
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBa5Cu8In2S12: a quaternary semiconductor with a unique 3D copper-rich framework and ultralow thermal conductivity†
Hua
Lin
ab,
Hong
Chen
ab,
Yu-Jun
Zheng
abc,
Yu-Kun
Chen
ab,
Ju-Song
Yu
abc and
Li-Ming
Wu
 *ab
*ab
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, People's Republic of China
bKey Laboratory of Optoelectronic Materials Chemistry and Physics, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, People's Republic of China. E-mail: liming_wu@fjirsm.ac.cn; Tel: +86-591-63173130
cUniversity of Chinese Academy of Sciences, Beijing 100039, People's Republic of China
First published on 31st January 2017
Abstract
A novel quaternary sulfide, Ba5Cu8In2S12 (1), has been successfully synthesized via a high-temperature solid-state reaction. It contains Cu8S10S4/2 clusters as basic building blocks, which are connected to one another by discrete In3+ ions to generate a 3D copper-rich framework, where the Ba2+ cations reside. Interestingly, such large clusters that are fused by five crystallographically independent Cu sites with three different chemical environments result in the increase of phonon scattering, which is the crucial factor to the exceptionally low lattice thermal conductivity (ca. 0.28 W m−1 K−1 at 773 K) in 1.
Solid-state cuprous- and argentous-based chalcogenides have been extensively investigated for several decades not only for their promising applications in thermoelectric (TE) fields, such as Cu2−xSe,1 Cu7PSe6,2 AgCuSe,3 AgSbTe24 and CsAg5Te3,5 but also for their intriguing architectures and topologies, for instance, monovalent Cu/Ag can create various asymmetric local environments to the chalcogenides due to the lack of crystal field stabilization, forming linear, trigonal, and tetrahedral geometries. On the other hand, group 13-containing chalcogenides containing MQ4 tetrahedra (e.g., M = Ga, In) serve as the smallest structural units and have also attracted intensive research. From the point of view of crystallography, these structural units are always linked together by sharing corners or edges to form 1D chains, 2D layers, and 3D networks. Also, most of them show exciting TE properties. For instance, InTe, a mixed-valent 1D compound, exhibits an ultralow lattice thermal conductivity (κL ≈ 0.4 W m−1 K−1) in high-quality crystalline ingots.6 2D layered In4PbxSnySe3 is an exceptional candidate for mid-temperature TE materials with a high figure of merit ZT value of 1.4 at 733 K.7 CuGaTe2 and CuInTe2 show high-efficiency TE performances based on the 3D diamond-like frameworks.8
To date, a large number of ternary A/TM/Q and A/M/Q (A = cations; TM = coinage metals; M = group 13 metals; and Q = chalcogenides) systems with diverse crystal structures have been explored and synthesized. However, the existing examples for quaternary compounds based on the A/TM/M/Q systems are limited to only K2TMM3Se6 (TM = Cu, Ag; M = Ga, In),9 CsAgGa2Se4,10 Ba7AgGa5Se15,11 Ba2AgInS4,12 Ba4TMGa5Q12 (TM = Cu, Ag; Q = S, Se),12,13 Ba4TMMSe6 (TM = Cu, Ag; M = Ga, In; Q = S, Se),14 Pb8TMIn17S34 (TM = Cu, Ag, Au),15 RE2CuInQ5 (RE = La–Nd, Sm; Q = S, Se),16 La4Ag2In4S13,17 and RE3CuGaQ7 (RE = La–Nd; Q = S, Se) compounds.18 Moreover, all of them are not coinage metal-rich compounds, i.e., the ratio of TM/M ≤ 1. In recent years, a large family of natural minerals called tetrahedrites has received worldwide intensive investigation due to their unique structural characteristics (e.g., large Cu/Sb ratio and different coordination geometries of Cu atoms) and their promising thermoelectric applications. For example, the pure synthetic tetrahedrite Cu12Sb4S13 has a ZT value of approximately 0.6 at 700 K; the values of transition-metal-substituted tetrahedrite compounds are in the range of 0.7–1.1.19 In this communication, we report a novel quaternary semiconductor, Ba5Cu8In2S12 (1), with a unique 3D copper-rich framework (Cu/In = 4) and unusual Cu coordination that exhibits ultralow thermal conductivity.
Air-stable 1 was prepared via a solid-state reaction by heating of a mixture of elements Ba, Cu, In, and S at 1223 K (for details, see the ESI†). The thermal stability of 1 was investigated by TG-DTA analysis on a NETZSCH STA 449C simultaneous analyser. As shown in Fig. 1a, compound 1 has excellent thermal stability and shows no obvious weight loss within the entire experimental temperature range. Moreover, the DTA data show only one sharp endothermic peak at around 968 K in the heating curve and one sharp exothermic peak at 902 K in the cooling curve, which indicates that 1 is a congruently melting compound. In order to further verify its thermal behaviour, the powder X-ray diffraction (PXRD) patterns recorded before and after DTA indicated the full recovery of compound 1 (Fig. 1b).
 | ||
| Fig. 1 Measurements on 1: (a) TG and DTA (inset) diagrams. (b) PXRD of simulated (black), as-synthesized (red), and DTA products (blue). | ||
Single-crystal XRD analysis reveals that 1 crystallizes in the monoclinic space group C2/c (no. 15) with a = 14.584(2) Å, b = 16.369(2) Å, c = 10.509(2) Å, β = 100.14(2)° and Z = 4.‡ In the asymmetric unit, the structure of 1 consists of 15 crystallographically independent sites, which include three Ba sites, one In site, six S sites and five Cu sites (the corresponding Cu5 atom splits into three sites, Cu5A, Cu5B, and Cu5C, with occupancies of 17(4)%, 42(2)%, and 41(2)%, respectively). As shown in Fig. 2, the 3D copper-rich Cu–In–S open framework was constructed by 1D [Cu8S12]16− chains (dashed area in Fig. 2) of vertex sharing S atoms. The channels along the c-direction were occupied by Ba2+ cations.
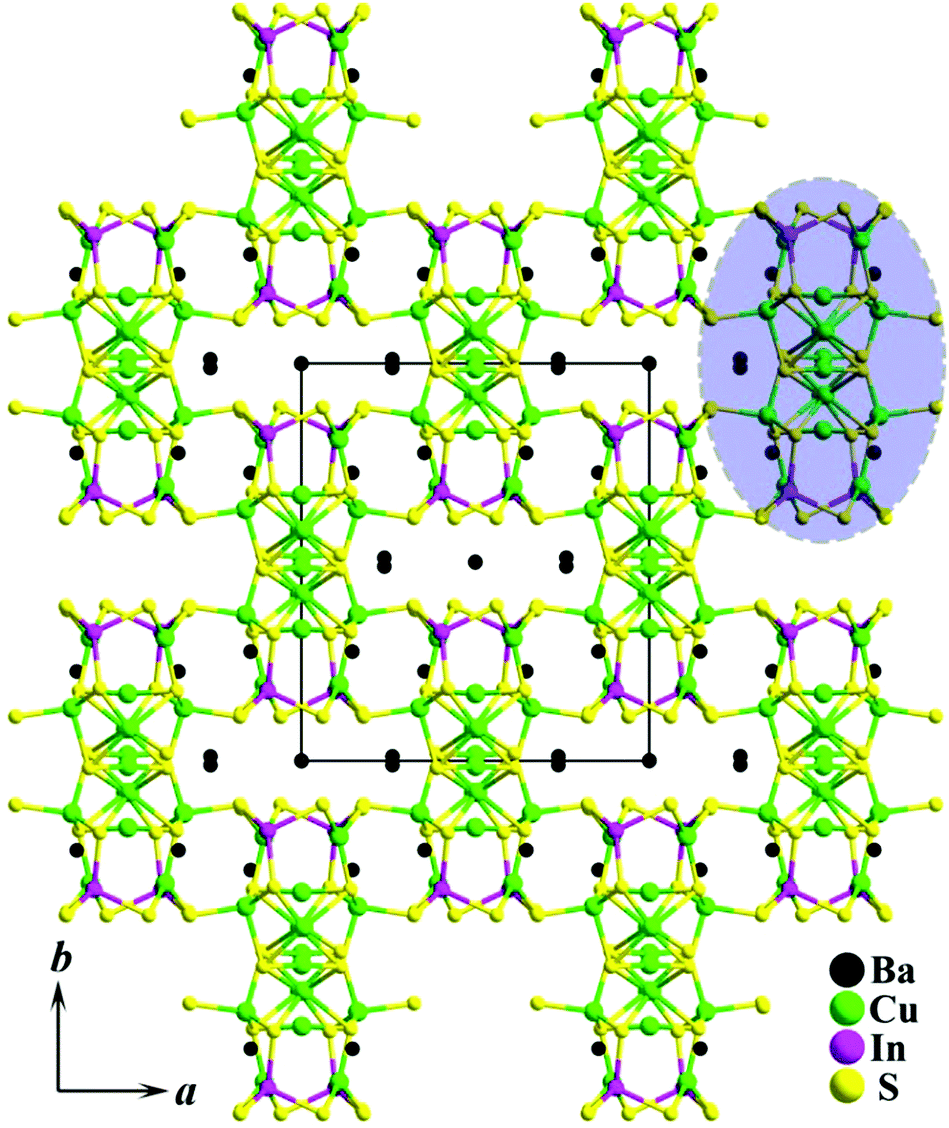 | ||
| Fig. 2 3D framework structure of 1 viewed down the c-axis with the unit cell marked (1D [Cu8S12]16− chain is outlined by a dashed area; for details, see Fig. 3). | ||
The critical building block of 1 is the Cu8S10S4/2 cluster, as shown in Fig. 3a, which consists of three types of coordination modes. The coordination geometry of Cu atoms is shown in Fig. 3b, where Cu1 and Cu5 are 3-fold coordinated in an approximate plane triangle, Cu2 and Cu4 are 2-fold coordinated in an approximate line, and Cu3 is 4-fold coordinated in a distorted tetrahedron. Each cluster is interconnected with other two neighbouring clusters by sharing four S6 atoms, which gives rise to 1D [Cu8S12]16− chains along the 〈100〉 direction with a width of 13.6 Å (Fig. 3c). These chains are further linked together by discrete In3+ ions to form a 3D open framework. The Cu atoms show normal Cu–S distances, ranging from 2.142(4) to 2.574(4) Å and the In atom is four-fold coordinated in a distorted tetrahedral sphere with In–S bond distances of 2.444(4)–2.510(4) Å. There are three crystallographically independent Ba atomic sites with two different chemical environments, as shown in Fig. S1 (ESI†). Both Ba1 and Ba3 exhibit a square prismatic coordination with eight Ba–S bonds of distances between 3.191(4) and 3.596(6) Å, whereas the Ba2 atom is surrounded by six close S atoms at the corners of a distorted triangular prism with Ba–S distances of 3.057(4)–3.319 (4) Å and two capping S atoms at a further distance of 3.712(4)–3.921 (4) Å. Such an interesting coordination geometry of the Ba2 atom is also observed in Ba12In4S19 and Ba4M2S8 (M = Ga, In).20
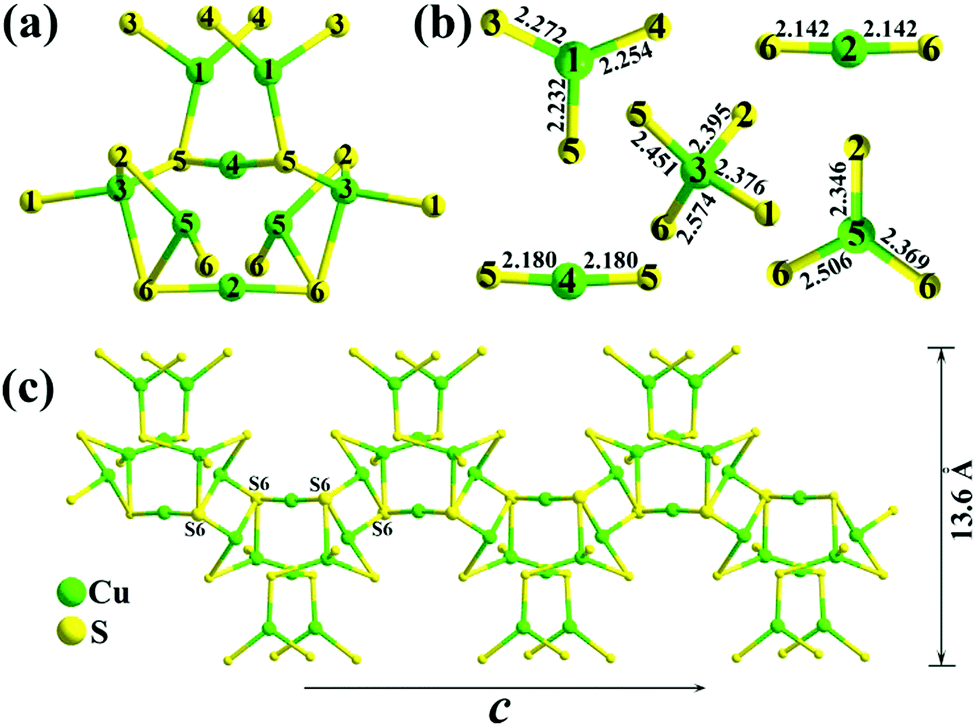 | ||
| Fig. 3 (a) Cu8S10S4/2 cluster with the atom number marked. (b) Coordination geometry of Cu atoms with Cu–S distances. (c) 1D [Cu8S12]16− chain along the 〈100〉 direction with the width shown in the right side. The split site of atom Cu5 is omitted for a better view. | ||
According to the solid-state optical absorption spectrum at room temperature, the energy gap (Eg) of 1 is estimated to be 2.14 eV (Fig. 4a), which is in agreement with its dark-red colour and suggests a semiconductor behaviour. To understand the nature of compound 1, the band structures together with the projected density of state (PDOS) calculations have been studied. As shown in Fig. 4b, the electronic structure calculations reveal an indirect Eg of 1.09 eV for 1, which is smaller than the measured value due to the well-known tendency of these calculations to underestimate the value of Eg. The PDOS between +5.0 and −7.0 eV, with the Fermi level at 0 eV, is shown in Fig. 4c. The major contribution to the valence band maximum (VBM) is from the S-3p and Cu-3d states, while the conduction band minimum (CBM) mainly consists of S-3p, In-5s, Cu-3d and Ba-5d states. Therefore, the optical absorption in 1 is mainly ascribed to the charge transitions from S-3p states to In-5s, Cu-3d and Ba-5d states.
 | ||
| Fig. 4 (a) Solid-state optical absorption spectrum of 1. (b) The calculated band structure of 1. (c) The projected DOS of 1 (the orbitals with minor contributions are omitted for clarity). (d) Thermal conductivity as a function of the temperature for a hot-pressed polycrystalline sample of 1 during two thermal cycles. | ||
The thermal conductivity (κtotal) and electrical conductivity (σ) of 1 have been measured on hot-pressed polycrystalline pellets, the relative densities of which are more than 98% of the theoretical value. As shown in Fig. 4d, the κtotal value decreases with increasing temperature from 0.39 W m−1 K−1 at 300 K to 0.28 W m−1 K−1 at 773 K. This decrease in κtotal with increasing temperature indicates that the phonon contribution to the conductivity is predominant. This is consistent with the very low thermal diffusivity (D) as a function of temperature (Fig. S2, in the ESI†) and the insulating character of the electrical transport properties (e.g., 1.75 × 10−10 S cm−1 at 300 K for 1; for details, see Fig. S3 in the ESI†). The thermal conductivity of 1 is one of the lowest reported for crystalline materials. Such a value is more interesting to compare with the exceptional TE materials in the range of 300–800 K, such as the highest ZT value in the SnSe crystal, which exhibits an ultralow κL value (0.25–0.68 W m−1 K−1), is due to the strong anharmonicity of chemical bonds in this layered compound,21 phonon-liquid electron-crystal (PLEC) materials Cu2−xS (0.30–0.55 W m−1 K−1)22 and Cu7PSe6 (0.23–0.34 W m−1 K−1),2 and typical phonon-glass electron-crystal (PGEC) materials, e.g. multiple-filled skutterudites (Ba0.08La0.05Yb0.04Co4Sb12, 1.61–0.97 W m−1 K−1),23 Zintl phases (Yb14MnSb11,24 0.61–0.47 W m−1 K−1, and Gd117Co56Sn112, 0.3–0.5 W m−1 K−1),25 clathrates (Ba8Ga16Ge30,26 0.20–0.46 W m−1 K−1, and Ba8Au16P30,27 0.15–0.45 W m−1 K−1), tetrahedrites (Cu12−xZnxSb4S12,19a 0.15–1.02 W m−1 K−1, and Cu12Sb4S12Se,19f 0.31–0.72 W m−1 K−1), highly disordered compound Zn4Sb3 (0.27–0.42 W m−1 K−1),28 semiconducting sulfosalt LiPbSb3S6 (0.21–0.24 W m−1 K−1)29 and Ag9TlTe5 (<0.25 W m−1 K−1).30 In addition, the ultralow thermal conductivity of 1 appears to be similar in origin to the recently reported CsAg5Te35 and likely results from the concerted rattling of Cu atoms with different chemical environments in the structure.
In summary, the first quaternary 3D copper-rich sulfide framework, Ba5Cu8In2S12, in the A/TM/M/Q system (A = cations; TM = coinage metals; M = group 13 metals; Q = chalcogenides) has been discovered. The remarkable structural feature is the novel Cu8S10S4/2 clusters with the Cu atoms in three different chemical environments. More interestingly, such an extraordinary group of Cu atoms in the structure results in an ultralow lattice thermal conductivity (ca. 0.28 W m−1 K−1) at around 800 K. This new insight will shed useful light on the search and design of promising thermoelectric materials with exceedingly low thermal conductivities.
This work was supported by the National Natural Science Foundation of China (21171168, 21225104, 21233009, 21301175, 21571020 and 91422303) and the Natural Science Foundation of Fujian Province (2015J01071).
Notes and references
- H. L. Liu, X. Shi, F. F. Xu, L. L. Zhang, W. Q. Zhang, L. D. Chen, Q. Li, C. Uher, T. Day and G. J. Snyder, Nat. Mater., 2012, 11, 422 CrossRef CAS PubMed.
- K. S. Weldert, W. G. Zeier, T. W. Day, M. Panthofer, G. J. Snyder and W. Tremel, J. Am. Chem. Soc., 2014, 136, 12035 CrossRef CAS PubMed.
- S. Ishiwata, Y. Shiomi, J. S. Lee, M. S. Bahramy, T. Suzuki, M. Uchida, R. Arita, Y. Taguchi and Y. Tokura, Nat. Mater., 2013, 12, 512 CrossRef CAS PubMed.
- T. J. Zhu, H. L. Gao, Y. Chen and X. B. Zhao, J. Mater. Chem. A, 2014, 2, 3251 CAS.
- H. Lin, G. J. Tan, J. N. Shen, S. Q. Hao, L. M. Wu, N. Calta, C. Malliakas, S. Wang, C. Wolverton and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2016, 55, 11431 CrossRef CAS PubMed.
- M. K. Jana, K. Pal, U. V. Waghmare and K. Biswas, Angew. Chem., Int. Ed., 2016, 55, 7792 CrossRef CAS PubMed.
- Z. S. Lin, L. Chen, L. M. Wang, J. T. Zhao and L. M. Wu, Adv. Mater., 2013, 25, 4800 CrossRef CAS PubMed.
- (a) T. Plirdpring, K. Kurosaki, A. Kosuga, T. Day, S. Firdosy, V. Ravi and G. J. Snyder, Adv. Mater., 2012, 24, 3622 CrossRef CAS PubMed; (b) R. H. Liu, L. L. Xi, H. L. Liu, X. Shi, W. Q. Zhang and L. D. Chen, Chem. Commun., 2012, 48, 3818 RSC.
- H. W. Ma, G. C. Guo, M. S. Wang, G. W. Zhou, S. H. Lin, Z. C. Dong and J. S. Huang, Inorg. Chem., 2003, 42, 1366 CrossRef CAS PubMed.
- D. J. Mei, W. L. Yin, K. Feng, L. Bai, Z. S. Lin, J. Y. Yao and Y. C. Wu, J. Solid State Chem., 2012, 186, 54 CrossRef CAS.
- W. L. Yin, R. He, K. Feng, W. Y. Hao, J. Y. Yao and Y. C. Wu, J. Alloys Compd., 2013, 565, 115 CrossRef CAS.
- W. L. Yin, K. Feng, D. J. Mei, J. Y. Yao, P. Z. Fu and Y. C. Wu, Dalton Trans., 2012, 41, 2272 RSC.
- S. M. Kuo, Y. M. Chang, I. Chung, J. I. Jang, B. H. Her, S. H. Yang, J. B. Ketterson, M. G. Kanatzidis and K. F. Hsu, Chem. Mater., 2013, 25, 2427 CrossRef CAS.
- J. L. Yao, Z. X. Wang, J. van Tol, N. S. Dalal and J. A. Aitken, Chem. Mater., 2010, 22, 1647 CrossRef CAS.
- C. K. Wang and S. C. Lee, Inorg. Chem., 2006, 45, 1415 CrossRef PubMed.
- M. R. Huch, L. D. Gulay, I. D. Olekseyuk and A. Pietraszko, J. Alloys Compd., 2006, 425, 230 CrossRef CAS.
- L. D. Gulay, M. Daszkiewicz and M. R. Huch, J. Solid State Chem., 2008, 181, 2626 CrossRef CAS.
- (a) X. Zhang, W. Chen, D. J. Mei, C. Zheng, F. H. Liao, Y. T. Li, J. H. Lin and F. Q. Huang, J. Alloys Compd., 2014, 610, 671 CrossRef CAS; (b) A. K. Iyer, B. W. Rudyk, X. S. Lin, H. Singh, A. Z. Sharma, C. R. Wiebe and A. Mar, J. Solid State Chem., 2015, 229, 150 CrossRef CAS.
- (a) X. Lu, D. T. Morelli, Y. Xia, F. Zhou, V. Ozolins, H. Chi, X. Zhou and C. Uher, Adv. Energy Mater., 2012, 3, 342 CrossRef; (b) X. Lu and D. T. Morelli, Phys. Chem. Chem. Phys., 2013, 15, 5762 RSC; (c) J. Heo, G. Laurita, S. Muir, M. A. Subramanian and D. A. Keszler, Chem. Mater., 2014, 26, 2047 CrossRef CAS; (d) R. Chetty, A. Bali, M. H. Naik, G. Rogl, P. Rogl, M. Jain, S. Suwas and R. C. Mallik, Acta Mater., 2015, 100, 266 CrossRef CAS; (e) X. Lu, D. T. Morelli, Y. Xia and V. Ozolins, Chem. Mater., 2015, 27, 408 CrossRef CAS; (f) X. Lu, D. T. Morelli, Y. X. Wang, W. Lai, Y. Xia and V. Ozolins, Chem. Mater., 2016, 28, 1781 CrossRef CAS.
- J. W. Liu, P. Wang and L. Chen, Inorg. Chem., 2011, 50, 5706 CrossRef CAS PubMed.
- L. D. Zhao, S. H. Lo, Y. S. Zhang, H. Sun, G. J. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373 CrossRef CAS PubMed.
- Y. He, T. Day, T. S. Zhang, H. L. Liu, X. Shi, L. D. Chen and G. J. Snyder, Adv. Mater., 2014, 26, 3974 CrossRef CAS PubMed.
- X. Shi, J. Yang, J. R. Salvador, M. F. Chi, J. Y. Cho, H. Wang, S. Q. Bai, J. H. Yang, W. Q. Zhang and L. D. Chen, J. Am. Chem. Soc., 2011, 133, 7837 CrossRef CAS PubMed.
- S. R. Brown, S. M. Kauzlarich, F. Gascoin and G. J. Snyder, Chem. Mater., 2006, 18, 1873 CrossRef CAS.
- D. C. Schmitt, N. Haldolaarachchige, Y. M. Xiong, D. P. Young, R. Y. Jin and J. Y. Chan, J. Am. Chem. Soc., 2012, 134, 5965 CrossRef CAS PubMed.
- E. S. Toberer, M. Christensen, B. B. Iversen and G. J. Snyder, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075203 CrossRef.
- J. Fulmer, O. I. Lebedev, V. V. Roddatis, D. C. Kaseman, S. Sen, J.-A. Dolyniuk, K. Lee, A. V. Olenev and K. Kovnir, J. Am. Chem. Soc., 2013, 135, 12313 CrossRef CAS PubMed.
- J. P. Lin, X. Q. Li, G. J. Qiao, Z. Wang, J. Carrete, Y. Ren, L. Z. Ma, Y. J. Fei, B. F. Yang, L. Lei and J. Li, J. Am. Chem. Soc., 2014, 136, 1497 CrossRef CAS PubMed.
- E. C. Agha, C. D. Malliakas, J. Im, H. Jin, L. D. Zhao, A. J. Freeman and M. G. Kanatzidis, Inorg. Chem., 2014, 53, 673 CrossRef CAS PubMed.
- K. Kurosaki, A. Kosuga, H. Muta, M. Uno and S. Yamanaka, Appl. Phys. Lett., 2015, 87, 061919 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data, experimental section and theoretical studies, together with additional figures and tables. CCDC 1504921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc09904a |
| ‡ Crystal data for 1 at 293(2) K: monoclinic; C2/c; Z = 4; a = 14.584(15) Å, b = 16.369(17) Å, c = 10.509(12) Å, β = 100.14(2)°, V = 2469(5) Å3. Collection and refinement data: dcalc = 4.867 g cm−3; μ = 17.386 mm−1; 9529 total reflections; 2835 unique reflections [Fo2 > 2σ(Fo2)]; GOF = 1.134; R1 = 4.79% and wR2 = 7.87% for I > 2σ(I). CCDC 1504921. |
| This journal is © The Royal Society of Chemistry 2017 |