
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast ring-opening of an intermediary α-stannyl-β-cyclopropylvinyl radical does not support formation of an α-stannylvinyl cation in the O-directed free radical hydrostannation of dialkyl acetylenes†
Hamish A.
Watson
,
Soraya
Manaviazar
,
Hannah G.
Steeds
and
Karl J.
Hale
 *
*
The School of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast BT9 5AG, Northern Ireland, UK. E-mail: k.j.hale@qub.ac.uk
First published on 15th November 2019
Abstract
O-directed hydrostannation of β-cyclopropyl propargyl alcohol 22 with stannanes and cat. Et3B in THF/H2O or PhMe/MeOH fails to deliver any detectable products of α-stannylvinyl cation capture. Instead only α-stannyl-β-cyclopropylvinyl radical intermediates can be detected, which undergo fast H-atom abstraction and/or cyclopropane ring-opening as a result of eliminative β-scission.
The O-directed, free radical hydrostannation of propargylically-oxygenated alkyl acetylenes with Ph3SnH/cat. Et3B and O2 in PhMe is a reaction of great synthetic worth for the highly stereocontrolled construction of (Z)-configured trisubstituted alkenes of high structural complexity.1,2 Given the superb performance of this reaction, in many extremely demanding settings,1,2 it is not at all surprising to find that there has been substantial interest in understanding the mechanistic origins of the high stereo- and regiocontrol that is typically observed.1–6
Our picture of these reactions1–3 (Scheme 1, mechanism 1) has them proceeding by a totally free radical mechanism in which an O-coordinated triphenylstannyl radical 31,3 preferentially adds to the α-carbon of the acetylene to give an inverting pair of bent stannylvinyl radicals 4, where lower hyperconjugative stabilisation in (E)-4, high internal A1,3- strain, and magnified steric repulsions collectively disfavour its H-atom abstraction from Ph3SnH. As a result, the more hyperconjugatively stabilised (Z)-invertomer (Z)-4 tends to preferentially react, since this does not experience such severe A1,3-strain, and it offers the least sterically hindered transition state for H-atom abstraction, which favours formation of the α-vinylstannane products (Z)-5. Although the initial O-directed addition can guide the O-coordinated tin radical to the β-acetylenic carbon of 3 to give (Z)-6, because this addition step is reversible, and the O-Sn complexation persists within the β-stannylvinyl radical adducts (Z)-6, this lengthens and weakens their C–Sn bonds, to inevitably promote their rapid dissociation back into 3. In contrast, the analogous elimination from the α-stannylvinyl radicals (Z)-4 is far less facile, due to the internal O–Sn coordination not persisting within such systems,1,3 with the result that H-atom abstraction is far more favourable, and the α-vinylstannanes of structure (Z)-5 typically predominate.
 | ||
| Scheme 1 The totally free radical O-directed mechanism for hydrostannation (mechanism 1), which operates at high Ph3SnH concentrations, and is experimentally supported.1–4 | ||
The above mechanistic analysis is one that has now been very strongly supported by X-ray crystallography,1,3 by Alabugin's computer-assisted DFT calculations,4 and by our own stannylvinyl radical trapping experiments with 8 (Scheme 2),3 which afforded a tandem β-addition/radical cyclisation product 10 at a range of stannane concentrations. Moreover, because the α:β addition step is reversible, and the regiochemistry changes significantly in favour of the O-directed α-addition product 9, as the stannane concentration increases (due to this promoting O–Sn complexation), this indicates that alkyne ground state polarity and electronic control cannot be the primary or exclusive determinants of product regiochemistry in these systems, for if these were solely governing outcome, then the regiochemistry of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 would never change as the stannane concentration altered; it would always remain constant. So, the O-directed free radical hydrostannation mechanism of Scheme 1 (mechanism 1) must be gaining the ascendancy over the non-directed, entirely free radical, addition/H-abstraction pathway (mechanism 2) as the Ph3SnH concentration rises.1,3
:10 would never change as the stannane concentration altered; it would always remain constant. So, the O-directed free radical hydrostannation mechanism of Scheme 1 (mechanism 1) must be gaining the ascendancy over the non-directed, entirely free radical, addition/H-abstraction pathway (mechanism 2) as the Ph3SnH concentration rises.1,3
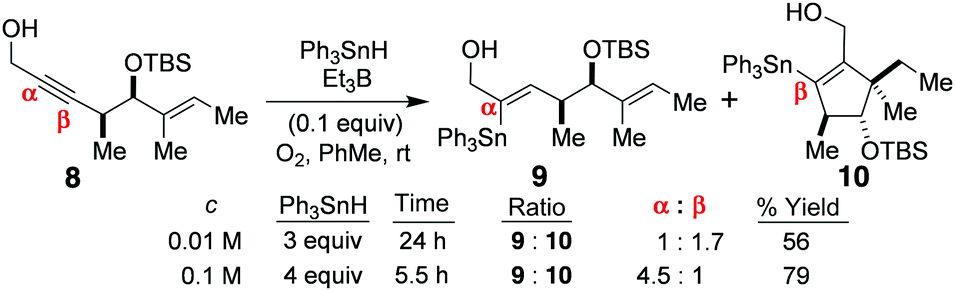 | ||
| Scheme 2 The changing regiochemistry at increased Ph3SnH concentrations, and the vinyl radical cyclisation product 10.3 | ||
Now despite the solidity of this work,3 a new mechanism was proffered in 2013–20145 (Scheme 3, mechanism 3); one where the α-vinylstannane products (Z)-15 were suggested to arise, not through a coordinately-controlled regioselective tin radical addition, but rather, through a non-O-directed, non-regioselective tin radical addition at either alkyne carbon of 11.
 | ||
| Scheme 3 The non-directed stannylvinyl cation mechanism5a,b for hydrostannation (mechanism 3) which is unsupported. | ||
In this respect, it was proposed that it would be of little real consequence if a mixture of α- and β-stannylvinyl radicals 13α and 13β ever arose, for these would both simultaneously undergo rapid single electron transfer (SET) to O2 present in the reaction medium, and this would lead to a pair of superoxide ion/α- and β-stannylvinyl cation ion pairs 14α and 14β, where the β-stannylvinyl cation would spontaneously undergo a 1,2-stannyl shift, to afford the more inductively stabilised α-stannylvinyl cation ion pair 14α exclusively. It was further suggested that the α-stannylvinyl cation in 14α would thereafter enjoy extra stabilisation from a dynamic partial bridging interaction with the tin substituent. It was also postulated that this α-stannylvinyl cation would subsequently engage in an ionic reduction with the excess stannane that was present, and that this would then afford the observed α-vinylstannane product (Z)-15, along with a tin cation 16, which itself would become involved in a second SET with the liberated superoxide ion, to return the O2 and a new R3Sn radical 17, which would further propagate the reaction chain.
A major problem1 with mechanism 35 is that vinyl radicals are known to add to O2 to form vinylperoxy radicals which then suffer a number of fates depending on the precise reaction conditions.1 Mechanism 3 also does not satisfactorily explain why the α:β-product regiochemistry (9:10) changes as the stannane concentration increases, when the alkyne 8 is hydrostannated (Scheme 2).3 This is because the tenets of mechanism 3 require the β-stannylvinyl cation to always spontaneously rearrange to its α-isomer,5 due to this being more stable, and the reaction being exclusively electronically controlled. Naturally, this would give 9 as the sole product. Yet, this is not what is observed, as evidenced by Scheme 2.3 Mechanism 35 is also incompatible with the observation of a β-stannylvinyl radical cyclisation product 10. The DFT calculations5a that accompany mechanism 3 also rule it out on simple thermodynamic grounds. In this regard, the postulated O2-mediated SET conversions of 13α to 14α and 13β to 14β (where R = H, and R1 = R2 = R3 = Me) are respectively associated with ΔE values of +47.27 kcal mol−1 and +45.2 kcal mol−1 for these two transitions in C6H6 at 25 °C (see Scheme 3). Such barriers are far too high to allow these conversions to proceed, as proposed, except through quantum mechanical tunnelling, but none of the traditional evidence needed to support such a mechanism has so far been provided by the proposers of mechanism 3. The fact also that the transition of 13α into 14α is higher in energy than the one for the transition of 13β into the more inductively destabilised 14β also seems discrepant. The conversion of 14β to 14α is also uphill to the tune of +2.1 kcal mol−1.5a Moreover, the rate accelerations that the mechanism 3 proposers have reported5c for the hydrostannations of alkyne 18 in polar solvents, over 90 min (Scheme 4), are likewise far too small to be considered supportive of ionic mechanism 3.1 Instead, they are much more aligned1 with an entirely free radical, O-directed, mechanism of the type shown in Scheme 1 (mechanism 1).1 Indeed, relative to THF, a net decrease in the reaction rate5c is observed for the hydrostannation of 18 in more polar solvents.
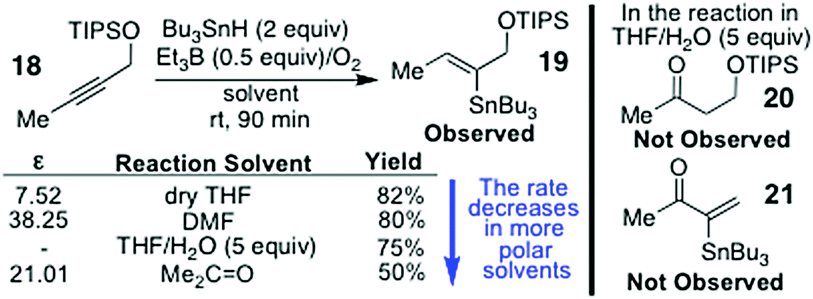 | ||
| Scheme 4 The polar solvent rate data of ref. 5c that is claimed to support mechanism 3. | ||
Now, despite the electronically-controlled stannylvinyl cationic ion pair hypothesis5 of Scheme 3 (mechanism 3) conflicting very strongly with the 2005 radical probe work of Scheme 2,1,3 and Alabugin's later 2015 experimental work and DFT calculations,4 which further confirmed an entirely free radical O-directed mechanism for these reactions (mechanism 1), still mechanism 35 remains active and unwithdrawn from the literature, despite several criticisms,1,4,6 with the consequence that its mechanistic principles are still continuing to be widely disseminated within the community. This is best illustrated by a recent 2019 hydrostannation review,7 where exclusivity of discussion and mechanistic primacy were given to mechanism 3 over mechanism 1 to explain the conversion of 11 into (Z)-15. However, as we have seen, mechanism 35 is at great odds with all experimental data gathered to date.1,3–5
Given this continuing propagation of a mechanism5 that has been significantly criticised1,4,6 (mechanism 35), over one that is much more reasonable and fully experimentally supported1,3,4 (mechanism 11–3), we felt it necessary to publish the results of our latest investigations into whether a stannylvinyl cation is a genuine intermediate in these reactions, since some in the field clearly seem unwilling to accept the unambiguous data that has been presented in Scheme 21,3 and elsewhere,4 which all rule against it.
Therefore, to test mechanism 3 further (Scheme 5), we purposely designed a β-cyclopropyl propargylic alcohol probe 22 that we believed would give rise to an exceptionally stable, yet still quite reactive, stannylvinyl cation intermediate 27, if it happened that mechanism 3 did hold true. It was reasoned that if 27 was indeed an authentic intermediate5 in the journey of 22 into vinylstannane 26, then this super-stabilised vinyl cation 27 would almost certainly react competitively with H2O and alcohols added to the reaction medium since it would be triply stabilised: (a) by neighbouring group participation from the ester carbonyl; (b) by hyperconjugation with the α-R3Sn substituent; and (c) by hyperconjugation with the adjacent cyclopropane ring. It should thus have sufficient longevity in solution to undergo nucleophilic trapping by: (1) externally added nucleophiles present in excess (e.g. H2O or MeOH) or (2) by the internal ester carbonyl to give a six-membered enol lactone (28), after subsequent nucleophilic attack on the resulting positively charged OMe group by the external nucleophile. A trapping of 27 with H2O would, of course, lead to ketone or stannyl enone products such as 31 and 30via29 (Scheme 5), or potentially bring about a competitive Stang–Muller elimination8 through an attack of the H2O on the R3Sn grouping.8a The latter would naturally return the starting acetylene 22 and lead to little overall reaction progression.
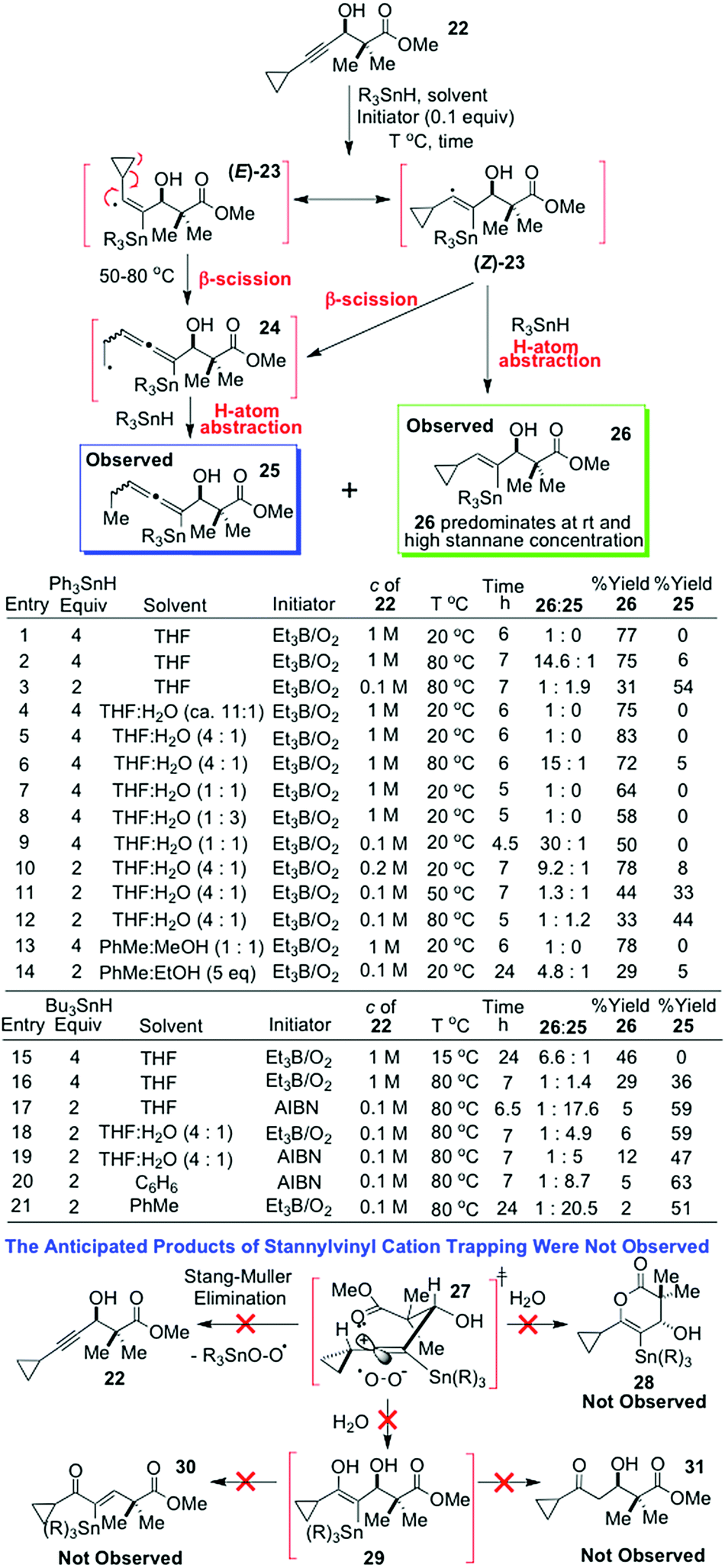 | ||
| Scheme 5 Our various hydrostannations of the probe 22. | ||
With regard to the feasibility of achieving an α-cyclopropyl vinyl cation trapping with H2O, the classic work of Bergman9 and Hanack10 had previously shown that although α-cyclopropyl vinyl cations benefit from significant charge stabilization from the cyclopropane ring, they still retain the ability to react rapidly with external nucleophiles such as H2O or acetate ion, either directly, or following internal 1,2-rearrangement. Muller's highly stable bis-silyl-vinyl cations, which have even been characterised by X-ray crystallography likewise react readily with nucleophiles, but by an eliminative nucleophilic attack on silicon to give an alkynylsilane.8b
Most importantly as well, however, the probe 22 would inevitably afford the α-stannylvinyl radical precursor of the postulated cation 27viz. radical 23, and if, by due modification of the hydrostannation conditions, we could identify a set of experimental circumstances where 23 could be induced to predominantly undergo fast H-atom abstraction from the stannane to give 26 while also undergoing concurrent vinyl radical eliminative β-scission of the cyclopropane ring to give the stannylhomoallenyl radical 24,11 and vice versa, where eliminative β-scission could be rendered more facile than concurrent stannylvinyl radical H-atom abstraction by 23, then this would unambiguously confirm that the proposed H-atom transfer must actually be proceeding through the α-stannylvinyl radical, and that 26 was not arising from an ionic reduction of the stannylvinyl cation 27 by the tin hydride. In essence, it would support mechanism 1 and rule out mechanism 3.
With this in mind, great experimental effort went into identifying conditions that would give this experimental read-out, and a summary of that effort can be found in the tabulated hydrostannation data of Scheme 5, which very clearly shows that only two primary products ever emerge from the free radical hydrostannation reactions of 22, and these are compounds 26 and 25, even when reactive nucleophiles such as H2O or alcohols are present in far greater excess than the stannane. Such observations only satisfactorily align with the intermediacy of a stannylcyclopropylvinyl radical,11 not a stannylvinyl cation,5 and these outcomes powerfully reconfirm the entirely free radical mechanism1,3 of such dialkyl acetylene hydrostannations (mechanism 1).1–4,6
Moreover, if one examines entry 10 in Scheme 5, one can see that the degree of conversion of the alkyne 22 into the hydrostannylated products 26 and 25 is very good indeed after 7 h at rt (86% overall yield, 26:25 = 9.2:1) in THF/H2O (4:1) at 0.2 M substrate concentration using 2 equiv. of Ph3SnH. The high yield observed indicates that a stannylvinyl cation of structure 27 cannot be a viable intermediate in these reactions, since such an α-stannylvinyl cation would almost certainly undergo significant facile elimination back to the starting acetylene8a in the presence of a strong nucleophile such as H2O or peroxide anion (Stang–Muller elimination8). However, only a minute amount of starting alkyne was ever present after 7 h at rt. In fact, when this same rt reaction was conducted with a much greater quantity of Ph3SnH (4 equiv.) in THF/H2O (4:1) at 1 M substrate concentration (entry 5), total product conversion occurred, with no starting acetylene 22 remaining at reaction end (see TLC in the ESI†). In this instance, 26 was obtained exclusively in 83% yield (entry 5). Additionally, no cyclopropyl ketone 31 or cyclopropyl stannyl enone β-elimination products such as 30 were ever detected in either of these two reactions from direct stannylvinyl cation capture by the excess H2O present. This outcome weighs heavily against the intermediacy of stannylvinyl cations5 in such hydrostannations. Also, no 1,2-shifted cyclobutenols9,10 were ever detected, nor primary stannylallenyl alcohols arising from direct competitive nucleophilic ring-opening of the cyclopropane ring in 27 with H2O.
The formation of only a minor quantity of the reduced allenyltin product 25 in entry 10, alongside the vinylstannane 26 is clearly only compatible with the intermediacy of a stannylvinyl radical intermediate 23 that primarily undergoes fast H-atom abstraction12 from the stannane at rt, alongside competitive radical induced eliminative opening of the cyclopropane ring with accompanying H-atom abstraction (see Scheme 5), a process well known for α-cyclopropylvinyl radicals.11 This point is perhaps much better illustrated by entry 11 where, when the hydrostannation reaction was performed at 0.1 M substrate concentration and lower Ph3SnH concentration (2 equiv.), in a mixture of THF/H2O (4:1) at 50 °C for 7 h, the ratio of 26:25 now changed very dramatically to 1.3:1. Clearly, working at higher dilutions and higher reaction temperatures with lower quantities of stannane quite profoundly affects the behaviour of the intermediary stannylvinyl radical 23, it now diverting it significantly down the pathway of competitive radical induced ring-opening, alongside stannylvinyl radical H-atom abstraction from the stannane. Indeed, heating at 80 °C with the same quantity of Ph3SnH (entry 12) promotes even more of the radical-induced β-scission event,11 as does performing the alkyne hydrostannation with Bu3SnH (entries 16–21) which typically affords 25 and 26 in much lower yields than does Ph3SnH.
So, to conclude, the combined observations recorded here, using a new type of vinyl radical clock,11 once more fail to support1,3,4,6 the intermediacy of an α-stannylvinyl cation5,7 in the O-directed free radical hydrostannation of alkyl acetylenes.1–6 Instead, the tabulated data of Schemes 5 and 2 only support the operation of mechanisms 1 and 2 in these reactions with the former massively predominating under the excess stannane conditions that we typically employ.1–4
Conflicts of interest
There are no conflicts to declare.Notes and references
- For a review on the rt O-directed hydrostannation with Ph3SnH/cat. Et3B in PhMe and its mechanism, see: K. J. Hale, S. Manaviazar and H. A. Watson, Chem. Rec., 2019, 19, 238 CrossRef CAS
.
-
(a) P. Dimopoulos, A. Athlan, S. Manaviazar, J. George, M. Walters, L. Lazarides, A. E. Aliev and K. J. Hale, Org. Lett., 2005, 7, 5369 CrossRef CAS
- P. Dimopoulos, J. George, D. A. Tocher, S. Manaviazar and K. J. Hale, Org. Lett., 2005, 7, 5377 CrossRef CAS
- K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165 CrossRef CAS
-
(a) M. S. Oderinde, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2013, 52, 11334 CrossRef CAS PubMed
- D. P. Curran and T. McFadden, J. Am. Chem. Soc., 2016, 138, 7741 CrossRef CAS PubMed
- M. Alami, A. Hamze and O. Provot, ACS Catal., 2019, 9, 3437 CrossRef CAS
-
(a) L. Williamson, P. J. Stang and A. M. Arif, J. Am. Chem. Soc., 1993, 115, 2590 CrossRef
- S. A. Sherrod and R. G. Bergman, J. Am. Chem. Soc., 1971, 93, 1925 CrossRef
- M. Hanack and T. Baessler, J. Am. Chem. Soc., 1969, 91, 2117 CrossRef CAS
-
(a) K. K. Milnes, S. E. Gottschling and K. M. Baines, Org. Biomol. Chem., 2004, 2, 3530 RSC
- L. J. Johnston, J. Lusztyk, D. D. M. Wayner, A. N. Abeywickreyma, A. L. J. Beckwith, J. C. Scaiano and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 4594 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05492h |
| This journal is © The Royal Society of Chemistry 2019 |