
Crystal-to-crystal photo-reversible polymerization mechanism of bis-thymine derivative†
Akihiro Udagawaa,
Priscilla Johnstonb,
Aya Sakonc,
Ryosuke Toyoshimac,
Hidehiro Uekusa c,
Hideko Koshimad,
Kei Saito*b and
Toru Asahi*ad
c,
Hideko Koshimad,
Kei Saito*b and
Toru Asahi*ad
aDepartment of Advanced Science and Engineering, Graduate School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
bSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia. E-mail: kei.saito@monash.edu
cDepartment of Chemistry and Materials Science, Tokyo Institute of Technology, Ookayama 2-12-1, Meguro-ku, Tokyo 152-8551, Japan
dResearch Organization for Nano & Life Innovation, Waseda University, 513 Wasedatsurumakicho, Shinjuku-ku, Tokyo 162-0041, Japan
First published on 2nd November 2016
Abstract
Solid-state photo-polymerization in crystals can produce stereoregular polymer molecules in environmentally friendly solvent-free systems. The polymerization mechanism of bis-thymine derivatives, such as dimethyl-3,3′-(3,3′-(butane-1,4-diyl)bis(5-methyl-2,4-dioxo-3,4-dihydropyrimidine-3,1(2H)-diyl))dipropanoate (1), known as unique molecules that can topochemically and reversibly polymerize in the crystalline state via [2 + 2]-cycloaddition reactions upon UV irradiation, remained to be solved. In this manuscript, the crystal structure of the polymeric photoproduct (1P) from a bis-thymine derivative 1 was determined using ab initio powder X-ray diffraction data and applied to investigate the polymerization mechanism of bis-thymine derivatives. The topochemical polymerization was found to be achieved via [2 + 2]-cycloaddition with the flexible butyl chain between thyminyl rings relieving the distortion of whole structure derived from cyclobutane formation. The crystal structure of 1P also showed that it polymerized stereoregularly with trans–anti cyclobutane conformations.
Introduction
Solid-state reactions have been known to be useful in the synthesis of complex molecules with controlled structure.1 The major advantages of solid-state reactions are that they are solvent-free and green chemical reactions that can produce stereo- and regio-specific products in high yields. Among all the solid-state reactions, photochemical [2 + 2]-cycloaddition of olefins to generate a cyclobutane, has been extensively investigated.2–8 In the 1960's, Schmidt postulated that for a [2 + 2]-cycloaddition to occur in the crystal-phase, the distance between the reactive olefins of adjacent molecules should be 3.5–4.2 Å.3 Since this concept was proposed, there have been many attempts to conduct various solid-state topochemical reactions, including [2 + 2]-cycloaddition, based on strategies to align reactants in the crystal lattice through the control of intermolecular interactions, such as hydrogen bonds, π–π interactions, cation–π interactions and template-directed systems.9–12Topochemical polymerization can proceed by photo- or thermally induced reactions to yield polymer in the crystal. Several reactions have been attempted, such as the 1,4-polymerization of diacetylenes13 and dienes,14 and the 1,6-polymerization of trienes,15 triacetylenes16 and quinodimethane17 to produce linear polymers. Examples of monomers undergoing topochemical polymerization via [2 + 2]-cycloaddition, however, are reported less numerous but include 2,5-distyrylpyrazine (DSP)18 and fluorinated distyrylbenzene.19 In these cases, the polymers are insoluble in common organic solvents and cannot be completely depolymerized upon photo-irradiation.
Thymine, one of the components of DNA, is well known to undergo reversible photo-dimerization and monomerization by UV irradiation via [2 + 2]-cycloaddition and the retro cleavage reaction, respectively.20 Recently, we have achieved photo-reversible topochemical polymerization of an n-butyl-linked bis-thyminyl molecule (dimethyl-3,3′-(3,3′-(butane-1,4-diyl)bis(5-methyl-2,4-dioxo-3,4-dihydropyrimidine-3,1(2H)-diyl))dipropanoate) (1) (Scheme 1).21 Although we determined the crystal structure of 1 by single-crystal X-ray diffraction, the crystal structure of the polymeric photoproduct (1P) could not be analyzed by repeated single-crystal X-ray diffraction structural analyses due to the macroscopic fracturing of the crystal during the polymerization. Because of this, the polymerization mechanism was remained to be solved. It is important to elucidate the polymerization mechanism to understand and extend the monomer structures for this type of topochemical polymerization. This photo-reversible polymer is anticipated to be useful for applications including novel re-usable or self-healing materials, and thus the crystal structure of 1P is important to obtain further ideas regarding the design of other monomer and polymer structures that can be reversibly polymerized and depolymerized in the crystalline state.22
 | ||
| Scheme 1 Photo-reversible polymerization of bis-thymine derivative (1) in the crystal. | ||
The crystal structures of powder samples can be determined by ab initio powder X-ray crystal structural analysis, if high quality diffraction pattern of samples with high crystallinity and less impurity are collected. The crystal structures of various molecules produced after topochemical reaction have recently been determined directly without recrystallization using powder X-ray diffraction data.23–26
In this study, we investigated the crystal structure of 1P using ab initio powder X-ray crystal structural analysis and compared the structure of 1 and 1P to study the mechanism of unique topochemical reversible polymerization.
Experimental
Preparation of bis-thymine derivative monomer 1 (dimethyl-3,3′-(3,3′-(butane-1,4-diyl)bis(5-methyl-2,4-dioxo-3,4-dihydropyrimidine-3,1(2H)-diyl))dipropanoate) crystal
The titled compound (1) was synthesized using a previously reported method.21 The single crystals were obtained by slowly evaporating the ethanol solution of 1 at room temperature.Preparation of 1P
Crystalline 1 was milled with a mortar and pestle. The resulting powder was spread into a thin layer on a Pyrex Petri dish, and irradiated (850 J cm−2) with an Ultraviolet Products CL1000M UV-crosslinker lamp that produced mid-range UV-wavelengths centered at 302 nm. The molecular weight of 1P was measured with gel permeation chromatography (GPC; TOSOH HLC-8220 instrument with CHCl3 as the eluent) as Mn 2.3 × 104 (Mw/Mn = 6.2). 1H-NMR (400 MHz, CDCl3): δH 1.36 (s, 6H), 1.61 (br. app. s, 4H), 2.60 (dt, J = 16.75, 5.56 Hz, 2H), 2.77 (dt, J = 16.84, 7.26 Hz, 2H), 3.25 (dt, J = 13.72, 7.03 Hz, 2H), 3.66 (s, 6H), 3.85 (br. t, 4H), 3.94 (s, 2H), 4.00 (dt, J = 14.27, 6.39 Hz, 2H). 13C-NMR (100 MHz, CDCl3): δC 14.34, 18.37, 25.63, 31.89, 32.15, 40.74, 43.82, 45.81, 51.84, 61.98, 151.36, 171.90.Powder X-ray diffraction measurement
The powder X-ray diffraction (XRD) data to compare the crystallinity of 1 and 1P were collected using a Bruker D8 Focus powder diffractometer with Cu Kα radiation (1.5418 Å) at 40 kV and 30 mA in the reflection mode with samples on glass substrates.To further study the crystal structure of 1P, a high quality powder XRD pattern for 1P was collected using a Rigaku SmartLab diffractometer with Cu Kα radiation (1.5418 Å) at 45 kV and 200 mA operating in transmission mode with a foil type sample holder (2θ range 3–70°; step size 0.01°; data collection time 12 h).
Ab initio crystal structure analysis of 1P from powder X-ray diffraction data
The powder X-ray diffraction pattern of 1P was indexed using the program DICVOL04 (ref. 27) (M19 = 43.5, F19 = 84.8), giving the following unit cell with monoclinic metric symmetry: a = 9.035 Å, b = 18.75 Å, c = 7.326 Å, β = 105.8° (V = 1194 Å3). From the systematic absences and consideration of the unit cell volume (corresponding to Z = 2), the space group was assigned as P21/c. The profile was fitted using the Pawley method28 in the program DASH29 and gave a good quality of fit (Rwp = 5.75%). The direct space simulated annealing structure solution calculation was carried out using the program DASH. The molecular model was taken from the previously reported crystal structure of methyl thymine dimer (CSD Ref code: CUPJAY).Subsequent Rietveld refinement was carried out using the GSAS program.30,31 In the Rietveld refinement, several restraints referred from the model structure used in the program DASH were applied to bond lengths and bond angles, and a global isotropic displacement parameter was also refined. The final Rietveld refinement gave the following parameters: a = 9.03640(25) Å, b = 18.7601(5) Å, c = 7.32887(12) Å, β = 105.8400(13); V = 1195.24(7) Å3; Rwp = 4.50%, Rp = 3.15%, RF2 = 8.71% (2θ range, 3.00–70.00°; 6699 profile points; 44 refined variables, the corresponding Rwp for the Le Bail fitting was 2.80%).
Results & discussions
The powder X-ray diffraction patterns of 1 and 1P are shown in Fig. 1. Different peak positions suggested that the crystal structure of 1 was changed upon UV irradiation to cause polymerization. The XRD patterns of both 1 and 1P exhibited sharp peaks which indicated that 1P maintained high crystallinity after photo-polymerization. The result of final Rietveld refinement gave good fitting to experimental high-quality diffraction data (Fig. 2). | ||
| Fig. 1 The powder X-ray diffraction patterns of 1 and 1P measured Cu Kα radiation (1.5418 Å) at 40 kV and 30 mA in reflection mode. | ||
 | ||
| Fig. 2 Results from powder X-ray diffraction analysis of 1P by Rietveld refinement: each plot shows the experimental powder X-ray diffraction profile (red + marks), the calculated powder X-ray diffraction profile (green solid line) and the difference profile (purple, lower line). Tick marks indicate peak positions. | ||
Comparison of crystal structures of 1 and 1P
The crystal structure data are summarized in Table 1, and the crystal structures of 1 and 1P are shown in Fig. 3. The structure of 1 was reported previously by our group (CSD Ref code: TIFZEP).21 In the crystal structure of 1, the reactive olefins in the thymine rings of adjacent monomer molecules were parallel stacked around inversion centre and separated by a distance of 4.213 Å (Fig. 3(a)), thereby satisfying the topochemical requirements for [2 + 2]-cycloaddition of 1.| 1 (monomer)21 | 1P (polymer) | |
|---|---|---|
| Single crystal | Powdered crystal | |
| Chemical formula | C22H30N4O8 | (C22H30N4O8)n |
| Crystal system | Monoclinic | Monoclinic |
| T/K | 100 | 293 |
| Space group | P21/c | P21/c |
| a/Å | 9.5800(19) | 9.03640(25) |
| b/Å | 16.550(3) | 18.7601(5) |
| c/Å | 7.6900(15) | 7.32887(12) |
| β/° | 112.13(3) | 105.8400(13) |
| V/Å3 | 1129.4(4) | 1195.24(7) |
| Z | 2 | 2 |
| Density/g cm−3 | 1.407 | 1.329 |
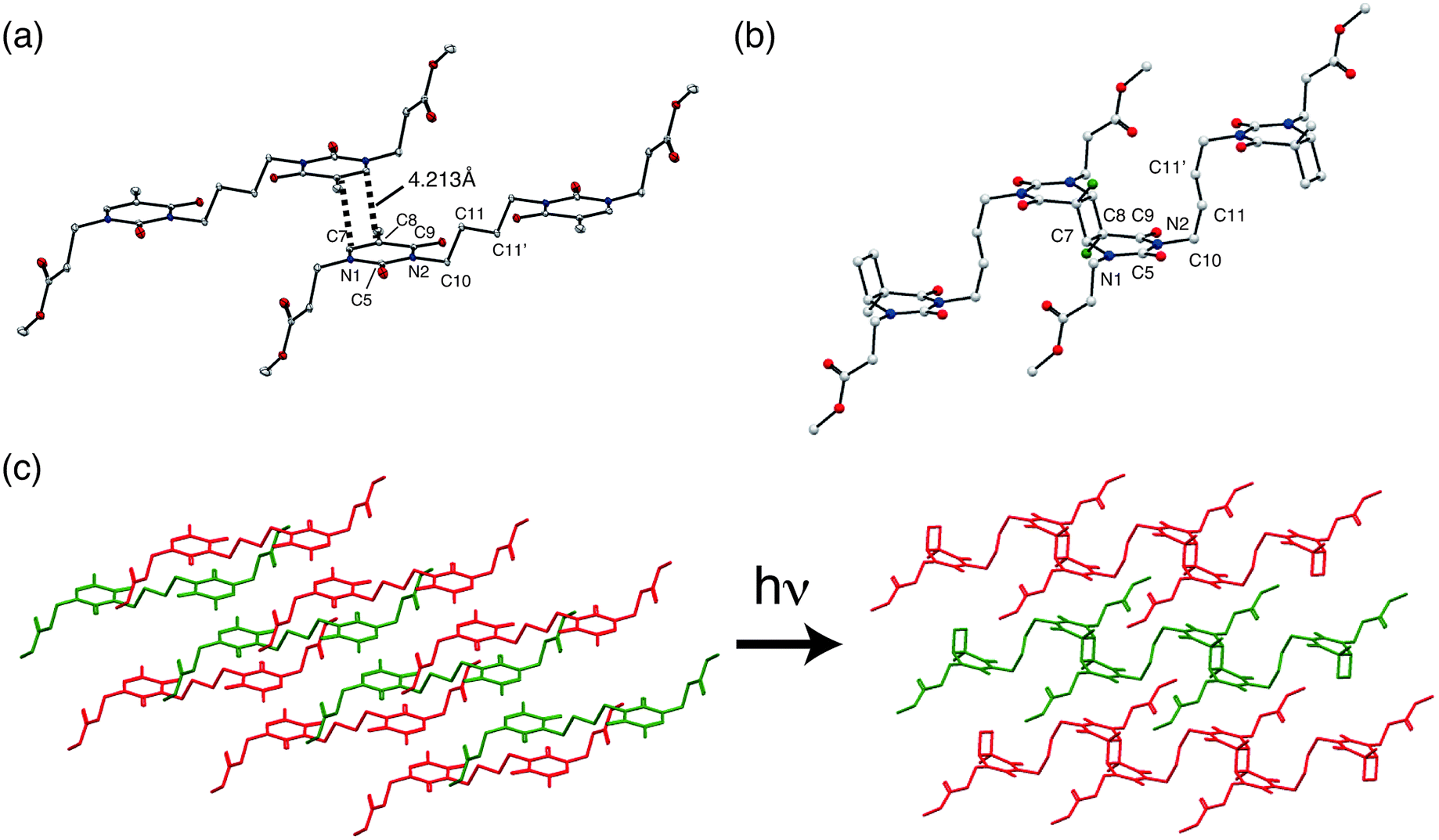 | ||
| Fig. 3 Molecular structures in the crystals of (a) 1 and (b) 1P. Each colored ellipsoid shows different atom: carbon (white), nitrogen (blue), oxygen (red) and methyl carbon related to stereochemistry (green); (c) schematic illustration of the topochemical polymerization of 1 to 1P. Hydrogen atoms were omitted for clarity. | ||
In the crystal structure of 1P, prepared by UV irradiation and [2 + 2]-cycloaddition of 1, cyclobutane rings were formed (Fig. 3(b)). Two thymine units can potentially form four cyclobutane ring isomers: cis–anti, trans–anti, cis–syn and trans–syn.32 It is noteworthy that all of the reacted thyminyl units in 1P afforded cyclobutanes that possessed trans–anti configurations, resulting from the stereoselectivity of the topochemical reaction. The conformational change of the butyl chain between the thyminyl rings was accompanied by shrinkage of the a axis. The torsion angles of N2–C10–C11–C11′ (Table 2) were found to be 178.65° and 78.60° in 1 and 1P, respectively, which corresponds anti (1) and gauche (1P) conformation change. This conformational change should help relieve the distortion at other sites arising from the formation of an almost square-shaped cyclobutane ring upon UV irradiation.
| 121 | 1P | |
|---|---|---|
| N2–C10–C11–C11′ | 178.65° | 78.6° |
| N2–C5–N1–C7 | 0.26° | 16.74° |
| C5–N1–C7–C8 | 1.14° | 20.43° |
| N1–C7–C8–C9 | 1.63° | 8.44° |
This is because [2 + 2]-cycloaddition reactions occur between two C (sp2)–C (sp2) double bonds, and convert to four C (sp3)–C (sp3) single bonds. Therefore in this case, thyminyl rings transformed from plane-like structure to cyclobutane-linked structures due to the change of stable bond lengths and bond angles of C (sp3) after UV irradiation. This conformational flexibility would be important for achieving high polymer molecular weights (1P possessed a Mn = ∼2.3 × 104 after 50 hours irradiation with UV light). This topochemical reaction is schematically illustrated by actual crystal structures of 1 and 1P (Fig. 3(c)).
Referring again to Table 1, the crystal system and space groups were same for 1 and 1P, although the lattice parameters were significantly changed. The axis lengths of a and c decreased by 5.6% and 4.7%, respectively; while the length of the b axis increased by 13.3% (Fig. 4(a)). Collectively, these changes resulted in an expansion of the unit cell volume by 5.8%. The change in each lattice parameter can be discussed based on the three parallel viewpoints along the lattice axes, focussing on the differences of the molecular structures of 1 and 1P, particularly in terms of the cyclobutane ring and the conformation of butyl chain between the thyminyl rings. From the view along the a axis, thyminyl rings of 1 were stacked parallel along the c axis, and the cyclobutane rings in 1P was clearly observed (Fig. 4(b) and (e)). The cyclobutane ring formation from 1 to 1P may cause the 4.7% shrinkage of the c axis length because the C–C single bond length (typically 1.5–1.6 Å) in cyclobutane ring12 is shorter than the distance of two thymines (4.213 Å). From the view along the b axis, the conformation of the butyl chain between thyminyl rings was changed from anti (1) to gauche (1P), resulting in the approach of the thyminyl rings (Fig. 4(c) and (f)). The 5.6% shrinkage of the length of the a axis can therefore be concluded to arise from this conformational change. Finally, from the view along the c axis, it was found that the reactive thyminyl rings were almost overlapped in 1, but had slid further along the a and b axes in 1P (Fig. 4(d) and (g)). Due to this difference, the lengths of the b axis increased in 13.3% from 1 to 1P. These changes in the lattice parameters were the likely cause of macroscopic cracks in the crystals, as previously reported.21
 | ||
| Fig. 4 (a) Schematic illustration of the change of lattice parameters; the crystal structure of 1 (b)–(d) and 1P (e)–(g); from the view along a, b and c axes, (b) and (e), (c) and (f), (d) and (g), respectively. | ||
The molecular weight of the photoproduct polymer should be corresponded to the macroscopic crystal size after UV irradiation, since this will be the limitation of the linear polymer elongating.33 In the reports by Nakanishi et al.34 the crystal was found to form cracks along the short axis of the unit cell corresponded to polymer chain growth direction upon photo irradiation. This is likely because there are only the weak van der Waals intermolecular interactions between the photoproduct polymer chains, which cannot stabilize crystal morphology. In our case, polymer chains of 1P grew along the a axis, and cannot strongly interact with other chains due to its molecular structure. In addition, space between the molecules without intermolecular interaction is observed perpendicular to the a axis. Therefore, the cracks could form parallel and perpendicular to the a axis upon UV irradiation. If the cracks form perpendicular to the a axis, the cracks can block the polymerization parallel to the a axis. This could be the reason of resulting the molecular weight saturated around 2.0 × 104. It is suggested that it is possible to achieve higher molecular weight by controlling the formation of cracks during the polymerization.
Theoretical calculation of the crystal structure of 1P
The crystal structure of 1P were also validated by theoretical calculations using the optimized crystal structure, referring to a previous report by Li et al.35 Geometry optimizations were carried out using CASTEP (Academic Release 6.1),36 via the interface with Materials Studio (Accelrys, 2011). The Perdew, Burke and Ernzerhof (PBE) exchange-correlation functional37 was applied, with the Grimme-06 semi-empirical dispersion correction.38 Integrals taken over the Brillouin zone were carried out on a Monkhorst–Pack grid39 with a maximum sample spacing of 0.05 Å−1.The optimized structure of 1P is shown in Fig. 5, and is almost identical to the structure elucidated from the experimental powder XRD pattern. The N2–C10–C11–C11′ torsion angles of the butyl chain were 78.63° and 62.79° in the experimental and calculated crystal structures, respectively. Accordingly, the crystal structure determined from powder diffraction data, including the molecular structure of 1P with the gauche conformation of butyl chain, was confirmed to be correct.
 | ||
| Fig. 5 Overlay of experimental structure (red) and optimized structure (blue). | ||
Conclusions
In this study, the crystal structure of bis-thymine polymer (1P) prepared by UV irradiation was successfully analyzed by ab initio structure determination from its powder XRD pattern. 1P was found to be stereoregular with trans–anti cyclobutane dimer conformations, and the change of lattice parameters was related to the structural changes of the molecule, such as the formation of cyclobutane ring and the conformation change of the flexible butyl linkage chain from anti (1) to gauche (1P). Finally, the crystal structure determined from powder diffraction data was verified by the theoretical calculation of optimized crystal structure of 1P.From this study, in addition to the previous exploration of photo-reactive bis-thyminyl derivatives, it was concluded that polymerization of thymine derivatives in the crystal should be achieved to be high molecular weight and high crystallinity when the reacting olefins of monomer molecules are parallel and closely stacked with a separation distance around 4.2 Å as well as the existence of flexible C (sp3) alkyl chain considered to relieve the distortion resulting from the formation of cyclobutane rings by UV irradiation. Although it is challenging to find molecules capable of photo-polymerization in the crystalline state, it may be realized in future by following this study, and by synthesizing other bis-thyminyl derivatives bearing flexible carbon chain.
Acknowledgements
This work was financially supported by the Leading Graduate Program in Science and Engineering, Waseda University and the cooperation of organization between Waseda University and JX Nippon Oil & Energy Corporation. This work was also supported by PRESTO, JST.Notes and references
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- G. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- M. D. Cohen, A. Elgavi, B. S. Green and Z. Ludmer, J. Am. Chem. Soc., 1972, 94, 6776–6779 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- R. Medishetty, I. H. Park, S. S. Lee and J. J. Vittal, Chem. Commun., 2016, 52, 3989–4001 RSC.
- Z. Yuan and F. Liang, Curr. Org. Chem., 2014, 18(15), 2016–2036 CrossRef CAS.
- Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed. Engl., 2015, 54, 11918–11928 CrossRef CAS PubMed.
- X. M. Cheng, Z. T. Huang and Q. Y. Zheng, Tetrahedron, 2011, 67, 9093–9098 CrossRef CAS.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS.
- S. Yamada, N. Uematsu and K. Yamashita, J. Am. Chem. Soc., 2007, 129, 12100–12101 CrossRef CAS PubMed.
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817–7818 CrossRef CAS.
- A. Sarkar, A. S. Okada, H. Nakanishi and H. Matsuda, Macromolecules, 1998, 31, 9174–9180 CrossRef CAS.
- T. Tanaka and A. Matsumoto, J. Am. Chem. Soc., 2002, 124, 9676–9677 CrossRef CAS PubMed.
- T. Hoang, J. W. Lauher and F. W. Fowler, J. Am. Chem. Soc., 2002, 124, 10656–10657 CrossRef CAS PubMed.
- J. Xiao, M. Yang, J. Lauher and F. Fowler, Angew. Chem., Int. Ed. Engl., 2000, 39, 2132–2135 CrossRef CAS.
- S. Nomura, T. Itoh, H. Nakasho, T. Uno, M. Kubo, K. Sada, K. Inoue and M. Miyata, J. Am. Chem. Soc., 2004, 126, 2035–2041 CrossRef CAS PubMed.
- M. Hasegawa and Y. Suzuki, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 813–815 CrossRef CAS.
- Y. Sonoda, M. Goto, S. Tsuzuki and H. Akiyama, J. Fluorine Chem., 2009, 130, 151–157 CrossRef CAS.
- N. Tohnai, Y. Inaki and M. Miyata, J. Photopolym. Sci. Technol., 1998, 11, 59–64 CrossRef CAS.
- P. Johnston, C. Braybrook and K. Saito, Chem. Sci., 2012, 3, 2301–2306 RSC.
- P. Johnston, D. Wheldale, C. Braybrook and K. Saito, Polym. Chem., 2014, 5, 4375–4384 RSC.
- M. Horiguchi, S. Okuhara, E. Shimano, D. Fujimoto, H. Takahashi, H. Tsue and R. Tamura, Cryst. Growth Des., 2008, 8, 540–548 CAS.
- K. Fujii, H. Uekusa, M. Fukano and H. Koshima, CrystEngComm, 2011, 13, 3197–3201 RSC.
- K. Fujii, H. Uekusa, N. Itoda, G. Hasegawa, E. Yonemochi, K. Terada, Z. Pan and K. D. M. Harris, J. Phys. Chem. C, 2010, 114, 580–586 CAS.
- F. Guo, J. Martí-Rujas, Z. Pan, C. E. Hughes and K. D. M. Harris, J. Phys. Chem. C, 2008, 112, 19793–19796 CAS.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- A. J. Florence, N. Shankland, K. Shankland, W. I. F. David, E. Pidcock, X. Xu, A. Johnston, A. R. Kennedy, P. J. Cox, J. S. O. Evans, G. Steele, S. D. Crosgrove and C. S. Frampton, J. Appl. Crystallogr., 2005, 38, 249–259 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- H. Koshima, K. Yamashita and T. Matsuura, Mol. Cryst. Liq. Cryst., 1998, 313, 303–308 CrossRef CAS.
- A. Matsumoto and K. Yokoi, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 3147–3155 CrossRef CAS.
- H. Nakanishi, M. Hasegawa and Y. Sasada, J. Polym. Sci., Part B: Polym. Lett., 1972, 10, 1537–1553 CAS.
- X. Li, A. D. Bond, K. E. Johansson and J. Van de Streek, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 784–789 CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
Footnote |
| † CCDC 1482780. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra24229d |
| This journal is © The Royal Society of Chemistry 2016 |