
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cation polymer-induced aggregation of water-soluble Au(I)–thiolate complexes and their photoluminescence properties†
Naiying
Hao
 ab,
Yitao
Cao
*b,
Ruili
Li
a,
Hongbin
Lin
b,
Huiting
Shan
b,
Tiankai
Chen
b,
Osburg Jin Huang
Chai
b,
Qiaofeng
Yao
b,
Xiaoqing
Chen
*a and
Jianping
Xie
*b
ab,
Yitao
Cao
*b,
Ruili
Li
a,
Hongbin
Lin
b,
Huiting
Shan
b,
Tiankai
Chen
b,
Osburg Jin Huang
Chai
b,
Qiaofeng
Yao
b,
Xiaoqing
Chen
*a and
Jianping
Xie
*b
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Engineering Drive 4, 117585, Singapore
First published on 23rd June 2022
Abstract
Au(I)–thiolate complexes are a new class of aggregation-induced emission (AIE) material. Here we demonstrate a new aggregation strategy of water-soluble Au(I)–thiolate complexes induced by cationic polymers at optimized pH values. The generated AIE shows longer wavelengths than the emission induced by other methods.
Au(I)–thiolate (SR) complexes that can be produced by the reduction of Au(III) to Au(I) by thiols have recently been proven to be a new class of aggregation-induced emission (AIE) materials.1,2 Researchers have designed several effective methods to induce the aggregation of Au(I)–SR complexes and generate interesting AIE features. For example, Luo et al. have found that the hydration shell of Au(I)–SR complexes can be disrupted by adding a counter solvent, resulting in charge neutralization and consequent aggregates of the complexes. Strong photoemission centered at 565 nm (yellow-emitting) was obtained from nonluminescent oligomeric Au(I)–GSH (GSH = L-glutathione in the reduced form) complexes by adding ethanol (95% by volume) into a water solution.3 Inorganic ions can also be employed to aggregate Au(I)–SR complexes due to the high affinity of electrostatic and coordination interactions between certain multivalent cations and anions from SRs in the complexes to form inter-and/or intracomplex cross-links. Based on this, dark yellow light (photoemission around 570 nm) can be observed under UV light in the mixture of Au(I)–GSH complexes and Cd2+ (with a Cd2+ to GSH ratio of 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2).3,4 These methods provide efficient strategies to obtain high luminescence in Au(I)–SR complexes, mostly with yellow color.
:2).3,4 These methods provide efficient strategies to obtain high luminescence in Au(I)–SR complexes, mostly with yellow color.
To further control the emission wavelength in AIE-type Au(I)–SR complexes, we need an in-depth investigation of the structural effects in aggregates. Metal NCs are ideal models in which metal(I)–SR complexes are anchored on metal cores to generate AIE properties.5–7 It is found that the AIE of long Au(I)–SR complexes on the Au(0) core can sustain strong luminescence.8,9 The emission wavelength of Au NCs can be adjusted from visible (red-emitting) to the near-infrared II (NIR-II) region by controlling the length of the Au(I)–SR motifs. The photoemission peaks located at 658, 660, 670, and 987 nm were obtained from the aggregates of [Au10(GSH)10], [Au15(GSH)13], [Au18(GSH)14], and [Au25(GSH)18]−, respectively. The existence of Au(0) cores in the AIE process of Au(I)–SR complexes make it possible to obtain different photoemission, while it requires a high capability in synthesis and purification of a series of metal nanocluster species. The development of a simpler method to obtain different photoemission from Au(I)–SR aggregates without Au(0) cores is still a challenge.
Here, we design a facile aggregation strategy to obtain significantly different AIE properties in Au(I)–SR complexes by using the electrostatic interaction between cationic polymer and anionic water-soluble Au(I)–SR complexes. Poly(diallyldimethylammonium chloride) (PDDA), a cationic polymer with quaternary ammonium groups, was chosen as the model cationic polymer in this research.10 GSH was used to synthesize nonluminescent oligomeric Au(I)–SR complexes.11 When GSH is completely deprotonated, the high negative charge density on the surface of the Au(I)–GSH complex allows it to be strongly attracted by PDDA, resulting in the formation of aggregates. Unlike yellow luminescence originating from aggregates induced by counter solvent and inorganic ions, the addition of PDDA to Au(I)–GSH complexes can lead to strong red luminescence (photoemission around 660 nm) and form homogeneous necklace-like aggregates.8 These interesting results show that PDDA might replace the role of Au(0) cores in the process of AIE of Au(I)–GSH complexes. It is worth mentioning that Au(I)–GSH complexes induced by counter solvent and cationic polymer are suitable for different pH values. We found that ethanol-induced AIE of Au(I)–GSH can only be observed below pH 11, while PDDA-induced AIE of Au(I)–GSH expands the pH value to 12. In addition, we further investigated the effect of PDDA concentration on luminescence and found that a sedimentation phenomenon occurred due to charge instability during the zeta potential of the mixture of PDDA and Au(I)–GSH complexes changing from negative to positive. The versatility of the cationic polymer-induced aggregation emission property was also demonstrated by using different oligomeric Au(I)-SR complexes and different cationic polymer solutions.
In this study, Au(I)–GSH complexes were synthesized by mixing GSH and HAuCl4 solutions at 25 °C for 5 min, followed by NaOH addition to bring pH to 12.4 ± 0.1 and aging for ∼15 h. The resultant solution was clear and light yellow (inset of Fig. 1A, left) with a weak absorption around 350 nm in the UV-vis region (Fig. 1A, red line). In the formation process of Au(I)–GSH complexes, Au(III) was reduced by GSH to Au(I). The Au+ ions have a coordination number of 2 and are bridged by sulfur in GSH, which can form the Au–S–Au (φ). Four ionizable functional groups (one ammonium group, two carboxylic acid groups, and one thiol group) in GSH molecule made it exhibit a different number of net charges under different pH values. There are more negative charges on GSH at higher pH. Larger Au–S–Au angle (φ) can be formed under higher pH due to the stronger repulsive interaction of negatively charged moieties of GSH. Thus, gold species dispersed well under high pH values.12,13 The absorption around 350 nm is an indication of metal–metal interaction, which is weak due to the high pH value (pH = 12).14 To unambiguously determine the formula of the complexes, we analyzed the purified Au(I)–GSH complexes by ESI-MS. As Fig. 1D shows, the main peaks in the mass spectra can be assigned to Au2(GSH)2, Au4(GSH)4, and Au11(GSH)11 combining a different number of Na+ ions (red, orange, and green rectangles) and each of them shows perfect fitting to the theoretical isotopic pattern (red, orange and green lines).
 | ||
| Fig. 1 (A) UV-vis absorption (red line) and photoemission (black line, λex = 365 nm) spectra of 2 mM Au(I)–GSH without PDDA. (Inset) Digital photos of Au(I)–GSH solution under visible (left) and UV (right) light. (B) UV-vis absorption (red line), photoexcitation (black, λem = 660 nm) and photoemission (black line with blue dot, λex = 365 nm; black line with orange dot, λex = 510 nm) spectra of Au(I)–GSH@PDDA (0.2 wt% PDDA in 2 mM Au(I)–GSH). (Inset) Digital photos of the mixture of PDDA and complexes under visible (left) and UV (right) light. (C) Typical TEM image of Au(I)–GSH@PDDA with corresponding elemental mapping by EDS. (D) ESI-MS analysis and corresponding theoretical isotopic patterns (colorful lines) of Au(I)–GSH complexes. | ||
The cationic polymer, PDDA, was then added to obtained Au(I)–GSH complexes to induce aggregation process. The high-resolution transmission electron microscopy (TEM) image of Au(I)–GSH@PDDA with corresponding elemental mappings (Fig. 1C) shows that homogeneous necklace-like aggregates were formed due to the combination of PDDA and GSH. Energy dispersive spectrometry (EDS) confirmed the nature of Au and PDDA. The addition of PDDA also changed the fluorescence property of Au(I)–GSH complexes. They are nearly nonluminescent (inset of Fig. 1A, right) under UV light with no signal in the emission spectrum of the Au(I)–GSH complexes (black line, λex = 365 nm), while Au(I)–GSH@PDDA displays strong red emission under UV light (inset of Fig. 1B, right). The broad emission band centered around 660 nm and multiple excitation peaks at 365 nm and 510 nm (Fig. 1B, black lines) are different from those of the PL spectra of PDDA solution (Fig. S1, ESI†), which can be assigned to electronic transition between the ligand and Au(I) of Au(I)–GSH aggregated by PDDA and the formation of Au(I)⋯Au(I).15
This AIE phenomenon occurs through electrostatic interaction between cationic polymer and Au(I)–SR complexes. GSH has four ionizable functional groups, resulting in four pKa values, pKa1 = 2.12, pKa2 = 3.53, pKa3 = 8.66, and pKa4 = 9.62.16 The charge states of Au(I)–SR complexes changed with the increase of pH value. Thus, the pH value could play an important role in the cationic polymer-induced AIE phenomenon. As shown in Fig. 2A and C, the mixture of PDDA and Au(I)–GSH complexes showed no luminescence at pH 10 under UV light, while it started to show weak fluorescence at pH 11. When the pH value reached 12, strong red light can be observed under UV light with an emission band around 660 nm (λex = 375 nm). Based on the pKa4 = 9.62 of GSH, the GSH ligands in the Au(I)–GSH complexes are expected to be nearly fully deprotonated into doubly charged anions at pH 12. The strong negative charge will facilitate the electrostatic interaction and induce the compact aggregation of Au(I)–GSH complexes on PDDA. The quantum yield (QY) of the luminescent aggregates at pH 12 reaches ∼6.52%.
 | ||
| Fig. 2 Photoemission spectra (λex = 375 nm) of Au(I)–GSH@PDDA (0.002 wt% PDDA in 2 mM Au(I)–GSH complexes) under different pH values (A) and corresponding digital photos under visible (top) and UV (bottom) light (C). Photoemission spectra (λex = 375 nm) of Au(I)–GSH complexes aggregated by ethanol (95%) under different pH values (B) and corresponding digital photos under visible (top) and UV (bottom) light (D). | ||
The AIE phenomenon induced by ethanol is caused by a different mechanism. As shown in Fig. 2B and D, the addition of 95% ethanol by volume to Au(I)–GSH complexes leads to bright yellow or orange luminescence under UV light, corresponding to emission peaks around 580–620 nm. This is because the addition of ethanol in water disrupted the hydration shell of Au(I)–GSH complexes, resulting in charge neutralization and consequent aggregates of complexes. In this case, the relatively lower charge density at low pH values (pH ≤ 10) is necessary to ensure a compact aggregation. These Au(I)–GSH aggregates induced by ethanol in a different pH range are expected to have a different structure from the one with a cationic polymer as the aggregation center, thus leading to different luminescence properties.
Besides, we also believe that the length of Au(I)–GSH complexes is another possible factor that determines the AIE properties. With increased pH, negative charge density on the Au(I)–GSH surface is increased, which will increase the intra-cluster repulsion in longer complexes and reduce their stability. Thus, higher pH conditions are expected to facilitate the formation of shorter Au(I)–GSH complexes. The aggregates formed by short Au(I)–GSH complexes and cationic polymer could endow emission maxima with red-shifts. This result is similar to our previous research that a shorter motif could endow emission maxima with red shifts in the AIE of Au NCs.8 We have tried to capture the differences in species at different pH values (Fig. S2, ESI†). Even though the characterization of high-pH samples was limited by the testing conditions of ESI-MS instrument that high pH would affect the degree of ionization of the samples, we do get some useful result that the dominant large Au(I)–GSH complexes disappeared at high pH values.
The luminescence intensity of Au(I)–GSH@PDDA increased monotonically with the increase of PDDA concentration and the changes are shown in the emission spectra (Fig. S3A, ESI†). At pH 12, with the increase of PDDA, the main peak in emission spectrum was blue-shifted a little due to the change of aggregation degree of Au(I)–GSH. The change of aggregation can be confirmed by TEM images. As shown in Fig. S3D (ESI†) (sample III, 0.002 wt% PDDA mixed with 2 mM Au(I)–GSH complexes), there are uniform aggregates formed with a diameter less than 20 nm, while bigger aggregates with a diameter around 50 nm can be observed in Fig. S3E (ESI†) (sample IV, 0.02 wt% PDDA mixed with 2 mM Au(I)–GSH complexes) and S2F (sample V, 0.2 wt% PDDA mixed with 2 mM Au(I)–GSH complexes). Although almost every sample with PDDA inside shows red light under UV light (Fig. S3B, ESI†), a sedimentation phenomenon can be observed in sample IV (0.02 wt% PDDA mixed with 2 mM Au(I)–GSH complexes), which is different from other samples. This is due to the instability of the charge. As shown in Fig. S3C, (ESI†) these Au(I)–GSH@PDDA samples were negatively charged in solutions for the PDDA concentration of ≤0.002 wt% due to the negative charge on the Au(I) complexes. When the concentration of PDDA was increased to 0.2 wt%, the sample displayed a strong positive charge due to the high content of the quaternary ammonium group in PDDA, and a chain-like structure was formed (Fig. 2F). Sample IV happened to be the transition from the negative charge state to the positive charge state. Thus, the sedimentation phenomenon caused by the instability of the surface charge can be observed. The molar ratio of Au to PDDA unit (DDA) was further explored in Fig. S4 and S5 (ESI†) to confirm the range of the sedimentation phenomenon and the result proved that the AIE property would not be influenced by the charge state too much. It illustrates that we can selectively synthesize luminescent Au(I)–SR complexes with different charges, which will further expand the applications of the cationic polymer AIE method in biosensing, environmental monitoring, etc.
The cationic polymer-induced aggregation emission property is also suitable for other cationic polymers and Au(I)–SR complexes. Polyethyleneimine (PEI) and poly[bis(2-chloroethyl)ether-alt-1,3-bis[3-(dimethylamino)propyl]urea]quaternized (Polyquaternium-2) were used for replacing PDDA to explore the AIE property of Au(I)–GSH complexes. Fig. 3A and B show the same peak at around 660 nm. The similar features of the spectra of luminescent aggregated complexes suggest that the emission from Au(I)–GSH complexes (pH 12) was derived from the AIE of Au(I)–SR complexes on cationic polymers. The multiple emission peaks in Fig. 3A are similar to those of luminous PEI (Fig. S6, ESI†). Similarly, the small peak around 450 nm in Fig. 3B should have originated from luminous polyquaternium-2 (Fig. S7, ESI†). Then GSH was replaced by 6-mercaptohexanoic acid (MHA) and 3-mercaptopropionic acid (MPA). As shown in Fig. 3C and D, a similar peak at ∼450 nm could be obtained, corresponding to that of luminous PDDA (Fig. S1, ESI†). Other peaks with multiple luminous points were due to the AIE of MHA–Au(I) complexes (pH 9) and MPA–Au(I) complexes (pH 8) induced by PDDA. As a result, bright red and pink light were observed under UV light. These results are different from the emission and UV spectra of Au(I)–MHA complexes and Au(I)–MPA complexes without PDDA (Fig. S8 and S9, ESI†). However, AIE properties of Au(I)–MHA@PDDA and Au(I)–MPA@PDDA showed different pH-dependence compared with that of Au(I)–GSH complexes. As shown in Fig. S10 and S11, (ESI†) the fluorescence intensities of Au(I)–MHA@PDDA and Au(I)–MPA@PDDA decreased with the increase of the pH value. Both MHA and MPA are monoacids with low pKa (pKa of MHA is 3.86, pKa of MPA is 4.34)17,18 and the complexes they form with Au dissolve completely only under alkaline conditions (Au(I)–MHA and Au(I)–MPA complexes respectively dissolve from pH 9 and 8). When pH increases to 8 or 9, MHA and MPA are nearly fully deprotonated. The increase of pH no longer has a significant effect on the negative charge density of MHA and MPA, but higher pH value increases the concentration of negative ions in solution. Au(I)–SR complexes have already reached the ionic product for binding to cationic polymer, so the increasing of negative ion concentration can reduce the binding of Au(I)–SR complexes with cationic polymer and therefore reduce the fluorescence intensities of samples. Although the AIE properties of different Au(I)–SR complexes have different pH-dependence because of the various properties of SR ligands, the above results unambiguously demonstrate the universality of the cationic polymer-induced aggregation emission of Au(I)–SR complexes.
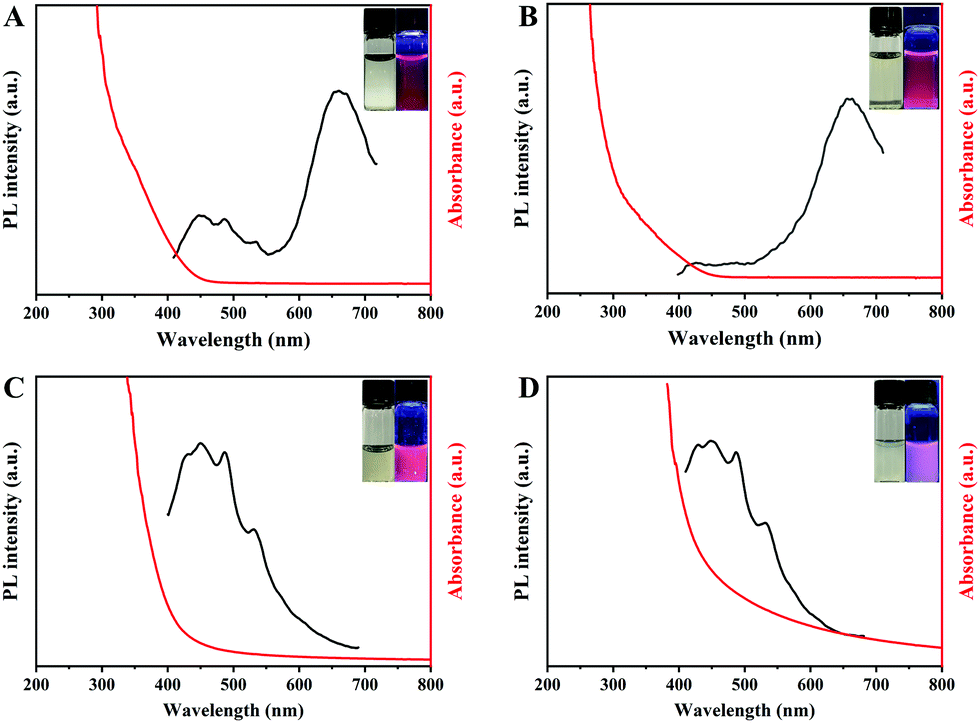 | ||
| Fig. 3 UV-vis adsorption (red lines), photoemission (black lines, λex = 365 nm) spectra and (insets) corresponding digital photos under visible (left) and UV (right) light of (A) Au(I)-GSH@PEI, (B) Au(I)–GSH@polyquaternium-2, (C) Au(I)–MHA@PDDA and (D) Au(I)–MPA@PDDA (0.02 wt% cationic polymer in 2 mM Au(I)–SR complexes). | ||
In summary, we have demonstrated that the cationic polymer-induced aggregation method is a simple and effective way to generate AIE phenomena in Au(I)–SR complexes. The resultant aggregates produce a completely different emission compared to the aggregates induced by ethanol. We found that the pH-dependent negative charge originating from the carboxyl groups of the ligands is the key to determine the AIE results. Furthermore, the concentration of the cationic polymer can be adjusted to obtain tunable charged AIE aggregates. The versatility and general utility of the cationic polymer-induced aggregation emission method have also been demonstrated by using oligomeric Au(I) complexes protected by different SR ligands and the use of different cationic polymers.
This work is financially supported by the Ministry of Education, Singapore (Academic Research Grant R-279-000-580-112 and R-279-000-538-114) and National Natural Science Foundation of China (21878339 and 22078369). N.H. gratefully acknowledges the financial support from China Scholarship Council.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138–1139 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/b105159h.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- T. Zhao, N. Goswami, J. Li, Q. Yao, Y. Zhang, J. Wang, D. Zhao and J. Xie, Small, 2016, 12, 6537–6541 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D. E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- K. Zheng, X. Yuan, K. Kuah, Z. Luo, Q. Yao, Q. Zhang and J. Xie, Chem. Commun., 2015, 51, 15165–15168 RSC.
- X. Kang, S. Wang, Y. Song, S. Jin, G. Sun, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2016, 55, 3611–3614 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, O. J. H. Chai, N. Ding, W. Xu, S. Zang and J. Xie, Angew. Chem., Int. Ed., 2020, 59, 9934–9939 CrossRef CAS PubMed.
- Y. Xiao, Z. Wu, Q. Yao and J. Xie, Aggregate, 2021, 2, 114–132 CrossRef.
- Z. Yang, X. Fan, W. Cheng, Y. Ding and W. Zhang, Anal. Chem., 2019, 91, 10295–10301 CrossRef CAS PubMed.
- N. Goswami, F. Lin, Y. Liu, D. T. Leong and J. Xie, Chem. Mater., 2016, 28, 4009–4016 CrossRef CAS.
- R. P. Brinas, M. Hu, L. Qian, E. S. Lymar and J. F. Hainfeld, J. Am. Chem. Soc., 2008, 130, 975–982 CrossRef CAS PubMed.
- X. Qi, W. Li, J. Gu, C. Guo and J. Zhang, RSC Adv., 2016, 6, 105110–105118 RSC.
- K. H. Leung, D. L. Phillips, Z. Mao, C. M. Che, V. M. Miskowski and C. K. Chan, Inorg. Chem., 2002, 41, 2054–2059 CrossRef CAS PubMed.
- Z. Wu, Y. Du, J. Liu, Q. Yao, T. Chen, Y. Cao, H. Zhang and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 8139–8144 CrossRef CAS PubMed.
- Q. Yao, Y. Yu, X. Yuan, Y. Yu, J. Xie and J. Y. Lee, Small, 2013, 9, 2696–2701 CrossRef CAS PubMed.
- A. Kudelski, A. Michota and J. Bukowska, J. Raman Spectrosc., 2005, 36, 709–714 CrossRef CAS.
- V. To, K. Masagounder and M. E. Loewen, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2021, 255, 110908 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supplementary figures. See DOI: https://doi.org/10.1039/d2cc02608b |
| This journal is © The Royal Society of Chemistry 2022 |