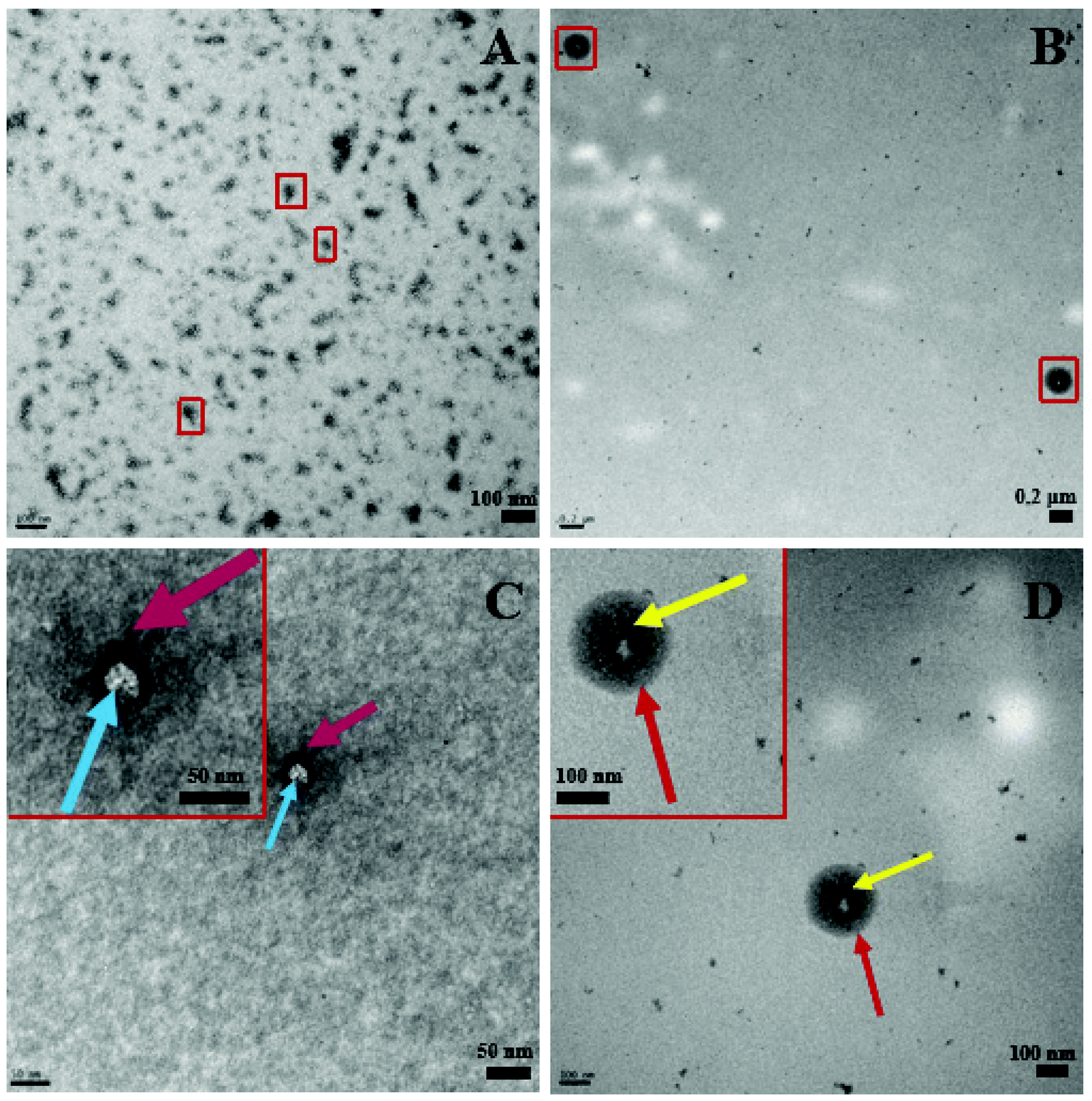 | ||
| Fig. 4 Room temperature TEM of LDBC (A and C) and IL-LDBC nanoparticles (B and D) show different morphologies. C and D show zoomed-in regions of nanoparticles shown in A and B, respectively. 2% UA negative stain shows the distinction between the polymer bulk and IL surface features. In C, the white LDBC core is marked by a blue arrow, and dark grey outwards PAMAM assembly is marked by the purple arrow. In D, IL-LDBC, similar LDBC morphology is shown as in C (yellow arrow), with a distinct dense IL coating built in around the LDBC surface (red arrow) indicated by the grey (IL, red arrow) vs. black (PAMAM, yellow arrow) contrast. When modified, IL-LDBCs are observed to have a larger size and higher dispersion (B) and less tendency to aggregate (A). | ||
To examine this hypothesis, IL-LDBC were examined for the extent of their intravenous biocompatibility compared with IL-PLGA NPs, and specifically, whether the IL assembled in a similar conformation on the outermost interfacing layer on both polymers to confer resistance to red blood cell (RBC) toxicity, once intravenously (IV) injected. IL-LDBCs conferred a three-fold reduction in hemolysis compared to LDBCs alone when tested in quadruplicate ex vivo against isolated BALB/c RBCs (Fig. 5). While PLGA NPs are not known to have significant hemolytic effects as they have been proven to be FDA-certified for in vivo biocompatibility, modification with IL caused IL-PLGA NPs to have a higher hemolysis rate, although still maintained well under 10%, a biocompatible level in accordance with previously published IL-PLGA NP values (<10%).33
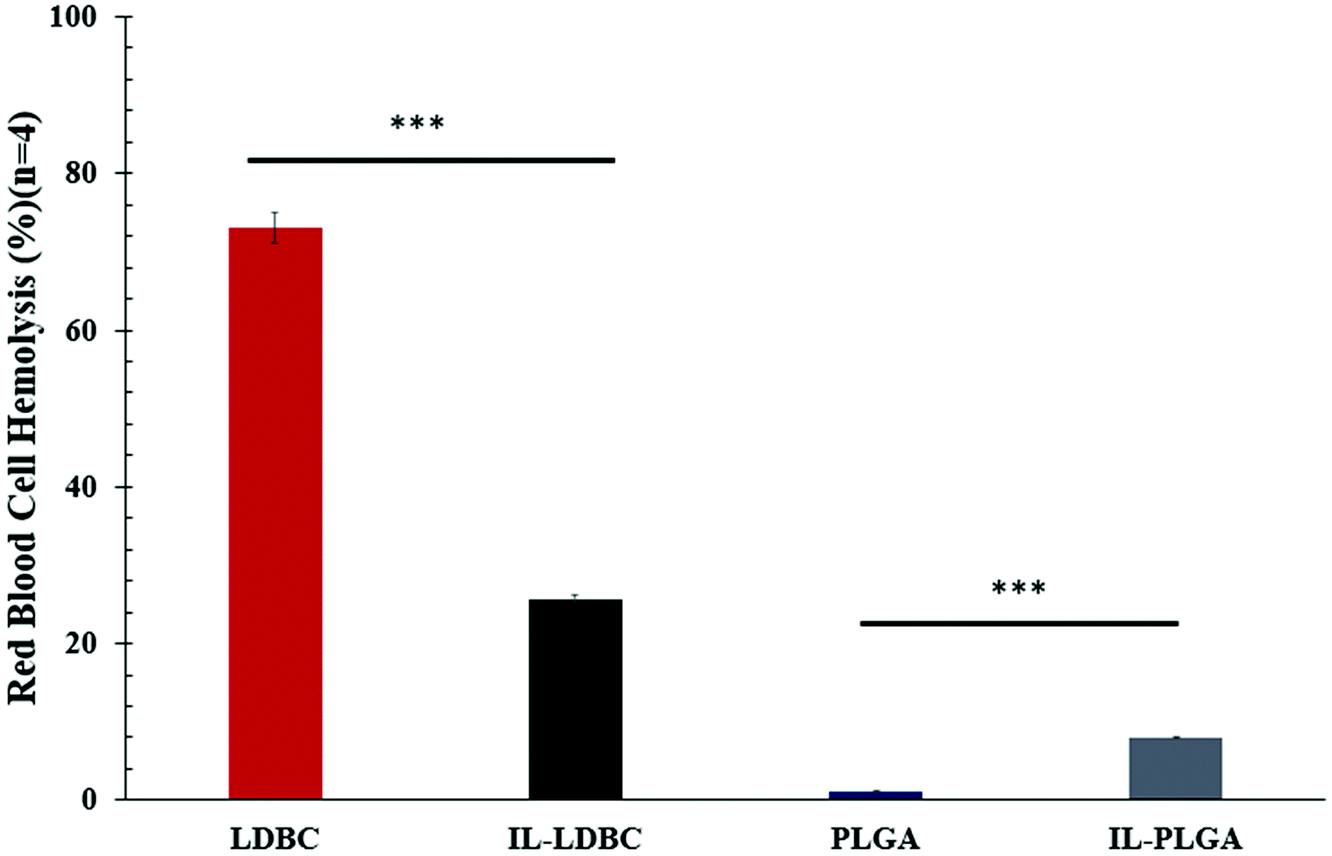 | ||
| Fig. 5 IL coating reduces the hemolytic nature of LDBCs. Average % red blood cell (RBC) hemolysis in response to NPs ex vivo with SEM (n = 4). *** denotes p < 0.001. Coating the LDBCs with CA2HA 1:1 significantly reduces hemolysis on RBC membranes roughly 3-fold, suggesting great compatibility with an intravenous-route of administration. IL-PLGA NPs remain under 10% hemolysis, which is acceptable for intravenous administration. | ||
Fluorescence-activated cell sorting (FACS) was used to qualitatively evaluate whether the IL was able to confer similar RBC “hitchhiking”36 properties to the LDBCs as previously seen in IL-PLGA NPs.33 Whole BALB/c mouse blood was mixed and incubated with NPs (1 mg mL−1 in 1× PBS pH 7.4) at a (1:10) (v/v) treatment dosage. Following this incubation, RBCs were isolated and washed three times in PBS prior to analysis. It was found that relative to PBS controls (Fig. 6A and D), bare LDBCs (Fig. 6B) had some native affinity for the RBC membrane, likely due to its amine (cationic) end groups. Bare PLGA NPs (Fig. 6E) showed no selectivity towards RBCs. In comparison, IL-LDBCs (Fig. 6C) conferred a dramatically higher level of affinity for RBC-binding while maintaining a lower level of hemolysis. This suggests that the assembly of the IL on the LDBC surface exposed similar structural moieties in the outermost coating to the IL-PLGA. Similar assembly of the anion on the outermost layer of the coating would then enable similar interactions to those observed between the IL-PLGA NPs (Fig. 6F and G) and RBCs in whole blood.
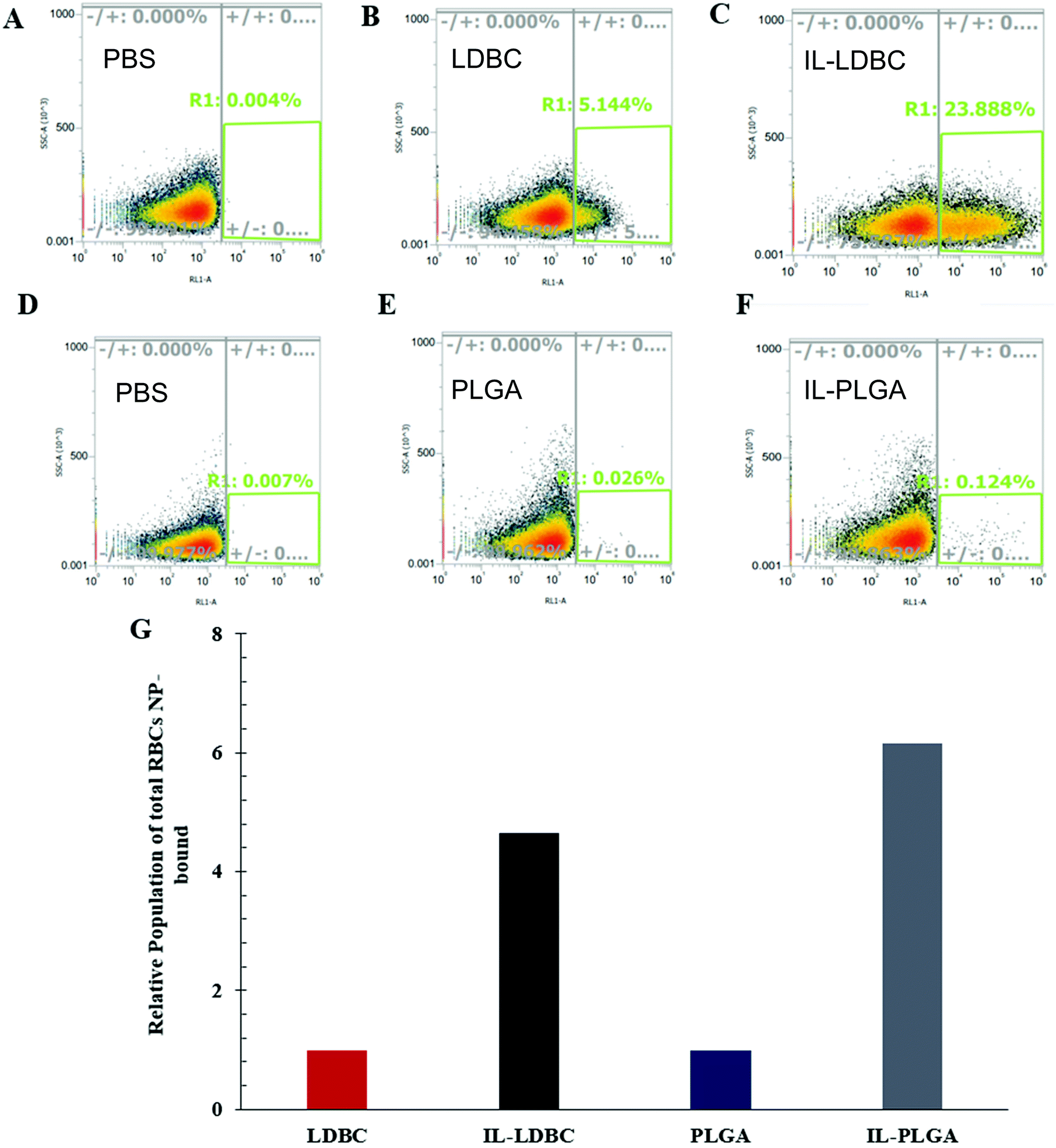 | ||
| Fig. 6 ILs confer hitchhiking capabilities onto both LDBCs and PLGA NPs. Live-cell FACS (A–C: PBS, LDBC, IL-LDBC; D–F: PBS, PLGA, IL-PLGA) and relative population (G) of incubated DiD-NPs qualitatively bound to red blood cells isolated from whole BALB/c mouse whole blood ex vivo. Data is represented as a relative increase of IL-LDBC and IL-PLGA to LDBC and PLGA NP normalized baseline values (baseline = 1), respectively. | ||
It is important to acknowledge that the process of membrane rupture may disturb hitchhiking events on RBCs, causing fluorescent NPs to be washed away. To the best of our knowledge, while there may be a small percentage of dead cells in Fig. 6, this is accounted for in our hemolysis assay showing biocompatibility, and, when held relative to a background subtraction of PBS and baseline of bare polymer NP, is negligible when considering the overall effect of hitchhiking in whole blood. Next, to quantify the degree of RBC hitchhiking in whole blood, whole blood treatment and isolation experiments were independently repeated in triplicate and analyzed via a fluorescent plate reader (Fig. 7). Surprisingly, it was discovered that IL-LDBC and IL-PLGA indeed spontaneously hitchhiked on to RBCs with comparative abilities (two-tailed t-test, p = 0.47), and respectively, at a significantly higher level than either LDBC (p = 0.00052) or PLGA NPs alone (p = 0.0062). The bare assemblies were primarily detected in the serum, with LDBCs demonstrating slightly higher RBC affinity, verifying the FACS screening discussed above. In contrast, IL-PLGA NPs were detected significantly less in serum than PLGA NPs (p = 0.0057).
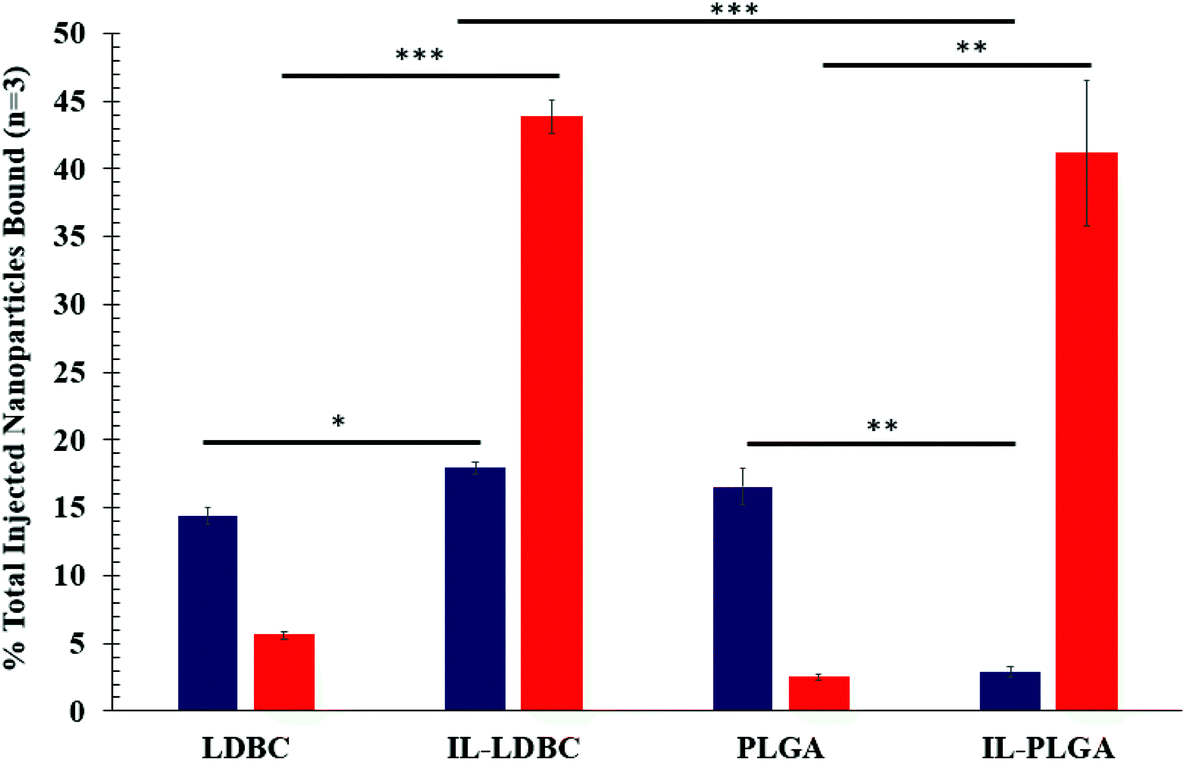 | ||
| Fig. 7 ILs confer the same relative hitchhiking abilities across both polymers but lose the ability to protect the NP from serum in the case of the LDBCs. Average % injected DiD-NPs in whole blood ex vivo bound to serum (navy) or red blood cells (red) with SEM (n = 3).*** is denoted as p < 0.001. ** = p < 0.01. * = p < 0.05. | ||
Interestingly, LDBC, IL-LDBC, and PLGA NPs did not have statistically comparable levels of detection in serum (one-way ANOVA: p = 0.0093; F-statistic value = 11.2). IL-LDBCs were also detected in the serum at a significantly higher rate compared to both LDBCs (p = 0.025) and the IL-PLGA NPs (p = 0.00082), but were at a comparable level to PLGA NPs (p = 0.24), which accumulated in serum in a statistically similar manner to LDBC NPs (p = 0.067). There are two possible interpretations for these results. First, IL-LDBCs were found qualitatively to bind RBCs less than IL-PLGA NPs by FACS when considered as a percentage of the RBC fraction itself. However, IL-LDBCs were quantitatively found equivalent to IL-PLGA NPs as a % injected dose (% ID). It is unlikely that IL-LDBCs followed the same pathway as the bare NP controls in serum (i.e., opsonization) due to their selective RBC hitchhiking behavior (and resistance to macrophage uptake, discussed later), as well as higher statistical significance in serum detection. Furthermore, it is possible that IL-PLGA NPs simply made more initial contact because of their size, and NPs without the strongest chemical affinity were washed away, explaining the lower percent ID in serum. The second explanation relies on the notion that the mechanism of RBC affinity is driven by differences in structural conformational assembly and serum protein-resistance. Specifically, the IL assembles in such a way on PLGA NP surfaces that the anionic unsaturated conformation on the interfacing layer is able to enforce serum protein-phobic properties long enough to maximize its affinity for RBCs (with any weakly bound NPs washed away from the RBC fractions), while the difference in the IL's conformational assembly on LDBCs still exposes moieties that drive strong RBC affinity, while masking others that determine protein phobicity, as shown in Scheme 2.33
 | ||
| Scheme 2 Chemical differences in the conformational assembly of choline hexenoate IL drives its selectivity to red blood cell membranes in whole mouse blood. Pictured: IL-PLGA NPs (right) have a “layered” coating of choline (green) and trans-2-hexenoic acid (blue). IL-PLGA NPs have a demonstrated higher selectivity for RBC hitchhiking as dominance of anion on the outermost interfacing layer drives protein resistance via its kinked trans-unsaturated chain (blue). In contrast, IL-LDBCs (left) have a “grafted” neutralized coating (yellow) with equal participation of anion and cation, driving its affinity to RBC membranes (via its carboxylic acid head and alkyl chain tail) while its ability to repel serum proteins is reduced due to masking of the double bond, likely due to its re-coordinated orientation with choline. | ||
In order to evaluate the impact of IL functionalization on cellular interactions, rhodamine B was encapsulated inside LDBC, IL-LDBC, PLGA, and IL-PLGA NPs, and each nanoassembly was evaluated for cell cytotoxicity and uptake differences in RAW 264.7 macrophages. No cytotoxicity was found at concentrations up to 50 μg mL−1 for any of the four NPs (Fig. 8A), but there were substantial differences among all four NP types in cellular uptake (Fig. 8B). Two feasible mechanisms that could explain the observed differences in cellular uptake are a charge-driven or a lipid-driven mechanism. For the charge-driven mechanism, PLGA NPs were determined to have a surface charge of approximately −20 mV due to the carboxylic acid functionalization on the surface, inhibiting passive uptake by the cells. However, LDBC NPs have a surface charge of ∼+33 mV due to their cationic amine surface, which resulted in a fast uptake process through the cell membrane, as anticipated.37
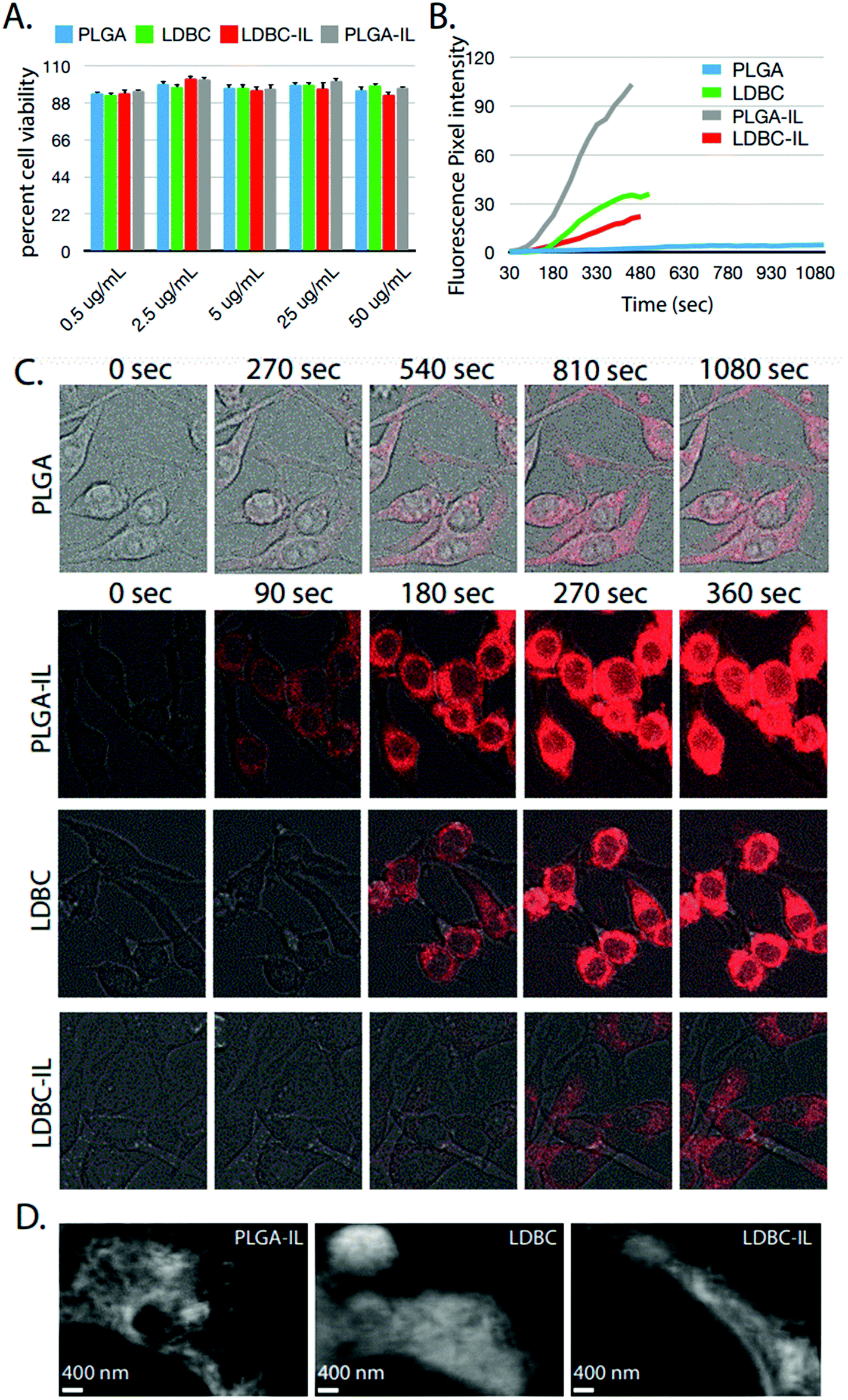 | ||
| Fig. 8 None of the NPs showed significant cell cytotoxicity but displayed significantly different rates and methods of cell entry and uptake. (A) Percent cell viability determined by LDH assay in Raw 264.7 macrophages. All materials and concentrations exhibited high biocompatibility with RAW cells. (B). Fluorescent intensity accumulating in cells over time-lapse imaging of RAW cells. (C) Images quantified in part B. Brightness gain for images of PLGA NPs was increased to permit visualization of fluorescence. IL modification of LDBCs reduced macrophage uptake due to anionic functionalization in the NP surface coating (D) STED imaging of particle fluorescence after 6 min of loading. Signal is associated with cell membranes, including the nuclear membrane. | ||
However, the second effect driving cellular uptake of NPs was uniquely determined by differences in the compositional assembly of the IL with the PLGA and LDBC polymers. Despite having a surface charge of −60 mV, IL-PLGA NPs penetrated the cell membranes aggressively over time, upending the traditional wisdom that the greater the negative charge, the slower the uptake.38 This may be explained by membrane lipid extraction, a phenomenon that has been invoked with ILs previously to explain their membrane permability.30,39 In contrast, IL-LDBC NPs had a slightly positive to neutral surface charge, which by typical cellular mechanisms would command robust cellular uptake.40 However, it was observed that IL-LDBCs bypassed the filopodia-mediated uptake observed with unmodified LDBCs41 or the endolysosomal pathway observed with DNA-coated iron oxide particles,42 and had a much more delayed uptake, beginning at 180 seconds (Fig. 8B and C).
It is possible that the differences observed between the two polymers is a result of a different conformation of ionic liquid at the interface of each. The outermost surface layer of the IL-LDBCs contains equal amounts of choline and trans-2-hexenoic acid by NMR spectroscopy. Exposure of the protons closest to the anion double bond may mediate a conformational assembly that exposes the double bond of the anion, which has been found to drive protein resistance and RBC affinity.33 However, the presence of equal amounts of choline in this outermost interfacing layer may change its uptake behavior despite neutralizing its surface. In comparison, the quantitatively determined higher ratio of anion to cation by NMR spectroscopy, as well as the zeta potential, suggested that IL interfacing layer in conformational assembly of IL-PLGA NPs was driven primarily by the anion, where we note comparatively high RBC affinity and greater selectivity with respect to serum compared to the IL-LDBCs (Scheme 2). It is possible that the mechanism of action for IL-PLGA NPs is related to lipid interactions with the RBC membrane when placed in whole blood, and that the interactions seen here with macrophages alone are simply a replication of that interaction with no other competing interactions, as monocytes are also present in whole blood and do not present any visible challenge to RBC membrane selectivity. As such, the presence of choline coordinating strongly in the interfacing layer may have ‘buffered’ the lipid extraction effect of the anion in IL-LDBCs, while in IL-PLGA NPs, the anion was exposed and may have been oriented in such a way that it was able to enter the RAW cell membrane, rather than simply being taken up and phagocytosed.43,44
This work provides evidence that the high precision tunability of ILs can be used to biofunctionalize polymeric nanomaterials. We have demonstrated that ILs are able to mediate monodisperse assembly of polymeric nanomaterials of diverse architectures. Additionally, our results show that ILs provide a means of control over the interactions of nanomaterials as they enter the blood stream and approach cell membranes. Future work, such as atomic force microscopy, small angle X-ray scattering, and small angle neutron scattering, among other techniques, is needed to explore the precise chemical mechanisms driving the interactions between polymer type and IL, red blood cell membranes and IL coatings.
Conclusions
We show that choline carboxylic-acid based ILs can electrostatically solvate LDBCs by direct dissolution, form stable and biocompatible IL-integrated LDBC and PLGA nanoparticles, and functionalize the NPs for RBC-hitchhiking. This work provides evidence of the use of biocompatible ILs to confer both stability and biological functionalization onto complex polymeric nanostructures. Future work will focus on the interactions that drive the processes reported here.Author contributions
Christine M. Hamadani: Investigation, methodology, visualization, formal analysis, funding, writing – original draft. Indika Chandrasiri: Investigation, writing – review & editing. Mahesh Loku Yaddehige: Investigation, writing – review & editing. Gaya S. Dasanayake: Investigation, writing – review & editing. Iyanuoluwani Owolabi: Investigation, writing – review & editing. Alex Flynt: Resources, supervision, writing – review & editing. Mehjabeen Hossain: Investigation, writing – review & editing. Lucy Liberman: Investigation, visualization, writing – review & editing. Timothy P. Lodge: Resources, supervision, funding, writing – review & editing. Thomas A. Werfel: Resources, supervision, writing – review & editing. Davita L. Watkins: Resources, supervision, writing – review & editing. Eden E. L. Tanner: Conceptualization, resources, supervision, funding, writing – original draft, project administration.Conflicts of interest
EELT is an inventor on a patent application that describes some of this technology.Acknowledgements
This work was supported by the College of Liberal Arts at the University of Mississippi, and the Sigma Xi Honor Society Student GIAR program. Cryo-TEM was carried out in the Characterization Facility, University of Minnesota using support from the Materials Research Facilities Network from the NSF through the MRSEC (Award Number DMR-2011401); the facility also receives support from the NSF through the NNCI (Award Number ECCS-2025124).References
- C. M. Dong and G. Liu, Linear-dendritic biodegradable block copolymers: From synthesis to application in bionanotechnology, Polym. Chem., 2013, 4, 46–52, 10.1039/c2py20441j.
- X. Fan, Y. Zhao, W. Xu and L. Li, Linear-dendritic block copolymer for drug and gene delivery, Mater. Sci. Eng. C, 2016, 62, 943–959, DOI:10.1016/j.msec.2016.01.044.
- I. Gitsov, Linear-dendritic block copolymers, Synth. Charact. Adv. Dendr. Macromol., 2002, 5, 45–87 CAS.
- F. Wurm and H. Frey, Linear-dendritic block copolymers: The state of the art and exciting perspectives, Prog. Polym. Sci., 2011, 36, 1–52 CrossRef CAS.
- I. Chandrasiri, D. G. Abebe, S. Gupta, J. S. D. Williams, W. D. Rieger, B. L. Simms, M. L. Yaddehige, Y. Noh, M. E. Payne, A. W. Fortenberry, A. E. Smith, J. Ilavsky, S. M. Grayson, G. J. Schneider and D. L. Watkins, Synthesis and characterization of polylactide-PAMAM Janus-type linear-dendritic hybrids, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1448–1459, DOI:10.1002/POLA.29409.
- Y. Liu, J. Wang, M. Zhang, H. Li and Z. Lin, Polymer-ligated nanocrystals enabled by nonlinear block copolymer nanoreactors: Synthesis, properties, and applications, ACS Nano, 2020, 14, 12491–12521, DOI:10.1021/ACSNANO.0C06936/ASSET/IMAGES/ACSNANO.0C06936.SOCIAL.JPEG_V03.
- Z. Li, M. Tang, S. Liang, M. Zhang, G. M. Biesold, Y. He, S. M. Hao, W. Choi, Y. Liu, J. Peng and Z. Lin, Bottlebrush polymers: From controlled synthesis, self-assembly, properties to applications, Prog. Polym. Sci., 2021, 116, 101387, DOI:10.1016/J.PROGPOLYMSCI.2021.101387.
- I. O. Lebedeva, E. B. Zhulina and O. V. Borisov, Self-Assembly of Linear-Dendritic and Double Dendritic Block Copolymers: From Dendromicelles to Dendrimersomes, Macromolecules, 2019, 52, 3655–3667 CrossRef CAS.
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches, Adv. Condens. Matter Phys., 2015, 2015, 1–22, DOI:10.1155/2015/151683.
- Z. Yang, A. H. Muller, C. Xu, P. S. Doyle, J. M. DeSimone, J. Lahann, F. Sciortino, S. Glotzer, L. Hong and D. A. Aarts, Janus particle synthesis, self-assembly and applications, 2012 Search PubMed.
- T. Nicolai, O. Colombani and C. Chassenieux, Dynamic polymeric micelles versus frozen nanoparticles formed by block copolymers, Soft Matter, 2010, 6, 3111, 10.1039/b925666k.
- S. L. Canning, G. N. Smith and S. P. Armes, A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly, Macromolecules, 2016, 49, 1985–2001, DOI:10.1021/ACS.MACROMOL.5B02602.
- I. Chandrasiri, D. G. Abebe, M. L. Yaddehige, J. S. D. Williams, M. F. Zia, A. Dorris, A. Barker, B. L. Simms, A. Parker, B. P. Vinjamuri, N. Le, J. N. Gayton, M. B. Chougule, N. I. Hammer, A. Flynt, J. H. Delcamp and D. L. Watkins, Self-Assembling PCL-PAMAM Linear Dendritic Block Copolymers (LDBCs for Bioimaging and Phototherapeutic Applications, ACS Appl. Bio Mater., 2020, 3, 5664–5677 CrossRef CAS PubMed.
- R. D. Rogers, CHEMISTRY: Ionic Liquids–Solvents of the Future?, Science, 2003, 302, 792–793, DOI:10.1126/science.1090313.
- P. Wasserscheid and T. Welton, Ionic Liq. Synth. (2nd Ed.), 2008 DOI:10.1002/9783527621194.
- K. R. Seddon, Ionic Liquids for Clean Technology, J. Chem. Technol. Biotechnol., 1997, 68, 351–356, DOI:10.1002/(SICI)1097-4660(199704)68:4<351::AID-JCTB613>3.0.CO;2-4.
- H. Zhang and H. Cui, Synthesis and characterization of functionalized ionic liquid-stabilized metal (gold and platinum) nanoparticles and metal nanoparticle/carbon nanotube hybrids, Langmuir, 2009, 25(5), 2604–2612, DOI:10.1021/la803347h.
- J. A. Widegren, A. Laesecke and J. W. Magee, The effect of dissolved water on the viscosities of hydrophobic room-temperature ionic liquids, Chem. Commun., 2005, 1610–1612, 10.1039/b417348a.
- H. V. Spohr and G. N. Patey, The influence of water on the structural and transport properties of model ionic liquids, J. Chem. Phys., 2010, 132, 234510, DOI:10.1063/1.3430561.
- W. Jiang, Y. Wang and G. A. Voth, Molecular dynamics simulation of nanostructural organization in ionic liquid/water mixtures, J. Phys. Chem. B, 2007, 111, 4812–4818, DOI:10.1021/jp0671421.
- T. S. de Almeida, A. Júlio, J. P. Mota, P. Rijo and C. P. Reis, An emerging integration between ionic liquids and nanotechnology: general uses and future prospects in drug delivery, Ther. Deliv., 2017, 8, 461–473, DOI:10.4155/tde-2017-0002.
- Z. He and P. Alexandridis, Nanoparticles in ionic liquids: Interactions and organization, Phys. Chem. Chem. Phys., 2015, 17, 18238–18261, 10.1039/c5cp01620g.
- H. Itoh, K. Naka and Y. Chujo, Synthesis of Gold Nanoparticles Modified with Ionic Liquid Based on the Imidazolium Cation, J. Am. Chem. Soc., 2004, 126, 3026–3027, DOI:10.1021/ja039895g.
- Z. Chen, P. A. FitzGerald, Y. Kobayashi, K. Ueno, M. Watanabe, G. G. Warr and R. Atkin, Micelle Structure of Novel Diblock Polyethers in Water and Two Protic Ionic Liquids (EAN and PAN), Macromolecules, 2015, 48, 1843–1851 CrossRef CAS.
- Y. He, Z. Li, P. Simone and T. P. Lodge, Self-assembly of block copolymer micelles in an ionic liquid, J. Am. Chem. Soc., 2006, 128, 2745–2750 CrossRef CAS PubMed.
- M. H. Zainal-Abidin, M. Hayyan, G. C. Ngoh and W. F. Wong, From nanoengineering to nanomedicine: A facile route to enhance biocompatibility of graphene as a potential nano-carrier for targeted drug delivery using natural deep eutectic solvents, Chem. Eng. Sci., 2019, 195, 95–106, DOI:10.1016/j.ces.2018.11.013.
- D. Zappi, S. Gabriele, L. Gontrani, D. Dini, C. Sadun, F. Marini and M. L. Antonelli, Biologically friendly room temperature ionic liquids and nanomaterials for the development of innovative enzymatic biosensors: Part II, Talanta, 2019, 194, 26–31, DOI:10.1016/j.talanta.2018.10.001.
- P. K. Kumar, M. Bisht, P. Venkatesu, I. Bahadur and E. E. Ebenso, Exploring the Effect of Choline-Based Ionic Liquids on the Stability and Activity of Stem Bromelain, J. Phys. Chem. B, 2018, 122, 10435–10444, DOI:10.1021/acs.jpcb.8b08173.
- E. E. L. Tanner, K. N. Ibsen and S. Mitragotri, Transdermal insulin delivery using choline-based ionic liquids (CAGE), J. Controlled Release, 2018, 286, 137–144, DOI:10.1016/j.jconrel.2018.07.029.
- E. E. L. Tanner, A. M. Curreri, J. P. R. Balkaran, N. C. Selig-Wober, A. B. Yang, C. Kendig, M. P. Fluhr, N. Kim and S. Mitragotri, Design Principles of Ionic Liquids for Transdermal Drug Delivery, Adv. Mater., 2019, 31, 1901103, DOI:10.1002/adma.201901103.
- A. Vaidya and S. Mitragotri, Ionic liquid-mediated delivery of insulin to buccal mucosa, J. Controlled Release, 2020, 327, 26–34, DOI:10.1016/j.jconrel.2020.07.037.
- A. Banerjee, K. Ibsen, T. Brown, R. Chen, C. Agatemor and S. Mitragotri, Ionic liquids for oral insulin delivery, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7296–7301, DOI:10.1073/pnas.1722338115.
- C. M. Hamadani, M. J. Goetz, S. Mitragotri and E. E. L. Tanner, Protein-avoidant ionic liquid (PAIL–coated nanoparticles to increase bloodstream circulation and drive biodistribution, Sci. Adv., 2020, 6, eabd7563, DOI:10.1126/sciadv.abd7563.
- E. E. L. Tanner, A. M. Curreri, J. P. R. Balkaran, N. C. Selig-Wober, A. B. Yang, C. Kendig, M. P. Fluhr, N. Kim and S. Mitragotri, Design Principles of Ionic Liquids for Transdermal Drug Delivery, Adv. Mater., 2019, 31, 1901103, DOI:10.1002/adma.201901103.
- B. C. Evans, C. E. Nelson, S. S. Yu, K. R. Beavers, A. J. Kim, H. Li, H. M. Nelson, T. D. Giorgio and C. L. Duvall, Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs., J. Visualized Exp., 2013 DOI:10.3791/50166.
- J. S. Brenner, D. C. Pan, J. W. Myerson, O. A. Marcos-Contreras, C. H. Villa, P. Patel, H. Hekierski, S. Chatterjee, J. Q. Tao, H. Parhiz, K. Bhamidipati, T. G. Uhler, E. D. Hood, R. Y. Kiseleva, V. S. Shuvaev, T. Shuvaeva, M. Khoshnejad, I. Johnston, J. V. Gregory, J. Lahann, T. Wang, E. Cantu, W. M. Armstead, S. Mitragotri and V. Muzykantov, Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude, Nat. Commun., 2018, 9(1), 2684, DOI:10.1038/s41467-018-05079-7.
- B. L. Simms, N. Ji, I. Chandrasiri, M. F. Zia, C. S. Udemgba, R. Kaur, J. H. Delcamp, A. Flynt, C. Tan and D. L. Watkins, Physicochemical properties and bio-interfacial interactions of surface modified PDLLA-PAMAM linear dendritic block copolymers, J. Polym. Sci., 2021, 59, 2177–2192, DOI:10.1002/POL.20210448.
- P. Foroozandeh and A. A. Aziz, Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles, Nanoscale Res. Lett., 2018, 13(1), 339, DOI:10.1186/S11671-018-2728-6.
- A. Banerjee, K. Ibsen, Y. Iwao, M. Zakrewsky and S. Mitragotri, Transdermal Protein Delivery Using Choline and Geranate (CAGE Deep Eutectic Solvent, Adv. Healthcare Mater., 2017, 6, 1601411, DOI:10.1002/adhm.201601411.
- L. Su and M. Noah, Deformation and poration of lipid bilayer membranes by cationic nanoparticles, Soft Matter, 2013, 9, 4969–4976, 10.1039/C3SM27578G.
- B. L. Simms, N. Ji, I. Chandrasiri, M. F. Zia, C. S. Udemgba, R. Kaur, J. H. Delcamp, A. Flynt, C. Tan and D. L. Watkins, Physicochemical properties and bio-interfacial interactions of surface modified PDLLA-PAMAM linear dendritic block copolymers, J. Polym. Sci., 2021, 59, 2177–2192, DOI:10.1002/POL.20210448.
- L. Zhang, X. Y. Tian, C. K. W. Chan, Q. Bai, C. K. Cheng, F. M. Chen, M. S. H. Cheung, B. Yin, H. Yang, W. Y. Yung, Z. Chen, F. Ding, K. C. F. Leung, C. Zhang, Y. Huang, J. Y. W. Lau and C. H. J. Choi, Promoting the Delivery of Nanoparticles to Atherosclerotic Plaques by DNA Coating, ACS Appl. Mater. Interfaces, 2019, 11, 13888–13904, DOI:10.1021/acsami.8b17928.
- J. Lin, L. Miao, G. Zhong, C. H. Lin, R. Dargazangy and A. Alexander-Katz, Understanding the synergistic effect of physicochemical properties of nanoparticles and their cellular entry pathways, Commun. Biol., 2020, 3, 1–10, DOI:10.1038/s42003-020-0917-1.
- C. Peetla and V. Labhasetwar, Biophysical characterization of nanoparticle endothelial model cell membrane interactions, Mol. Pharm., 2008, 5, 418–429, DOI:10.1021/mp700140a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr00538g |
| This journal is © The Royal Society of Chemistry 2022 |