
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst aromatic amine organocatalysed activation of α,β-unsaturated ketones†
Isaac G.
Sonsona
 a,
Eugenia
Marqués-López
a,
M. Concepción
Gimeno
b and
Raquel P.
Herrera
*a
a,
Eugenia
Marqués-López
a,
M. Concepción
Gimeno
b and
Raquel P.
Herrera
*a
aDepartamento de Química Orgánica, Laboratorio de Organocatálisis Asimétrica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), (CSIC-Universidad de Zaragoza), C/Pedro Cerbuna, No. 12, E-50009 Zaragoza, Spain. E-mail: raquelph@unizar.es
bDepartamento de Química Inorgánica Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) (CSIC-Universidad de Zaragoza), C/Pedro Cerbuna, No. 12, E-50009 Zaragoza, Spain
First published on 16th July 2019
Abstract
This work provides an unprecedented example of a chiral aromatic amine used to activate α,β-unsaturated ketones in asymmetric aminocatalysis. Chiral aromatic diamine VII has been efficiently employed, as a proof of concept, in the Michael addition reaction between benzylideneacetones (1a–f) and coumarins (2a–d). The reaction gives rise to warfarin derivatives 3 with promising results using this family of catalysts for the first time. The additional studies performed supported the bifunctional mode of activation of the chiral catalyst VII and the covalent nature of the interactions between the catalyst VII and benzylideneacetones 1.
Introduction
After the tragic effects caused by the prescription of thalidomide to pregnant women at the beginning of the 1960s, more efforts have been devoted to the synthesis and commercialisation of single enantiopure pharmaceuticals. The obtainment of a single enantiomer, as an optically active drug, is becoming progressively more vital for the treatment of many diseases. To achieve this pivotal goal, asymmetric catalysis has been employed as a powerful tool to develop new reactions affording enantioenriched compounds.1 In this respect, one of the most important aims of organocatalysis has also been the preparation of new chiral biologically active compounds or natural products.2Among the plethora of drug-based compounds, warfarin 3a is one of the most widely used anticoagulants due to its advantages such as oral administration, low price and permanent effect. Like other pharmaceuticals, it is prescribed worldwide as a racemate. However, it is well-known that the anticoagulant activity of the S enantiomer is about 5–8 times higher than that of the R enantiomer.3 Different catalytic asymmetric approaches towards the synthesis of optically active warfarin by means of enzymatic,4 metal based5 or organocatalytic procedures have been reported.6 Among the organocatalytic approaches, the first example was reported by Jørgensen's group using a chiral imidazolidine organocatalyst following the Michael addition of cyclic 1,3-dicarbonyl compounds to α,β-unsaturated enones.7 After this pioneering example, other organocatalytic procedures were reported with the main aim of obtaining better results for the enantiopure products. Some of these organocatalytic approaches have been developed using aliphatic amines as catalysts.8 It is important to cite some of these pioneering examples using aliphatic primary chiral amines such as those reported by Chin9 and Chen10 or more recently, by Zlotin's group.11
In contrast, the use of chiral aromatic amines to promote organocatalytic reactions has been eclipsed by the corresponding chiral aliphatic amines, mainly due to the conjugation between the nitrogen lone pair and the aromatic ring. Consequently, this conjugation is responsible for their less nucleophilic behavior in comparison with aliphatic amines.12 In fact, there are scarce organocatalytic examples where a chiral aromatic amine promotes the reaction by itself.13,14 In addition to all reported examples for the preparation of warfarin and its derivatives, to the best of our knowledge there is not any example of a chiral aromatic amine used as an organocatalyst for the synthesis of these interesting molecules. Moreover, none of these previous examples using aromatic amines was involved in the activation of α,β-unsaturated ketones.
Hence, based on our continuous search for the discovery of new organocatalysts and catalytic reactions,15 we have studied this alternative as a totally unexplored approach.
Results and discussion
To test the model process depicted in Scheme 1, simple chiral amine structures, non containing in their skeletons a proline, an amino-cinchona or a DPEN moiety as is usual, were selected as promising catalysts. Interestingly, these simple amines have been overlooked so far in the literature for this process. | ||
| Scheme 1 Screening of catalysts in the Michael addition between 4-hydroxycoumarin (2a) and benzylideneacetone (1a). | ||
To our delight, all catalysts, except II, promoted the Michael addition reaction affording final desired product 3a (Scheme 1). Primary amines exhibited higher reactivity in comparison with catalyst VII (as expected). Although catalyst VII was not the most reactive, it was surprisingly the most enantioselective. To the best of our knowledge this is the first time that this aromatic amine has been used to activate ketone electrophiles in asymmetric aminocatalysis.16 Therefore, with these promising results of enantioselectivity in hand, a deep exploration of different parameters, in the previous model reaction, was carried out using catalyst VII (Tables S1 and S2, ESI†).
The study of the reaction conditions such as the amount of solvent or equivalents of reagents 1a and 2a, and catalyst VII loading, afforded smooth variations in both the yield and enantioselectivity of the reaction (Table S1, ESI†). In general, the concentration of the reaction led to better reactivity without impairing the enantioselectivity of the process (Table S1, entries 9–12, ESI†). Variations in the amount of coumarin 2a did not afford an appreciable improvement neither in the reactivity nor in the enantioselectivity of the process. The exploration of a vast number of different solvents did not afford better results in comparison with those obtained with THF (Table S2, ESI†). With the best reaction conditions in hand (Table S1, entry 11), the scope of the reaction was explored for different benzylideneacetones (1a–f) and coumarins (2a–d) (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Prod. | Yieldb (%) | eec (%) |
| a To a mixture of catalyst VII (20 mol%, 0.02 mmol) and coumarins 2a–d (0.2 mmol) in THF (100 μl), benzylideneacetones 1a–f (0.1 mmol) were added at room temperature. b After isolation by column chromatography. c Determined by chiral HPLC analysis. | |||||
| 1 | H (1a) | H (2a) | 3a | 68 | 64 |
| 2 | 4-Cl (1b) | H (2a) | 3b | 76 | 64 |
| 3 | 4-Br (1c) | H (2a) | 3c | 85 | 66 |
| 4 | 4-CN (1d) | H (2a) | 3d | 92 | 50 |
| 5 | 4-Me (1e) | H (2a) | 3e | 62 | 58 |
| 6 | H (1a) | Cl (2b) | 3f | 57 | 67 |
| 7 | H (1a) | Br (2c) | 3g | 53 | 67 |
| 8 | H (1a) | Me (2d) | 3h | 25 | 61 |
| 9 | 4-Cl (1b) | Br (2c) | 3i | 25 | 62 |
| 10 | 4-Me (1e) | Cl (2b) | 3j | 20 | 62 |
| 11 | 4-OMe (1f) | Cl (2b) | 3k | 52 | 54 |
| 12 | 4-OMe (1f) | Br (2c) | 3l | 40 | 54 |
In all cases, the Michael addition reaction took place smoothly giving rise to the desired final products 3 with moderate to good yields and with promising enantioselectivities. This proof of concept is well accounted for a variety of coumarins 2a–d and benzylideneacetones 1a–f, and the crude products of the reactions are very clean. Although the values of enantioselectivity did not show a clear correlation with the electronic environment of the reagents, the reactivity suggests a dependence on the electronic environment in the aromatic ring of coumarins 2 and benzylideneacetones 1. Therefore, deactivated benzylideneacetones 1e, f (with electron-donating groups) led to lower reactivities (entries 5, 10–12). Furthermore, coumarin 2d, with a methyl group, seems to decrease the reactivity of the process (entry 8). The absolute configuration was determined to be R by comparison of the optical rotation of products 3a and 3b with those reported in the literature.6b,17 Therefore, the same stereochemical outcome was assumed for all products 3.
Additionally, we have found some inconsistences regarding the sign of the values previously reported for the optical rotation of some of these products. In order to also support the absolute configuration of our products, single crystals were grown from adduct 3c′ and the stereochemical outcome was determined to be also R for this product (Fig. 1).18 It is worth noting that product 3c was crystallised as its pseudo-diastereomeric hemiketal form 3c′.
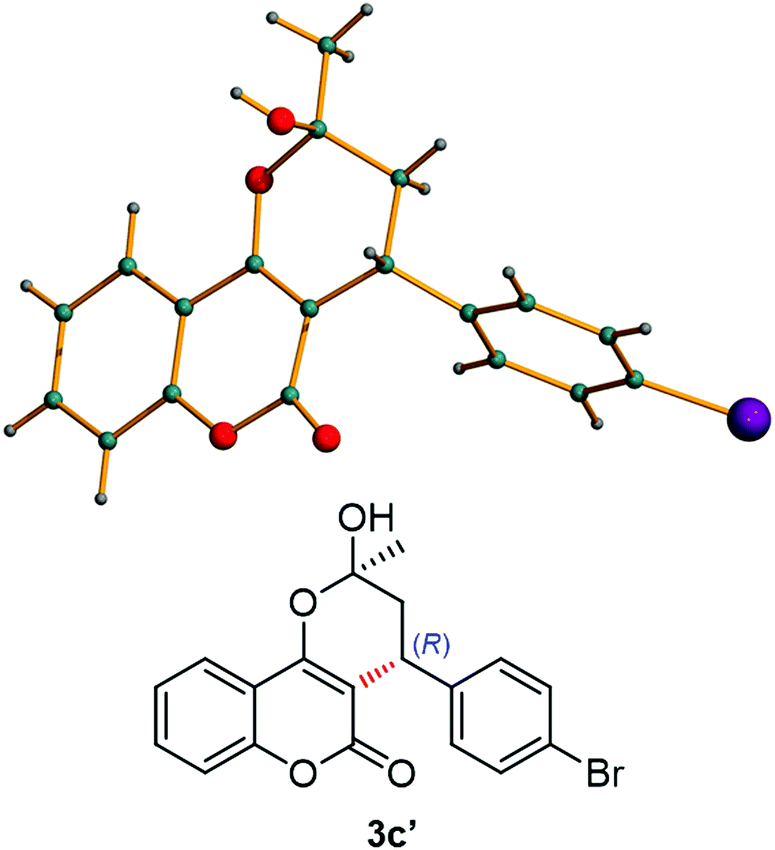 | ||
| Fig. 1 X-ray crystal structure of (R)-3c′. | ||
In order to gain insights into the mode of activation by the catalyst VII in this Michael reaction, additional experiments were performed. First, ESI-MS analysis was carried out (Fig. 2). ESI-MS has been considered as an important technique for mechanistic studies of organic reactions.19 The presence of plausible intermediates in the solution of the reaction mixture was analysed directly (VII (20 mol%, 0.02 mmol), coumarin 2a (0.15 mmol) and benzylideneacetone 1a (0.1 mmol), in THF (100 μl) at room temperature) (Fig. 2).
 | ||
| Fig. 2 Cationic ESI spectrum of the reaction mixture. | ||
In the cationic ESI spectrum recorded directly from the solution to the gas phase, several important mass peaks related to some plausible intermediates of the reaction, such as the imine between catalyst VII and benzylideneacetone 1a (m/z 413.2, Int. 1, and m/z 451.2, Int. 1′) were found. The resulting enamine intermediates (Int. 2 and Int. 3), generated after coumarin 2a attacks Int. 1, were also found. From this observation, it can be assumed that catalyst VII gives rise an imine with benzylideneacetone 1a before the attack of the nucleophile 2a.
It is remarkable that the formation of a diimine intermediate with the catalyst (Int. 4) was not observed in the ESI-MS spectrum, which could be due to the formation of an imine with each NH2 group in the catalytic structure, as previously observed20 or hypothesised9 by other authors, using primary aliphatic amines.
Furthermore, some additional reactions were also performed in order to understand the role of the second NH2 group in the catalyst (Scheme 2).
 | ||
| Scheme 2 Study of the bifunctional role of the catalyst VII. | ||
Some conclusions could be made from the results shown in Scheme 2: (1) first, the second NH2 group present in VII could not be involved in the generation of a second imine in agreement with the results found in the ESI-MS analysis (Int. 1 and Int. 1′, Fig. 2). (2) Interestingly, the second NH2 group in catalyst VII could play an important role in the enantioselection process, since when catalyst VIII is used, the enantioselectivity dramatically collapses and a racemic mixture is obtained. (3) The reaction seems to be mainly activated by an aminocatalytic approach more than hydrogen bond activation alone, which agrees with the intermediates (Int. 1–3) found in the ESI-MS spectra (Fig. 2). Hence, when catalyst IX (which is more acidic than VII and VIII) was employed the process did not work. (4) Moreover, the addition of an external Brønsted acid did not enhance the reactivity of the process when using VII. In the case of using catalyst VII alone, iminium ions would be formed after deprotonation of the coumarin substrate (Fig. 3). (5) A strange behaviour was observed with the low reactivity displayed by catalyst VIII, just with one NH2 group in its structure, since a higher reactivity could be initially expected. Thus, it seems that the OH group is not involved in the enantioselection process and more importantly, it likely inhibits the reaction. This finding could be explained by assuming an intramolecular hydrogen bonding between RO-H⋯NH2R. This would make the NH2 group in VIII less nucleophilic than in catalyst VII, avoiding the formation of the initial imine (Int. 1 and Int. 1′, Fig. 2) and therefore, obstructing the reaction. A similar intramolecular hydrogen bonding, although weaker, between RHN-H···NH2R would also be possible.
 | ||
| Fig. 3 Plausible TS to explain the bifunctional role of the catalyst. | ||
Based on all these experiments, VII would act as a bifunctional catalyst,21 activating at the same time the electrophile, by the generation of an imine or iminium ion, and driving the attack of the nucleophile (Fig. 3).
According to the above outcomes, a stereochemical model is proposed in Fig. 3.22 The approach of the coumarin 2 to the in situ generated imine (Int. 1) would be driven by an RHN–H⋯−OR′ bond, resulting in the addition to the re face (lower face) of the imine (or iminium ion, Fig. 3). Moreover, we could not ignore the possibility of an intramolecular hydrogen bond between the second amino group and the iminium ion, giving rise to a more rigid transition state. Hence, the enantiomer obtained would agree with the absolute configuration (R) found in final products 3.
Conclusions
In conclusion, we have developed the first example of a chiral aromatic amine used as an organocatalyst to activate α,β-unsaturated ketones in aminocatalysis. This Michael addition reaction is efficiently catalysed by the simple diamine catalyst VII, which can be easily accessed in both enantiomeric forms from commercially available sources. Final warfarin derivatives 3 are obtained with good results, as a proof of concept. The additional ESI-MS and experimental studies performed supported a plausible bifunctional mode of activation by catalyst VII. The detection of important and crucial intermediates of the reaction strongly supports the role of the diamine catalyst VII in the activation mechanism via aminocatalysis. This work contributes to the scarcely developed field of aromatic amines as organocatalysts and opens a door for further studies in this area of research.Experimental
General experimental methods and instrumentation
Starting materials 1a and 2a–d, as well as amine catalysts I–IX, are commercially available and were employed as received without further treatment or purification. In the case of solvent, commercial tetrahydrofuran (THF, HPLC grade) was transferred to a new recipient and molecular sieves (4 Å) were added.All reactions were performed at room temperature under ambient conditions. The reactions were monitored by thin-layer chromatography (TLC) using aluminum sheets recoated with silica gel and a fluorescent indicator (60 F254, 0.2 mm). Compounds were visualised at 254 nm by using UV light. Products 3 were isolated by flash chromatography using silica gel (0.06–0.2 nm) as the stationary phase and a mixture of commercial dichloromethane and ethyl acetate as an eluent.
The chiral HPLC analysis of products 3 was performed using a Waters 600 system, with a Daicel ChiralPak IC column as the stationary phase and a mixture of commercial n-hexane and isopropyl alcohol as an eluent. The specific rotation of products 3 was determined using a Jasco P-1020 polarimeter, in acetonitrile of HPLC grade as the solvent. The absolute configuration of products 3a and 3b was assigned comparing their specific rotation with those reported in the literature.
The NMR spectra of the reagents and products were recorded at 300 MHz (Bruker ARX300 spectrometer) or 400 MHz (Bruker AV400 spectrometer), in chloroform-d (CDCl3) or dimethyl sulfoxide-d6 ((CD3)2SO) as the deuterated solvent. The infrared spectra of the starting materials and products were obtained employing attenuated total reflection infrared (ATR-FTIR) spectroscopy using a PerkinElmer FTIR spectrometer equipped with a universal ATR sampling accessory. The HRMS analysis of the reagents and products was performed using a MicroTof-Q mass spectrometer and electrospray (ESI) as the ionisation method. The melting point of the reagents and products was determined using a Gallenkamp MPD 350 BM 2.5 device.
Synthesis of benzylideneacetone derivatives 1b–f
The substrates 1b–f were synthesised from the corresponding commercial aldehydes 4b–f and the phosphonium ylide 5, which was previously prepared following the procedure reported in the literature using commercially available reagents (Scheme S1, ESI†).24 The respective yields after purification are shown in Scheme S1 (ESI†). The ratio (E)/(Z) of products 1b–f was determined by 1H-NMR spectroscopy, using DMSO-d6 as the solvent.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 as an eluent. The mixture of isomers obtained was washed with cold n-hexane, affording 320 mg of compound 1b (71% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was further purified by recrystallisation in methanol, affording 203 mg of compound 1c (36% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was purified by recrystallisation in methanol, affording 230 mg of compound 1d (54% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was purified by flash chromatography using n-hexane/diethyl ether 9:1 as an eluent, affording 205 mg of compound 1e (51% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was purified by recrystallisation in methanol, affording 191 mg of compound 1f (43% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was washed with cold n-hexane, affording 320 mg of compound 1b (71% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was further purified by recrystallisation in methanol, affording 203 mg of compound 1c (36% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was purified by recrystallisation in methanol, affording 230 mg of compound 1d (54% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was purified by flash chromatography using n-hexane/diethyl ether 9:1 as an eluent, affording 205 mg of compound 1e (51% yield, (E)/(Z) > 99:1).
:2 as an eluent. The mixture of isomers obtained was purified by recrystallisation in methanol, affording 191 mg of compound 1f (43% yield, (E)/(Z) > 99:1).
General procedure for the enantioselective synthesis of warfarin derivatives 3a–l catalysed by the chiral aromatic diamine VII
To a mixture of benzylideneacetone derivative 1a–f (0.1 mmol) and catalyst VII (5.74 mg, 0.02 mmol, 20 mol%) in THF (100 μl) at room temperature, the corresponding 4-hydroxycoumarin 2a–d (0.2 mmol, 2 eq.) was added. The reaction mixture was stirred at room temperature for three days, leading to the final desired product with the lack of by-products. After this reaction time, the residue obtained was purified by flash chromatography using the gradient from CH2Cl2:AcOEt, 99:1 to CH2Cl2:AcOEt, 94:6 as the eluent, affording the corresponding pure product 3. The different yields and enantioselectivities obtained are collected and compared in Table 1 of the manuscript.
:20, flow rate 1 ml min−1, λ = 282.5 nm): τmajor = 16.7 min; τminor = 24.3 min. [α]23D = +8.7 ± 0.1 (c 0.75, acetonitrile, 64% ee). {lit.,6b [α]23D = −9.4 (c 0.4, acetonitrile, 83% ee, S-3a)}.
:20, flow rate 1 ml min−1, λ = 279.4 nm): τmajor = 10.5 min; τminor = 21.3 min. [α]25D = −6.1 ± 0.1 (c 0.60, acetonitrile, 64% ee). {lit.,17 [α]25D = −8.8 (c 0.27, acetonitrile, 79% ee, R-3b)}.
:20, flow rate 1 ml min−1, λ = 279.4 nm): τmajor = 11.7 min; τminor = 23.0 min. [α]23D = −10.0 ± 0.1 (c 0.60, acetonitrile, 66% ee).
:20, flow rate 1 ml min−1, λ = 250.0 nm): τmajor = 20.8 min; τminor = 26.5 min. [α]23D = −10.9 ± 0.1 (c 1.37, acetonitrile, 50% ee). M.p. 105–107 °C. IR (cm−1) 3353 (OH), 2226 (CN), 1680 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1608 (CO), 1381, 1068, 759, 727, 563. In CDCl3, the product was found to exist in a fast equilibrium between its open chain form 3d (≈10 mol%) and both pseudo-diastereomeric hemiketals 3d′ (≈30 mol% and ≈60 mol%). 1H-NMR (300 MHz, CDCl3) δ 1.72 (s, 0.9H3d′[a], RCH3), 1.77 (s, 1.8H3d′[b], RCH3), 1.92 (dd, J1 = 10.4 Hz, J2 = 8.9 Hz, 0.6H3d′[a], RCH2R′), 2.32 (s, 0.3H3d, RCH3), 2.37–2.49 (m, 1.2H3d′[b], RCH2R′), 3.01 (br s, 0.3H3d′[a], ROH), 3.23–3.33 (m, 0.1H3d, RCH2R′), 3.33 (br s, 0.6H3d′[b], ROH), 3.86 (dd, J1 = 14.5 Hz, J2 = 8.0 Hz, 0.1H3d, RCH2R′), 4.19–4.24 (m, 0.6H3d′[b] + 0.3H3d′[a], RCHR′R′′), 4.68–4.71 (m, 0.1H3d, RCHR′R′′), 7.22–7.24 (m, 0.1H3d, Ar-H), 7.27–7.37 (m, 2.4H3d′[b] + 1.2H3f′[a], Ar-H), 7.41–7.43 (m, 0.2H3d, Ar-H), 7.50–7.60 (m, 1.8H3d′[b] + 0.9H3d′[a] + 0.4H3d, Ar-H), 7.81–7.83 (m, 0.6H3d′[b], Ar-H), 7.86–7.88 (m, 0.3H3d′[b], Ar-H), 7.95–7.98 (m, 0.1H3d, Ar-H), 9.64 (br s, 0.1H3d, ROH). 13C-NMR (75 MHz, CDCl3) δ 28.2, 28.4, 35.1, 35.2, 35.8, 39.6, 42.0, 44.9, 98.9, 100.0, 101.0, 103.3, 110.4, 110.5, 115.5, 115.8, 116.5, 116.8, 116.9, 119.1, 122.9, 123.0, 124.0, 124.2, 128.1, 128.5, 129.1, 132.0, 132.1, 132.3, 132.4, 132.7, 148.4, 149.3, 153.1, 159.4, 161.3. HRMS (ESI+) calcd for [NaC20H15NO4]+ ([M + Na]+) 356.0899; found 356.0893.
:20, flow rate 1 ml min−1, λ = 279.4 nm): τmajor = 17.3 min; τminor = 33.2 min. [α]23D = +3.3 ± 0.2 (c 0.37, acetonitrile, 58% ee). {lit.,6a [α]25D = +16.6 (c 1.0, dichloromethane, 96% ee, (R)-3e)}.
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 13.1 min; τminor = 18.3 min. [α]23D = −22.1 ± 0.1 (c 0.69, acetonitrile, 67% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 13.7 min; τminor = 18.9 min. [α]23D = −24.3 ± 0.1 (c 1.00, acetonitrile, 67% ee).
:20, flow rate 1 ml min−1, λ = 271.0 nm): τmajor = 20.0 min; τminor = 30.7 min. [α]23D = −4.9 ± 0.1 (c 0.31, acetonitrile, 61% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 8.2 min; τminor = 13.6 min. [α]24D = −35.2 ± 0.1 (c 0.34, acetonitrile, 62% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 12.2 min; τminor = 19.4 min. [α]24D = −25.8 ± 0.2 (c 0.20, acetonitrile, 62% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 17.3 min; τminor = 31.6 min. [α]23D = −27.9 ± 0.1 (c 0.77, acetonitrile, 54% ee). IR (cm−1) 3353 (OH), 1694 (CO), 1614 (CO), 1510, 1241, 824, 730, 535. In CDCl3, the product was found to exist in a fast equilibrium between its open chain form 3k (≈20 mol%) and both pseudo-diastereomeric hemiketals 3k′ (≈40 mol%). 1H-NMR (400 MHz, CDCl3) δ 1.68 (s, 1.2H3k′, RCH3), 1.74 (s, 1.2H3k′, RCH3), 2.00 (dd, J1 = 14.0 Hz, J2 = 11.5 Hz, 0.4H3k′[a], RCH2R′), 2.04 (s, 0.2H3k, RCH3), 2.29 (s, 0.4H3k, RCH3), 2.38 (dd, J1 = 14.2 Hz, J2 = 6.8 Hz, 0.4H3k′[b], RCH2R′), 2.47 (dd, J1 = 14.1 Hz, J2 = 6.9 Hz, 0.4H3k′[b], RCH2R′), 2.54 (dd, J1 = 14.2 Hz, J2 = 3.0Hz, 0.4H3k′[a], RCH2R′), 3.11 (br s, 0.4H3k′, ROH), 3.25–3.30 (m, 0.4H3k′ + 0.2H3k, ROH + RCH2R′), 3.75–3.86 (m, 1.2H3k′[a] + 1.2H3k′[b] + 0.6H3k, ROCH3), 4.09–4.15 (m, 0.4H3k′ + 0.2H3k, RCHR′R′′ + RCH2R′), 4.24 (dd, J1 = 6.6 Hz, J2 = 2.8 Hz, 0.4H3k′, RCHR′R′′), 4.63 (dd, J1 = 10.4 Hz, J2 = 2.1 Hz, 0.2H3k, RCHR′R′′), 7.12–7.31 (m, 2H, Ar-H), 7.12–7.31 (m, 3H, Ar-H), 7.41–7.44 (m, 0.4H3k′ + 0.2H3k, Ar-H), 7.51 (dd, J1 = 8.8 Hz, J2 = 2.5 Hz, 0.4H3k′, Ar-H), 7.78 (d, J1 = 2.5 Hz, 0.4H3k′, Ar-H), 7.86 (d, J1 = 2.5 Hz, 0.4H3k′, Ar-H), 7.91 (d, J1 = 2.5 Hz, 0.2H3k, Ar-H), 9.56 (br s, 0.2H3k, ROH). 13C-NMR (100 MHz, CDCl3) δ 28.0, 28.5, 33.4, 34.7, 40.7, 42.6, 55.4, 55.4, 99.5, 101.0, 102.4, 113.8, 114.3, 114.9, 117.8, 118.2, 118.3, 122.5, 122.8, 123.7, 128.1, 128.2, 129.3, 129.3, 129.7, 131.6, 131.8, 132.1, 132.8, 134.8, 157.5. HRMS (ESI+) calcd for [NaC20H17ClO5]+ ([M + Na]+) 395.0662; found 395.0657.
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 19.5 min; τminor = 36.9 min. [α]24D = −34.3 ± 0.1 (c 0.61, acetonitrile, 54% ee).
O), 1608 (CO), 1381, 1068, 759, 727, 563. In CDCl3, the product was found to exist in a fast equilibrium between its open chain form 3d (≈10 mol%) and both pseudo-diastereomeric hemiketals 3d′ (≈30 mol% and ≈60 mol%). 1H-NMR (300 MHz, CDCl3) δ 1.72 (s, 0.9H3d′[a], RCH3), 1.77 (s, 1.8H3d′[b], RCH3), 1.92 (dd, J1 = 10.4 Hz, J2 = 8.9 Hz, 0.6H3d′[a], RCH2R′), 2.32 (s, 0.3H3d, RCH3), 2.37–2.49 (m, 1.2H3d′[b], RCH2R′), 3.01 (br s, 0.3H3d′[a], ROH), 3.23–3.33 (m, 0.1H3d, RCH2R′), 3.33 (br s, 0.6H3d′[b], ROH), 3.86 (dd, J1 = 14.5 Hz, J2 = 8.0 Hz, 0.1H3d, RCH2R′), 4.19–4.24 (m, 0.6H3d′[b] + 0.3H3d′[a], RCHR′R′′), 4.68–4.71 (m, 0.1H3d, RCHR′R′′), 7.22–7.24 (m, 0.1H3d, Ar-H), 7.27–7.37 (m, 2.4H3d′[b] + 1.2H3f′[a], Ar-H), 7.41–7.43 (m, 0.2H3d, Ar-H), 7.50–7.60 (m, 1.8H3d′[b] + 0.9H3d′[a] + 0.4H3d, Ar-H), 7.81–7.83 (m, 0.6H3d′[b], Ar-H), 7.86–7.88 (m, 0.3H3d′[b], Ar-H), 7.95–7.98 (m, 0.1H3d, Ar-H), 9.64 (br s, 0.1H3d, ROH). 13C-NMR (75 MHz, CDCl3) δ 28.2, 28.4, 35.1, 35.2, 35.8, 39.6, 42.0, 44.9, 98.9, 100.0, 101.0, 103.3, 110.4, 110.5, 115.5, 115.8, 116.5, 116.8, 116.9, 119.1, 122.9, 123.0, 124.0, 124.2, 128.1, 128.5, 129.1, 132.0, 132.1, 132.3, 132.4, 132.7, 148.4, 149.3, 153.1, 159.4, 161.3. HRMS (ESI+) calcd for [NaC20H15NO4]+ ([M + Na]+) 356.0899; found 356.0893.
:20, flow rate 1 ml min−1, λ = 279.4 nm): τmajor = 17.3 min; τminor = 33.2 min. [α]23D = +3.3 ± 0.2 (c 0.37, acetonitrile, 58% ee). {lit.,6a [α]25D = +16.6 (c 1.0, dichloromethane, 96% ee, (R)-3e)}.
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 13.1 min; τminor = 18.3 min. [α]23D = −22.1 ± 0.1 (c 0.69, acetonitrile, 67% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 13.7 min; τminor = 18.9 min. [α]23D = −24.3 ± 0.1 (c 1.00, acetonitrile, 67% ee).
:20, flow rate 1 ml min−1, λ = 271.0 nm): τmajor = 20.0 min; τminor = 30.7 min. [α]23D = −4.9 ± 0.1 (c 0.31, acetonitrile, 61% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 8.2 min; τminor = 13.6 min. [α]24D = −35.2 ± 0.1 (c 0.34, acetonitrile, 62% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 12.2 min; τminor = 19.4 min. [α]24D = −25.8 ± 0.2 (c 0.20, acetonitrile, 62% ee).
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 17.3 min; τminor = 31.6 min. [α]23D = −27.9 ± 0.1 (c 0.77, acetonitrile, 54% ee). IR (cm−1) 3353 (OH), 1694 (CO), 1614 (CO), 1510, 1241, 824, 730, 535. In CDCl3, the product was found to exist in a fast equilibrium between its open chain form 3k (≈20 mol%) and both pseudo-diastereomeric hemiketals 3k′ (≈40 mol%). 1H-NMR (400 MHz, CDCl3) δ 1.68 (s, 1.2H3k′, RCH3), 1.74 (s, 1.2H3k′, RCH3), 2.00 (dd, J1 = 14.0 Hz, J2 = 11.5 Hz, 0.4H3k′[a], RCH2R′), 2.04 (s, 0.2H3k, RCH3), 2.29 (s, 0.4H3k, RCH3), 2.38 (dd, J1 = 14.2 Hz, J2 = 6.8 Hz, 0.4H3k′[b], RCH2R′), 2.47 (dd, J1 = 14.1 Hz, J2 = 6.9 Hz, 0.4H3k′[b], RCH2R′), 2.54 (dd, J1 = 14.2 Hz, J2 = 3.0Hz, 0.4H3k′[a], RCH2R′), 3.11 (br s, 0.4H3k′, ROH), 3.25–3.30 (m, 0.4H3k′ + 0.2H3k, ROH + RCH2R′), 3.75–3.86 (m, 1.2H3k′[a] + 1.2H3k′[b] + 0.6H3k, ROCH3), 4.09–4.15 (m, 0.4H3k′ + 0.2H3k, RCHR′R′′ + RCH2R′), 4.24 (dd, J1 = 6.6 Hz, J2 = 2.8 Hz, 0.4H3k′, RCHR′R′′), 4.63 (dd, J1 = 10.4 Hz, J2 = 2.1 Hz, 0.2H3k, RCHR′R′′), 7.12–7.31 (m, 2H, Ar-H), 7.12–7.31 (m, 3H, Ar-H), 7.41–7.44 (m, 0.4H3k′ + 0.2H3k, Ar-H), 7.51 (dd, J1 = 8.8 Hz, J2 = 2.5 Hz, 0.4H3k′, Ar-H), 7.78 (d, J1 = 2.5 Hz, 0.4H3k′, Ar-H), 7.86 (d, J1 = 2.5 Hz, 0.4H3k′, Ar-H), 7.91 (d, J1 = 2.5 Hz, 0.2H3k, Ar-H), 9.56 (br s, 0.2H3k, ROH). 13C-NMR (100 MHz, CDCl3) δ 28.0, 28.5, 33.4, 34.7, 40.7, 42.6, 55.4, 55.4, 99.5, 101.0, 102.4, 113.8, 114.3, 114.9, 117.8, 118.2, 118.3, 122.5, 122.8, 123.7, 128.1, 128.2, 129.3, 129.3, 129.7, 131.6, 131.8, 132.1, 132.8, 134.8, 157.5. HRMS (ESI+) calcd for [NaC20H17ClO5]+ ([M + Na]+) 395.0662; found 395.0657.
:20, flow rate 1 ml min−1, λ = 272.2 nm): τmajor = 19.5 min; τminor = 36.9 min. [α]24D = −34.3 ± 0.1 (c 0.61, acetonitrile, 54% ee).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a 2018 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation. The authors also thank Ministerio de Economía, Industria y Competitividad (MINECO-FEDER CTQ2016-75816-C2-1-P and CTQ2017-88091-P), Universidad de Zaragoza (JIUZ-2017-CIE-05) and Gobierno de Aragón-Fondo Social Europeo (Research Group E07_17R) for financial support of their research. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- (a) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, Berlin-Heidelberg, 2nd edn, 2004 Search PubMed; (b) V. Caprio and J. M. J. Williams, Catalysis in Asymmetric Synthesis, Wiley-Blackwell, Oxford, 2nd edn, 2009 Search PubMed.
- (a) R. M. de Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 2575–2600 CrossRef CAS; (b) E. Marqués-López, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138–1167 RSC; (c) E. Marqués-López and R. P. Herrera, in Comprehensive Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2013, pp. 1359–1383 Search PubMed; (d) J. Alemán and S. Cabrera, Chem. Soc. Rev., 2013, 42, 774–793 RSC; (e) B.-F. Sun, Tetrahedron Lett., 2015, 56, 2133–2140 CrossRef CAS.
- For selected examples, see: (a) A. Breckenridge and M. L’E Orme, Life Sci., 1972, 11, 337–345 CrossRef CAS; (b) A. Breckenridge, M. Orme, H. Wesseling, R. J. Lewis and R. Gibbons, Clin. Pharmacol. Ther., 1974, 15, 424–430 CrossRef CAS; (c) R. A. O'Reilly, N. Engl. J. Med., 1976, 295, 354–357 CrossRef; (d) L. B. Wingard, R. A. O'Reilly and G. Levy, Clin. Pharmacol. Ther., 1978, 23, 212–217 CrossRef CAS; (e) V. Farina, J. T. Reeves, C. H. Senanayake and J. J. Song, Chem. Rev., 2006, 106, 2734–2793 CrossRef CAS.
- For selected examples, see: (a) B.-H. Xie, Z. Guan and Y.-H. He, J. Chem. Technol. Biotechnol., 2012, 87, 1709–1714 CrossRef CAS; (b) K. Sano, S.-i. Saito, Y. Hirose, Y. Kohari, H. Nakano, C. Seki, M. Tokiwa, M. Takeshita and K. Uwai, Heterocycles, 2013, 87, 1269–1278 CrossRef CAS; (c) W.-L. Liu, N.-S. Yang, Y.-T. Chen, S. Lirio, C.-Y. Wu, C.-H. Lin and H.-Y. Huang, Chem. – Eur. J., 2015, 21, 115–119 CrossRef CAS.
- For pioneering examples, see: (a) A. Robinson and H.-Y. Li, Tetrahedron Lett., 1996, 37, 8321–8324 CrossRef CAS; (b) N. Halland, T. Velgaard and K. A. Jørgensen, J. Org. Chem., 2003, 68, 5067–5074 CrossRef CAS; (c) Y. Tsuchiya, Y. Hamashima and M. A. Sodeoka, Org. Lett., 2006, 8, 4851–4854 CrossRef CAS.
- For selected recent examples, see: (a) J. Dong and D.-M. Du, Org. Biomol. Chem., 2012, 10, 8125–8131 RSC; (b) T. Shi, Z. Guo, H. Yu, J. Xie, Y. Zhong and W. Zhu, Adv. Synth. Catal., 2013, 355, 2538–2543 CrossRef CAS; (c) J. Kumagai, Y. Kohari, C. Seki, K. Uwai, Y. Okuyama, E. Kwon and H. Nakano, Heterocycles, 2015, 90, 1124–1134 CrossRef CAS; (d) M. Işik, H. U. Akkoca, İ. M. Akhmedov and C. Tanyeli, Tetrahedron: Asymmetry, 2016, 27, 384–388 CrossRef; (e) V. Modrocká, E. Veverková, M. Mečioravá and R. Šebesta, J. Org. Chem., 2018, 83, 13111–13120 CrossRef PubMed; (f) R.-Q. Mei, X.-Y. Xu, Y.-C. Li, J.-Y. Fu, Q.-C. Huang and L.-X. Wang, Tetrahedron Lett., 2011, 52, 1566–1568 CrossRef CAS . For a review, see: ; (g) A. Ricci, Org. Chem., 2014, 2014, 531695 Search PubMed.
- N. Halland, T. Hansen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 4955–4957 CrossRef CAS.
- For selected reviews, see: (a) A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS; (c) L.-W. Xu and Y. Lu, Org. Biomol. Chem., 2008, 6, 2047–2053 RSC; (d) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807–1821 RSC; (e) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC; (f) L.-W. Xu, L. Li and Z. H. Shi, Adv. Synth. Catal., 2010, 352, 243–279 CrossRef CAS and references cited in.
- H. Kim, C. Yen, P. Preston and J. Chin, Org. Lett., 2006, 8, 5239–5242 CrossRef CAS PubMed.
- J.-W. Xie, L. Yue, W. Chen, W. Du, J. Zhu, J.-G. Deng and Y.-C. Chen, Org. Lett., 2007, 9, 413–415 CrossRef CAS.
- A. S. Kucherenko, A. A. Kostenko, G. M. Zhdankina, O. Y. Kuznetsova and S. G. Zlotin, Green Chem., 2018, 20, 754–759 RSC.
- For a pivotal recent comprehensive summary, see: J. Lv, Q. Zhang, M. Cai, Y. Han and S. Luo, Chem. – Asian J., 2018, 13, 740–753 CrossRef CAS.
- For scarce examples using chiral primary aromatic amines, see: (a) A. Sakakura, K. Suzuki, K. Nakano and K. Ishihara, Org. Lett., 2006, 8, 2229–2232 CrossRef CAS; (b) T. Kano, Y. Tanaka, K. Osawa, T. Yurino and K. Maruoka, Chem. Commun., 2009, 1956–1958 RSC; (c) P. Galzerano, G. Bencivenni, F. Pesciaioli, A. Mazzanti, B. Giannichi, L. Sambri, G. Bartoli and P. Melchiorre, Chem. – Eur. J., 2009, 15, 7846–7849 CrossRef CAS PubMed; (d) T. Baba, J. Yamamoto, K. Hayashi, M. Sato, M. Yamanaka, T. Kawabata and T. Furuta, Chem. Sci., 2016, 7, 3791–3797 RSC.
- For scarce examples using chiral secondary aromatic amines, see: (a) T. Kano, Y. Tanaka and K. Maruoka, Org. Lett., 2006, 8, 2687–2689 CrossRef CAS PubMed; (b) T. Kano, Y. Tanaka and K. Maruoka, Tetrahedron, 2007, 63, 8658–8664 CrossRef CAS.
- (a) J. V. Alegre-Requena, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2016, 358, 1801–1809 CrossRef CAS; (b) J. V. Alegre-Requena, E. Marqués-López and R. P. Herrera, Chem. – Eur. J., 2017, 23, 15336–15347 CrossRef CAS; (c) J. V. Alegre-Requena, E. Marqués-López and R. P. Herrera, ACS Catal., 2017, 7, 6430–6439 CrossRef CAS.
- Only in a Diels Alder reaction reported by Ishigara's group, VII was the catalyst of choice (ref. 13a).
- Z. Dong, L. Wang, X. Chen, X. Liu, L. Lin and X. Feng, Eur. J. Org. Chem., 2009, 5192–5197 CrossRef CAS.
- CCDC 1913255 (3c′) contains the supplementary crystallographic data for this paper†.
- For selected examples, see: (a) C. A. Müller and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3363–3366 CrossRef; (b) C. A. Müller, C. Markert, A. M. Teichert and A. Pfaltz, Chem. Commun., 2009, 1607–1618 RSC; (c) V. M. Williams, J. R. Kong, B. J. Ko, Y. Mantri, J. S. Brodbelt, M.-H. Baik and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 16054–16062 CrossRef CAS; (d) Reactive Intermediates, ed. L. S. Santos, Wiley-VCH, Weinheim, 2010 Search PubMed; (e) E. Marqués-López, R. P. Herrera, T. Marks, W. C. Jacobs and M. Christmann, Synthesis, 2013, 1016–1028 CrossRef.
- H.-M. Yang, L. Li, K.-Z. Jiang, J.-X. Jiang, G.-Q. Lai and L.-W. Xu, Tetrahedron, 2010, 66, 9708–9713 CrossRef CAS.
- (a) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785–795 CrossRef CAS; (b) S. J. Connon, Chem. Commun., 2008, 2499–2510 RSC; (c) I. G. Sonsona, E. Marqués-López and R. P. Herrera, Beilstein J. Org. Chem., 2016, 12, 505–523 CrossRef CAS.
- M. Leven, J. M. Neudörfl and B. Goldfuss, Beilstein J. Org. Chem., 2013, 9, 155–165 CrossRef CAS.
- T. Stern, S. Rückbrod, C. Czekelius, C. Donner and H. A. Brunner, Adv. Synth. Catal., 2010, 352, 1983–1992 CrossRef CAS.
- J. Vicente, M. T. Chicote and I. Saura-Llamas, J. Chem. Ed., 1993, 70, 163–164 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1913255. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj02392e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |