
Multifunctional FePt–Au heterodimers: promising nanotheranostic agents for dual-modality MR/CT imaging diagnosis and in situ cancer therapy†
Jinlong Wangab,
Ludan Yuebc,
Zunfu Hud,
Zhichao Daib,
Yafei Qie,
Xiuwen Zheng*b,
Zhongfang Li*a and
Dexin Yu*e
aSchool of Chemical Engineering, Shandong University of Technology, Zibo 255000, Shandong, P.R. China. E-mail: zhfli@sdut.edu.cn
bSchool of Chemistry & Chemical Engineering, Linyi University, Linyi 276000, Shandong, P.R. China. E-mail: zhengxiuwen@lyu.edu.cn
cCollege of Chemistry, Chemical Engineering & Materials Science, Shandong Normal University, Jinan 250000, Shandong, P.R. China
dCollege of Chemistry & Molecular Engineering, Qingdao University of Science & Technology, Qingdao 266000, Shandong, P. R. China
eQilu Hospital of Shandong University, Jinan 250000, Shandong, P.R. China. E-mail: ydx0330@sina.com
First published on 26th October 2016
Abstract
We report the synthesis of multifunctional FePt–Au hybrid nanoparticles via a simple hydrothermal approach and their potential application in cancer dual-modality MR/CT imaging diagnosis and simultaneous in situ therapy. After conjugation with meso-2,3-dimercaptosuccinic acid (DMSA) and SH–PEG–FA, the FePt–Au HNPs present high biostability in physiological solutions and successfully target folate acid (FA) receptor-positive cancer cells such as MCF-7, HeLa and HepG2. As a pH-sensitive agent, the as-prepared FePt–Au–DMSA/PEG–FA HNPs exhibit high cytotoxicity to the targeted cancer cells due to the generation of many reactive oxygen species (ROS) induced by the released Fe within the cells. The corresponding half-maximum inhibitory concentration value (IC50, Fe) is about 3.0 μg mL−1. MR images and CT scans in vitro and in vivo demonstrate that the HNPs hold great potential as a dual-modality MR/CT imaging contrast agent for high-accuracy early-stage diagnosis of cancer. In addition, in vivo anti-tumor and histological studies indicate that the tumor growth is significantly inhibited after treating with HNPs with no observable toxicity found in the other tissues. Therefore, the FePt–Au–DMSA/PEG–FA HNPs are a promising multifunctional nanotheranostic agent for dual-modality MR/CT imaging diagnosis and in situ cancer therapy.
Introduction
In the past few years, one of the most important emerging areas in cancer nanotechnology is theranostics, which integrates the functions of diagnostic imaging and therapy into a single nanosystem.1–6 Compared to traditional separate nanomaterials, nanotheranostics have the potential to overcome the undesirable differences that exist in the biodistribution and selectivity that currently occurs between distinct imaging and therapeutic agents.7,8 Based on this, researchers are devoted to explore novel hybrid nanoparticles (HNPs) such as FePt–Fe2O3,9 FePt–Fe3O4,10 Fe3O4@NaYF4:Yb/Er,11 Au–Fe2O3,12 Au–Fe3O4 (ref. 13 and 14) and Fe3O4 Core@hybrid@Au,15 which combines different components to realize the ultimate potential of diagnosis and simultaneous treatment of disease with few side-effects. Once immobilized with a targeting agent such as folic acid (FA) or transferrin (Tf), these smart HNPs will easily target the tumor lesion location and present their clinical behavior at the molecular level.6,9In response to the demand for diagnoses in clinical settings, multi-modality imaging has been widely studied,5,16 especially the upconversion nanoparticles,17,18 which holds promising prospects to achieve high-accuracy early-stage diagnosis of tumors. It would be optimal if the nanoparticles could induce apoptosis of cancer cells in situ while achieving a diagnosis. Recently, mono-dispersed FePt nanoparticles (NPs) with a chemically disordered face-centered cubic (fcc) structure have attracted considerable interest for their potential clinical applications in both cancer diagnoses and therapy.19–22 Based on the excellent superparamagnetism and X-ray attenuation properties, FePt NPs have the potential to be used as a contrast agent for dual-modality T2-weight magnetic resonance (MR) imaging and computed tomography (CT) imaging diagnoses.23,24 On the other hand, FePt NPs also hold potential applications in both chemodynamic therapeutics25 and hyperthermia therapeutics26 due to the high production of ROS induced by the released Fe within cancer cells27 or the photothermal effect activated by a near infrared (NIR) femtosecond laser, respectively. However, further development of FePt NPs in the clinic has been impeded because of the limitations of biocompatibility, stability and large amount of Pt required for CT contrast imaging.
As is well known, Au NPs have been widely used in various biomedical applications,28–30 particularly as a contrast agent for CT imaging diagnoses because of their good biocompatibility and high X-ray attenuation coefficient.31,32 On this point, the introduction of Au NPs could effectively improve the stability of FePt NPs in physiological environments and simultaneously reduce the large amount of single Pt required for CT imaging in clinic.33,34 Herein, combining the merits of fcc-FePt (i.e., pH-sensitive and chemodynamic therapy) and Au (i.e., easy to be functionalized by mercapto group-containing biocompatible polymers), we designed a novel heterodimer of FePt–Au HNPs as a potential dual-modality nanotheranostic agent for MR/CT imaging diagnosis and in situ cancer therapy.
Results and discussion
Synthesis, preparation and characterization
The synthesis and surface modification procedure of HNPs is illustrated in Scheme 1A. To obtain uniform FePt–Au HNPs and prevent the nucleation of isolated Au, a typical seed-mediated growth procedure35 was used wherein FePt nanocubes were used as seeds due to their higher specific surface areas compared to that of spherical structures. In addition, an Au(I)–OAm complex was selected as the Au precursor because of its high thermal stability over the alternative Au(III) precursor (HAuCl4). As shown in Fig. 1A and B, the FePt nanocubes (A) and FePt–Au HNPs (B) show low size dispersion and have a mean size about 10 and 20 nm, respectively. The insets in Fig. 1A and B show the HRTEM images of individual FePt and FePt–Au HNPs, respectively. It is clear that the lattice fringes are coherently extended from the FePt seeds to the Au domain, suggesting epitaxial growth between single crystalline FePt and polycrystalline Au. STEM and energy-dispersive spectroscopy (EDS) mapping analyses (Fig. S1†) further confirm the formation of the heterodimers of the FePt–Au HNPs and the uniform distribution of elements (i.e., Fe and Pt dispersed in a cupped cubic shape; Au dispersed in spherical shape). The magnetic properties of the FePt seeds and FePt–Au HNPs were determined at room temperature (298 K) (Fig. 1C). The saturation magnetization value of the FePt–Au HNPs (Ms = 11.8 emu g−1) is smaller than that of the FePt seeds (Ms = 18.4 emu g−1), which may be attributed to the interfacial interactions between FePt and Au.34,36 XRD patterns of FePt seeds and FePt–Au HNPs displayed in Fig. 1D indicate that the position and relative intensity of all diffraction peaks coincide with the standard fcc-FePt (JCPDS 29-0716)37 and Au (JCPDS 04-0784)38 powder diffraction patterns and no obvious peaks for impurities could be found. Moreover, the localized surface plasmon resonance (LSPR) of FePt–Au HNPs shows a red shift compared to that of the Au nanocrystals, as shown in the UV-vis absorption spectra (Fig. S2†), which is mainly attributed to the electron transport across the interface between FePt and Au NPs.35 All these results indicate that mono-dispersed fcc FePt–Au HNPs have been successfully synthesized and EDS analysis revealed that the composition of the sample was Fe22Pt25Au53, which is consistent with the ICP-MS results of Fe23Pt26Au51. | ||
| Scheme 1 (A) Schematic for the synthesis and surface modification procedure of the FePt–Au–DMSA/PEG–FA HNPs. (B) Illustration of the possible mechanism of the FePt–Au–DMSA/PEG–FA HNPs inducing the production of high ROS. | ||
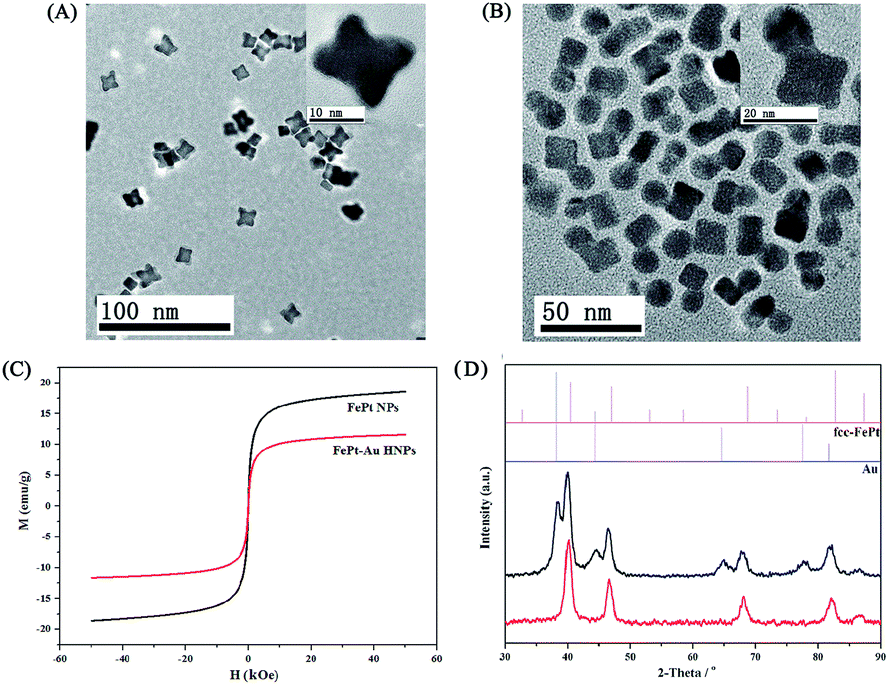 | ||
| Fig. 1 TEM images of (A) FePt seeds and (B) FePt–Au HNPs (inset: HRTEM image of individual FePt and FePt–Au HNPs). (C) The magnetic properties of FePt and FePt–Au HNPs (D) XRD patterns of FePt seeds and corresponding FePt–Au HNPs. | ||
For biological applications, we carried out a ligand-exchange procedure (Fig. S3†) by DMSA to transfer the as-synthesized hydrophobic FePt–Au HNPs (coated with OA and OAm) into aqueous solutions taking advantage of the strong Au–S, Pt–S linkages and high affinity of iron for the carboxylate group.23,24,39 The targeting agent of SH–PEG–FA was subsequently conjugated onto the surface of the FePt–Au HNPs through the robust Au–S linkage, which further improved the biocompatibility and stability (Fig. S4†) of the HNPs and simultaneously introduced the targeting capability toward the FA receptor-positive tumor cells. However, a slight coagulation phenomenon appeared after the FePt–Au HNPs were modified by DMSA and SH–PEG–FA (Fig. S5†), which is mainly attributed to the hydrogen bonds between the COOH groups of the DMSA ligands39 and the magnetism of the FePt–Au NPs. As shown in the FT-IR spectra (Fig. S6†), the absorption band at ∼2900 cm−1 in the production of the FePt–Au–DMSA/PEG–FA HNPs (i.e., FePt–Au–FA HNPs) is attributed to the CH vibration in PEG, indicating that SH–PEG–FA has been successfully conjugated onto the surface of the FePt–Au–DMSA HNPs. On the other hand, the average hydrodynamic diameter of the FePt–Au–DMSA HNPs after conjugation with SH–PEG–FA increased by nearly 53 nm (Fig. S7†) mainly due to the long-chain of PEG while the corresponding zeta potential decreased by almost −12 mV (Fig. S8†) as a result of the negatively charged FA. These changes in size and zeta potential also suggest that SH–PEG–FA has been successfully bonded onto the FePt–Au–DMSA HNPs.
Cytotoxicity assay and cell imaging
It is well known that the uptake of Fe NPs in cellular systems will lead to the release of Fe in an acidic environment, which will further catalyze intracellular hydrogen peroxide (H2O2), transforming it into reactive oxygen species (ROS) and resulting in the death of cells due to their strong oxidation reactions.25,27The cytotoxicity of the FePt–Au–FA HNPs was evaluated by the WST-1 assay using 5 different cells (i.e., MCF-7, HeLa, HepG2, BRL-3A and L02 cells). As shown in Fig. 2A, the HNPs present high cytotoxicity to FA receptor-positive tumor cells (i.e., MCF-7, HeLa and HepG2) while there was a negligible impact to the normal cells (i.e., BRL-3A and L02). The corresponding value of the half-maximum inhibitory concentration (IC50, Fe) is approximately 3.0 μg mL−1. Moreover, the in vitro cellular uptake assay of the FePt–Au–FA HNPs toward MCF-7 and LO2 cells (Fig. S9†) also revealed the high targeting efficiency of the HNPs toward the FA receptor-positive tumor cells. To investigate the cytotoxicity changes with time, a WST-1 assay was conducted using MCF-7 cells wherein the cells were treated with FePt–Au–FA HNPs for different times (i.e., 2, 4, 6, 12, 24 and 48 h) but at the same Fe concentrations of IC50. As shown in Fig. 2B, the cell viability dramatically decreases with time as expected, indicating that improved therapeutic effect is observed with long time periods. Moreover, almost no toxicity could be found on the FePt–Au–DMSA/PEG–COOH HNPs toward the FA receptor-positive MCF-7 cells, which is consistent with the results of the in vitro cellular uptake assay of the FePt–Au–DMSA/PEG–COOH HNPs (Fig. S10†), indicating the high targeting efficiency of the FA-modified FePt–Au HNPs. The possible mechanism of the HNPs inducing the production of ROS is shown in Scheme 1B. When the HNPs enter the cancer cells by receptor-mediated endocytosis, Fe will be released by the degradation of the HNPs in the acidic lysosome (pH ∼ 4.8) and then it reacts with H2O2 produced by the mitochondria to generate ROS and Fe3+ through the Fenton reaction.25,40,41 To further verify the behavior of Fe from FePt under an acidic environment, an experiment was designed to measure the release of Fe from the FePt–Au–FA HNPs in the PBS buffer at pH 4.8 and pH 7.38. The corresponding samples were removed from the reservoir solution at different time intervals (i.e., 3, 6, 12, 24, 48 and 72 h) and were measured by ICP-MS. It is clear that the concentration of Fe released at pH 4.8 increases with the time (Fig. 2C) with a 25% release of Fe at 12 h and 43% release of Fe at 72 h, whereas almost no Fe was released at pH 7.38 throughout the tests. The changes of TEM images toward the FePt–Au HNPs before and after the Fe releasing tests at pH 4.8 (insets in Fig. 2C) further confirmed the behavior of the release of Fe from FePt. On the other hand, Fe inside the cancer cells was visualized by Prussian blue staining, which is a standard protocol widely used to examine cellular uptake of Fe NPs. As shown in Fig. 2D, the FePt–Au–FA HNPs existed in almost all of the MCF-7 cells. These labeled cells show clusters of dense blue granules in the cytoplasm, indicating the existence of Fe3+. Moreover, the presence of high ROS induced by the released Fe within the MCF-7 cells at various incubation times (i.e., 2, 4, 6, 12 and 24 h) of the FePt–Au–FA HNPs were detected using a commercially available fluorescence probe DCFH–DA. As shown in Fig. 2E, the fluorescence intensity of DCF (i.e., reaction product of the probe with ROS) was gradually increased with time, demonstrating the generation of ROS with the release of Fe. In addition, a strong green fluorescence signal from the MCF-7 cells after treating with the FePt–Au–FA HNPs and DCFH–DA could be observed clearly in Fig. 2F and G. Based on the results of the Fe release tests, Prussian blue staining and ROS studies, we conclude that the cytotoxicity of the FePt–Au–FA HNPs was mainly triggered by the released of Fe within the cancer cells. Moreover, to evaluate the cytotoxicity of the HNPs, tLyp-1,42,43 a type of novel tumor homing and penetrating peptide, was also used instead of FA as a targeting agent. The data from the WST-1 assay and ROS studies performed on FePt–Au–DMSA/PEG–tLyp-1 HNPs (Fig. S11†) show similar results as those obtained for the FePt–Au–FA HNPs, indicating the universality of the cytotoxicity of the HNPs.
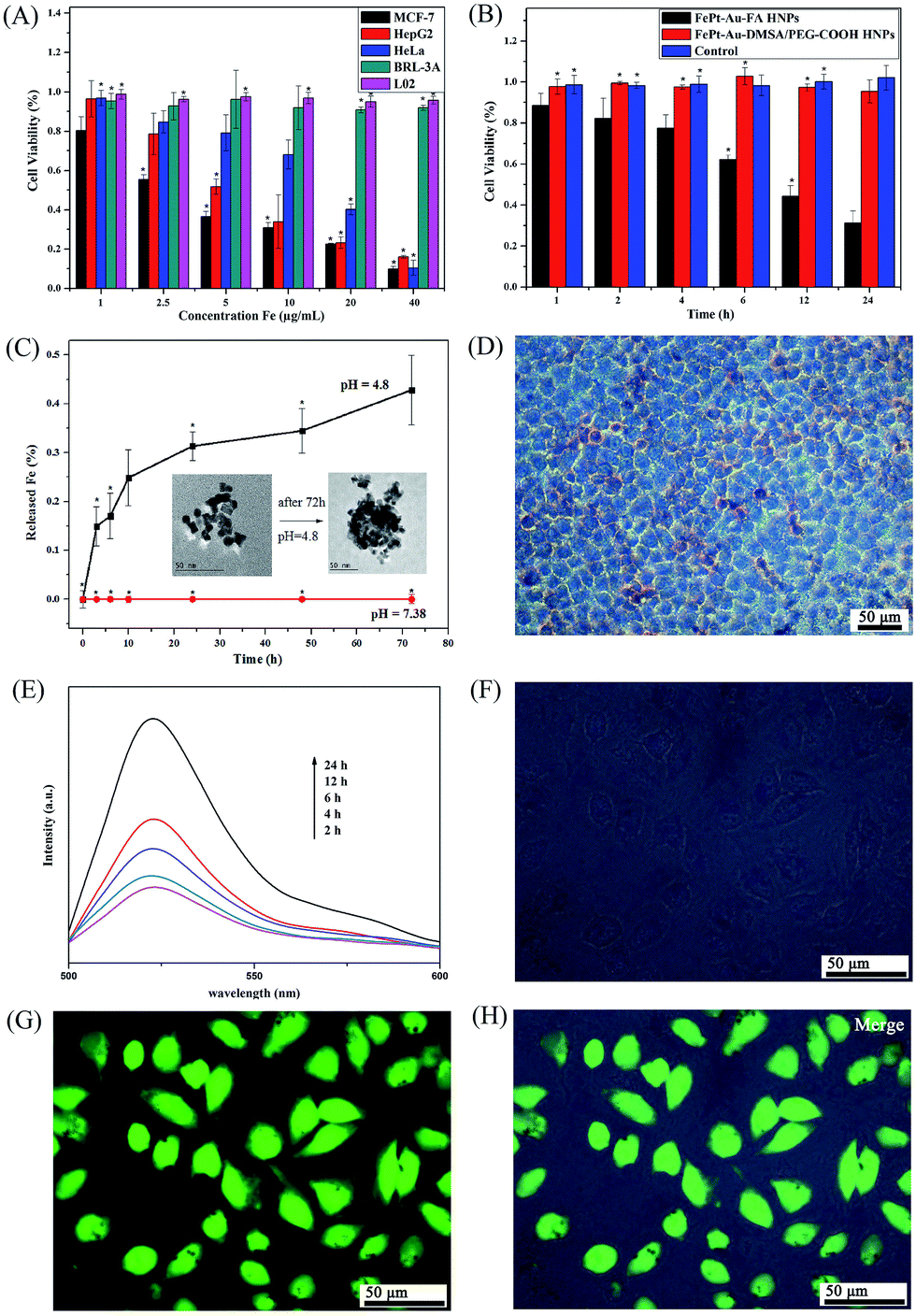 | ||
| Fig. 2 (A) Viabilities of different cell lines treated with FePt–Au–FA HNPs for 6 h. (B) Viabilities of MCF-7 cells treated with FePt–Au–FA HNPs and FePt–Au–DMSA/PEG–COOH HNPs for a different number of hours at the same Fe concentration of 3.0 μg mL−1. Error bars were based on quartet samples. The asterisks indicate P < 0.05. (C) Situations of Fe releasing from FePt–Au HNPs over time in PBS (pH = 4.8 and 7.38, respectively). Insets are the TEM images of the FePt–Au HNPs before and after the releasing tests for Fe in pH 4.8. (D) Photomicrographs of Prussian blue staining of MCF-7 cells treated with FePt–Au–FA HNPs HNPs for 6 h at an Fe concentration of 3.0 μg mL−1. (E) Time-dependent fluorescent intensity from DCFH–DA labeled MCF-7 cells after being treated with FePt–Au–FA HNPs at an Fe concentration of 3.0 μg mL−1. (F–H) Typical green fluorescent images of DCFH–DA labeled MCF-7 cells after being treated with FePt–Au–FA HNPs for 6 h at an Fe concentration of 3.0 μg mL−1. | ||
In vitro MR/CT imaging study
Fig. 3A shows the linear correlation of the T2 relaxation rates (1/T2) against the Fe concentration (mM) of the FePt–Au–FA HNPs in PBS. The calculated relaxation rate (r2) is 43.8 mM−1 s−1. | ||
| Fig. 3 T2 relaxation rates (1/T2) plotted against the Fe concentration of the FePt–Au–DMSA HNPs treated with (A) no cells and (B) MCF-7 cells for 6 h (inset is the corresponding T2 weighted MR images). (C) The value of 1/T2 changes with time performed on the MCF-7 cells treated with FePt–Au–DMSA HNPs at an Fe concentration of 5.0 μg mL−1 (inset is the corresponding T2 weighted MR images). (D) X-ray attenuation assay of the FePt–Au–FA HNPs and FePt–DMSA–PEG–FA HNPs treated with MCF-7 cells for 6 h at different Pt concentrations (insets are the corresponding CT contrast images). | ||
Based on the high T2 relaxation, the FePt–Au–FA HNPs hold great potential to be used as a T2 weighted MR contrast agent. The in vitro MRI results are shown in Fig. 3B wherein it can be seen that the T2 signal intensity significantly decreased and the corresponding T2-weighted MR image darkened with an increase in the Fe concentration. The r2 value in this case was calculated to be 29.2 mM−1 s−1. It is interesting that the dependence of the relaxation rate on the Fe concentration was less linear in the case of the cells compared to the case of PBS, which we attributed to the variations of Fe release that occurred in the cancer cells. To verify this hypothesis, we treated the MCF-7 cells with FePt–Au–FA HNPs for different time (i.e., 3, 6, 12, 24 and 48 h) but at the same Fe concentration of 5.0 μg mL−1. From Fig. 3C, we can see that the T2 signal intensity gradually increased and the corresponding T2-weighted MR image brightened with an increase in time, demonstrating the degradation of the HNPs in the cancer cells, which is also consistent with the results of Fe release test (Fig. 2C, the released Fe concentration increases with time at pH 4.8) and ROS studies (Fig. 2E, the fluorescence intensity of the DCF increases with time).
To explore the potential applications of the HNPs as CT imaging contrast agents, the X-ray attenuation property of the formed FePt-based HNPs was studied in the presence and absence of Au. As shown in Fig. 3D, it is clear that the intensity of the CT images as well as the corresponding Hounsfield units (HU) values of both the FePt–Au–FA HNPs and FePt–DMSA/PEG–FA HNPs linearly increased with increasing Pt content. However, FePt–Au–FA HNPs exhibited much higher X-ray attenuation potency than that of the FePt–DMSA/PEG–FA HNPs at the same Pt concentration, which could be ascribed to the synergistic effect between Pt and Au. Therefore, the large amount of single Pt required for CT imaging in the clinic would be significantly reduced, indicating the great potential applications of the FePt–Au–FA HNPs as a CT contrast agent for cancer diagnosis.
In vivo MR/CT imaging study
Encouraged by the high dual-modality MR/CT contrast efficiency presented in vitro, the potential applications of the FePt–Au–FA HNPs as a contrast agent for in vivo dual-modality MR/CT imaging have been explored. As shown in Fig. 4A, the tumor region (indicated by the red circle) dramatically darkened after intratumor injection of the HNPs, indicating the great potential of the HNPs to be used as a contrast agent for T2-weighted MR imaging diagnoses. However, the tumor region started to become bright and the corresponding MR signal intensity (i.e., the corresponding data points are marked with a red dot) almost recovered at 24 h (Fig. 4B), which was mainly attributed to the consumption of the HNPs within the tumor cells and the further metabolism process of the HNPs undergoing in the tumor region. On the other hand, from Fig. 4C, we can see that the HNPs could target and accumulate in the tumor region (marked in a red circle) with renal clearance (marked in a red rectangle) after intravenous injection of the HNPs mainly due to the targeting function of FA, the long circulation ability provided by the high negative potential (−20 mV) and the enhanced permeability and retention effect (EPR) of cancerous tumors. In addition, as a result of the Fe release within the cancer cells and further in vivo metabolism, the HNPs could also be completely cleared from the tumor region after 24 h. | ||
| Fig. 4 (A) T2-weighted MR images of a 4 T1 tumor-bearing mouse taken before injection (0 h), as well as 0.5 h, 1 h, 2 h and 24 h after intratumor injection of FePt–Au–FA HNPs (50 μL in PBS, Fe concentration, 500 μg mL−1), and (B) the corresponding alteration of the acquired MR signal intensity. (C) T2-weighted MR images of a 4 T1 tumor-bearing mouse taken before injection (0 h), and 2 h and 24 h after intravenous injection of FePt–Au–FA HNPs (50 μL in PBS, Fe concentration, 500 μg mL−1). In vivo tumor (D) CT and (E) the corresponding 3D restructured images of the 4 T1 tumor-bearing mouse taken before (0 h) and after (0.5 h) intratumor injection of FePt–Au–FA HNPs (100 μL in PBS, Pt concentration, 5 mg mL−1 and Au concentration, 5 mg mL−1), respectively. (F) The T2-weighted MR imaging performed on the same mouse before (0 h) and (0.6 h) after intratumor injection of the HNPs. (G) The corresponding alterations of the acquired MR/CT image signal intensity after intratumor injection of the HNPs in the tumor area. (H) In vivo tumor volume changes of 4 T1 tumor-bearing mice after various treatments: (a) PBS intratumor injection; (b) FePt–Au–FA HNPs (Fe concentration, 500 μg mL−1) intratumor injection; and (c) FePt–Au–FA HNPs (Fe concentration, 500 μg mL−1) intravenously injected through the tail vein. Error bars were based on quartet samples. Inset shows the typical tumor images after the mice were treated for 12 days. | ||
Next, the in vivo CT imaging capability of the FePt–Au–FA HNPs was explored by intratumor injection of these HNPs into a tumor-bearing mouse. As shown in Fig. 4D and E, the injection of the HNPs produced a significant CT contrast enhancement in the tumor region (indicated by the red circle) compared with the image at 0 h. The results of the T2-weighted MR imaging (Fig. 4F) performed on the same mouse is consistent with the CT scans. The corresponding alterations of the acquired MR/CT signal intensity in the tumor area are shown in Fig. 4G (the corresponding data points are marked with a red dot in Fig. 4D and F). With respect to the MR and CT imaging before administration, a significant reduction in the T2-weighted signal intensity of 15.6% and a considerable 2050% contrast enhancement of the HU value were observed toward the tumor region. Overall, these findings indicate that the as-prepared FePt–Au–FA HNPs could potentially be used as an in vivo contrast agent for cancer dual-modality MR/CT imaging diagnosis.
In vivo anti-tumor effect and histological study
The therapeutic effect of the FePt–Au–FA HNPs was quantitatively evaluated by observing the tumor growth rates. The relative changes in tumor volume with time are plotted and shown in Fig. 4H. It is clear that the relative tumor volume injected with PBS exhibits a trend toward rapid growth compared with the others injected with the HNPs solution. In addition, the tumor growth of mice with an intratumor injection of HNPs was more severely inhibited than the others with an intravenous injection, which we attributed to the relatively low transport efficiency through the vessels and organs. Moreover, the tumor images of the three groups (inset in Fig. 4H) after treatment for 12 days further indicate that the tumor treated with the HNPs by an intratumor injection had the best outcomes resulting from the treatment.The in vivo cytotoxicity of other tissues induced by nanomaterials is of great concern before being used in practical applications. To further systemically evaluate the toxicity of the FePt–Au–FA HNPs in vivo, a pathological assay (H&E staining) was performed on the tumors and the other major organs (heart, liver, spleen, lung and kidney) of the treated mice. As shown in Fig. S12,† all the major organs maintained their typical structural phenotypes and no apparent histopathological abnormalities or lesions were observed, whereas the tumor tissues treated with the FePt–Au–FA HNPs showed a typical cell death response such as extensive fragmentation, nuclear shrinkage and deeper cytoplasmic staining. Moreover, neither death nor obvious weight loss was observed in the treated mice.
Conclusions
In summary, we developed a heterodimer of FePt–Au HNPs as a multifunctional nanotheranostic agent for in situ MR/CT guided cancer therapy. The conjugation of biocompatible SH–PEG–FA endows FePt–Au HNPs with high stability in physiological solutions and the ability to target FA receptor-rich cancer cells such as MCF-7, HeLa and HepG2. As a pH-sensitive agent, the as-prepared FePt–Au–FA HNPs exhibited high cytotoxicity to the targeted tumor cells with a negligible impact to normal cells (BRL-3A and L02), which was attributed to the excessive production of ROS induced by the released Fe within the cancer cells. MR images and CT scans in vitro and in vivo indicated that the HNPs hold great potential as a dual-modality contrast agent for MR/CT imaging for the highly accurate early-stage diagnosis of cancer. Notably, the introduction of Au NPs makes FePt–Au–FA HNPs exhibit a much higher X-ray attenuation potency than that of FePt–DMSA/PEG–FA HNPs at the same Pt concentration, which would significantly reduce the large amount of single Pt needed for CT imaging in the clinic. Moreover, in vivo anti-tumor and histological studies showed that the HNPs could have high inhibitory efficacy to the tumor growth with almost no occurrence of histopathological abnormalities or lesions in the major organs of the mice. These results highlight the great potential of the multifunctional FePt–Au–FA HNPs in future clinical theranostic applications.Acknowledgements
This study was financially supported by the Natural Science Foundation of China (21375057) and the Shandong Province Natural Science Foundation (ZR2012HM012).Notes and references
- C. Li, Nat. Mater., 2014, 13, 110–115 CrossRef CAS PubMed.
- L. Cheng, S. Shen, S. Shi, Y. Yi, X. Wang, G. Song, K. Yang, G. Liu, T. E. Barnhart, W. Cai and Z. Liu, Adv. Funct. Mater., 2016, 26, 2185–2197 CrossRef CAS PubMed.
- Z. Zhang, C. Liu, J. Bai, C. Wu, Y. Xiao, Y. Li, J. Zheng, R. Yang and W. Tan, ACS Appl. Mater. Interfaces, 2015, 7, 6211–6219 CAS.
- K. Yang, G. Yang, L. Chen, L. Cheng, L. Wang, C. Ge and Z. Liu, Biomaterials, 2015, 38, 1–9 CrossRef CAS PubMed.
- M. Chen, S. Tang, Z. Guo, X. Wang, S. Mo, X. Huang, G. Liu and N. Zheng, Adv. Mater., 2014, 26, 8210–8216 CrossRef CAS PubMed.
- Y. Jiao, Y. Sun, X. Tang, Q. Ren and W. Yang, Small, 2015, 11, 1962–1974 CrossRef CAS PubMed.
- S. S. Kelkar and T. M. Reineke, Bioconjugate Chem., 2011, 22, 1879–1903 CrossRef CAS PubMed.
- H. Chen, F. Liu, Z. Lei, L. Ma and Z. Wang, RSC Adv., 2015, 5, 84980–84987 RSC.
- Y. Liu, K. Yang, L. Cheng, J. Zhu, X. Ma, H. Xu, Y. Li, L. Guo, H. Gu and Z. Liu, Nanomedicine: Nanotechnology, Biology and Medicine, 2013, 9, 1077–1088 CrossRef CAS PubMed.
- J. S. Beveridge, M. R. Buck, J. F. Bondi, R. Misra, P. Schiffer, R. E. Schaak and M. E. Williams, Angew. Chem., 2011, 50, 9875–9879 CrossRef CAS PubMed.
- L. Zeng, L. Xiang, W. Ren, J. Zheng, T. Li, B. Chen, J. Zhang, C. Mao, A. Li and A. Wu, RSC Adv., 2013, 3, 13915 RSC.
- W. Gao, L. Ji, L. Li, G. Cui, K. Xu, P. Li and B. Tang, Biomaterials, 2012, 33, 3710–3718 CrossRef CAS PubMed.
- C. Xu and S. Sun, Dalton Trans., 2009, 5583–5591 RSC.
- H. Y. Zhao, S. Liu, J. He, C. C. Pan, H. Li, Z. Y. Zhou, Y. Ding, D. Huo and Y. Hu, Biomaterials, 2015, 51, 194–207 CrossRef CAS PubMed.
- W. Dong, Y. Li, D. Niu, Z. Ma, J. Gu, Y. Chen, W. Zhao, X. Liu, C. Liu and J. Shi, Adv. Mater., 2011, 23, 5392–5397 CrossRef CAS PubMed.
- J. Li, L. Zheng, H. Cai, W. Sun, M. Shen, G. Zhang and X. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 10357–10366 CAS.
- F. Liu, X. He, L. Liu, H. You, H. Zhang and Z. Wang, Biomaterials, 2013, 34, 5218–5225 CrossRef CAS PubMed.
- D. Ni, W. Bu, S. Zhang, X. Zheng, M. Li, H. Xing, Q. Xiao, Y. Liu, Y. Hua, L. Zhou, W. Peng, K. Zhao and J. Shi, Adv. Funct. Mater., 2014, 24, 6613–6620 CrossRef CAS.
- Y. Shi, M. Lin, X. Jiang and S. Liang, J. Nanomater. 2015, 467873.
- Z. Bao, M. He, H. Quan, D. Jiang, Y. Zheng, W. Qin, Y. Zhou, F. Ren, M. Guo and C. Jiang, RSC Adv., 2016, 6, 35124–35134 RSC.
- L. Wang, X. Zheng, W. Zhang, X. Quan, Q. Hu, W. Wu, P. Zong and M. Wu, RSC Adv., 2013, 3, 9042 RSC.
- X. Zheng, W. Chen, P. Cui, Z. Wang and W. Zhang, RSC Adv., 2014, 4, 58489–58494 RSC.
- S. Liang, Q. Zhou, M. Wang, Y. Zhu, Q. Wu and X. Yang, Int. J. Nanomed., 2015, 10, 2325–2333 CrossRef CAS PubMed.
- S. W. Chou, Y. H. Shau, P. C. Wu, Y. S. Yang, D. B. Shieh and C. C. Chen, J. Am. Chem. Soc., 2010, 132, 13270 CrossRef CAS PubMed.
- C. Xu, Z. Yuan, N. Kohler, J. Kim, M. A. Chung and S. Sun, J. Am. Chem. Soc., 2009, 131, 15346 CrossRef CAS PubMed.
- C. L. Chen, L. R. Kuo, S. Y. Lee, Y. K. Hwu, S. W. Chou, C. C. Chen, F. H. Chang, K. H. Lin, D. H. Tsai and Y. Y. Chen, Biomaterials, 2013, 34, 1128–1134 CrossRef CAS PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., 2016, 55, 2101–2106 CrossRef CAS PubMed.
- J. Conde, N. Oliva, Y. Zhang and N. Artzi, Nat. Mater., 2016, 15, 1128–1138 CrossRef CAS PubMed.
- X. Yan, J. Blacklock, J. Li and H. Möhwald, ACS Nano, 2012, 6, 111 CrossRef CAS PubMed.
- K. A. Rawat, H. Basu, R. K. Singhal and S. K. Kailasa, RSC Adv., 2015, 5, 19924–19932 RSC.
- H. Wang, L. Zheng, C. Peng, M. Shen, X. Shi and G. Zhang, Biomaterials, 2013, 34, 470–480 CrossRef CAS PubMed.
- H. Liu, Y. Xu, S. Wen, J. Zhu, L. Zheng, M. Shen, J. Zhao, G. Zhang and X. Shi, Polym. Chem., 2013, 4, 1788 RSC.
- Y. Zhang, Q. Wang, B. Ashall, D. Zerulla and G. U. Lee, Adv. Mater., 2012, 24, 2485–2490 CrossRef CAS PubMed.
- J. S. Choi, Y. W. Jun, S. I. Yeon, H. C. Kim, J. S. Shin and J. Cheon, J. Am. Chem. Soc., 2006, 128, 15982 CrossRef CAS PubMed.
- J. Zhu, J. Wu, F. Liu, R. Xing, C. Zhang, C. Yang, H. Yin and Y. Hou, Nanoscale, 2013, 5, 9141–9149 RSC.
- P. de la Presa, M. Multigner, M. P. Morales, T. Rueda, E. Fernández-Pinel and A. Hernando, J. Magn. Magn. Mater., 2007, 316, e753–e755 CrossRef CAS.
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543 CrossRef CAS PubMed.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. Sun, Nano Lett., 2005, 5, 379 CrossRef CAS PubMed.
- Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2003, 125, 4046 CrossRef CAS PubMed.
- M. A. Malvindi, V. D. Matteis, A. Galeone, V. Brunetti, G. C. Anyfantis, A. Athanassiou, R. Cingolani and P. P. Pompa, PLoS One, 2014, 9, e85835 Search PubMed.
- S. J. Soenen, U. Himmelreich, N. Nuytten and M. De Cuyper, Biomaterials, 2011, 32, 195–205 CrossRef CAS PubMed.
- L. Roth, L. Agemy, V. R. Kotamraju, G. Braun, T. Teesalu, K. N. Sugahara, J. Hamzah and E. Ruoslahti, Oncogene, 2012, 31, 3754–3763 CrossRef CAS PubMed.
- B. Xu, Y. Ju, Y. Cui, G. Song, Y. Iwase, A. Hosoi and Y. Morita, Langmuir, 2014, 30, 7789–7797 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and figures. See DOI: 10.1039/c6ra23645f |
| This journal is © The Royal Society of Chemistry 2016 |