
Fabrication of PES-based membranes with a high and stable desalination performance for membrane distillation†
Lixia Liuab,
Fei Shen*ab,
Bowu Zhangc,
Haiqing Jiangc,
Jingye Lic,
Jianquan Luoab,
Huanhuan Wuab,
Rashid Khanab and
Yinhua Wan*ab
aState Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: fshen@ipe.ac.cn; yhwan@ipe.ac.cn; Tel: +86-10-82544991
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China
First published on 27th October 2016
Abstract
Polyethersulfone (PES)-based membranes with a high and stable desalination performance for vacuum membrane distillation (VMD) were fabricated by spraying a dispersion containing hydrophobic-modified PES grafted copolymers onto porous PES membranes. The copolymer (PES-g-PFMA-C8) with different degrees of grafting (DG) was synthesized by grafting 1H,1H,2H,2H-perfluorodecyl methacrylate (FMA-C8) onto PES by simultaneous irradiation in a homogeneous system wherein the kinetics of the radiation-induced graft polymerization was studied. The copolymers, pristine PES membrane and fabricated PES-based membranes were also characterized by Fourier transform infrared spectroscopy, X-ray photoelectron spectroscopy and scanning electron microscopy. After spraying, the hydrophobicity of the membrane increased and its maximum pore size reduced, which enhanced its desalination stability. During an intermittent 80 h VMD desalination operation, a flux of approximately 50.5 kg m−2 h−1 and a stable salt rejection of 99.98% were achieved when a 3.5 wt% NaCl solution was treated at 70 °C under vacuum of 92 kPa by the PES-based membrane using the copolymer with a DG of 20.3%, whereas salt leakage obviously occurred since the 52nd hour of operation when the pristine PES membrane was used. These results indicate that the modified PES membrane developed in this study is a promising candidate for VMD desalination due to its comparable flux.
1 Introduction
Fresh water scarcity is a big challenge in the 21st century with the growing population and improved living standards. Fortunately, seawater desalination offers hope in producing clean and fresh water.1 Compared with the mainstream desalination technologies, such as multi-stage flash distillation, reverse osmosis and multiple-effect distillation, membrane distillation (MD) is gradually developing as a competitive alternative due to its relatively low operation temperature and pressure.2,3 However, no large-scale MD application has been implemented to date. In practice, maintaining a stable flux and avoiding pore wetting are crucial for the commercialization of MD.4,5 For this purpose, many efforts have been launched to design suitable and valuable membranes for MD,6–8 which include not only the utilization of conventional hydrophobic polymers, such as polypropylene (PP), polyvinylidene fluoride (PVDF) and polytetrafluoroethylene (PTFE),9,10 but also the development of viable surface hydrophobization strategies on hydrophilic membranes.11,12As a hydrophilic material, polyethersulfone (PES) has been widely used to prepare membranes for ultrafiltration and microfiltration due to its good thermal stability, chemical resistance, excellent processability, and convenience in controlling its porosity and morphology.13 Compared with hydrophobic polymers, the low thermal conductivity of PES is beneficial to diminish the membrane heat transfer by conduction, and thus improve the heat efficiency of the MD process.9 Therefore, PES-based membranes fabricated for MD have recently emerged.14–19 However, the hydrophilicity of PES limits its application in the field of MD. Several methods have been used to improve the hydrophobicity of PES-based membranes. Suk et al. synthesized a series of surface modification macromolecules (SMMs), incorporated them into the PES casting solution, and then fabricated PES-based membranes by non solvent induced phase separation (NIPS) technique.14,15 During the process of membrane formation, the hydrophobic segment of the SMMs could migrate to the top surface of the membrane, which increased the hydrophobicity of the membrane.16 The fabricated membranes were successfully used in the separation of a water–ethanol mixture through the vacuum membrane distillation (VMD) process. On the other hand, tetraethylorthosilicate (TEOS) and trimethylchlorosilane (TMSCl) were used as grafting agents for the surface treatment of PES membranes.17 The hydrophilic PES membranes were transformed into highly hydrophobic membranes with contact angles (CA) as high as 150°. CF4 plasma surface modification was also carried out to fabricate a PES-based hydrophobic membrane for MD.18 Using the modified flat sheet PES-based membrane, the direct contact membrane distillation (DCMD) stability test showed a water flux of 24.5 m−2 h−1 and almost 100% salt rejection when 4 wt% NaCl solution was treated at temperature of about 61.5 °C. Abdallah et al. developed a titanium oxide nanotubes/polyethersulfone (TNTs-PES) blend membrane for VMD water desalination.19 A maximum salt rejection of 98% and permeate flux of 15.2 kg m−2 h−1 at 7000 ppm feed salt concentration were obtained at 300 mbar vacuum pressure and 65 °C feed temperature.
The above studies verify that PES-based membranes could indeed be used in the MD process. The flux and stability of PES-based membranes are also comparable to those of membranes fabricated from inherently hydrophobic polymers. Besides the methods mentioned above, the direct hydrophobization of PES materials might be an alternative to utilize the merits of PES to fabricate PES-based membranes for MD.
As a well-known method for the modification of polymer materials, γ-ray radiation has the advantages of simplicity, non-selectivity, strong penetrability and more importantly, avoidance of chemical initiators.20,21 Since radiation technology has developed into a mature technology, the application of the γ-ray radiation modification technique in nuclear track membranes22 and crosslinked cables23 have been industrialized. This technique has also been reported to realize membrane hydrophilization or hydrophilic modification of materials for membrane preparation.24 However, as far as we know, no report about the utilization of this technique to PES hydrophobic modification for MD application has been published.
Herein, 1H,1H,2H,2H-perfluorodecyl methacrylate (denoted as FMA-C8) was chosen as the monomer for PES hydrophobization by γ-ray radiation graft polymerization in a homogeneous system, which could allow the monomer to access the substrate polymer more uniformly.25 Then, FMA-C8 grafted PES copolymers (PES-g-PFMA-C8) were sprayed on the pristine PES membrane for MD desalination. Additionally, the kinetics of the radiation-induced graft polymerization was studied. The synthesis and characterization of PES-g-PFMA-C8 and the modified PES membranes are investigated. The VMD desalination performance of the pristine and modified PES membranes is also evaluated and discussed.
2 Experimental
2.1 Materials and chemicals
PES (E6020P) was purchased from BASF, Germany. 1H,1H,2H,2H-Perfluorodecyl methacrylate (FMA-C8, 98%) was purchased from Fuxin Hengtong Fluorine Chemicals Co., Ltd., China. 4-Methoxyphenol was purchased from Tokyo Chemical Industry Company. Trichlorotrifluoroethane (F113, 99.5%) was purchased from Aladdin Industrial Corporation. Methanol (99.5%) and N,N-dimethyl acetamide (DMAc, 99%) was purchased from Sinopharm Chemical Reagent Co., Ltd., China. LiCl and poly-N-vinyl-2-pyrrolidone (PVP K30) of analytical grade were supplied by Xilong Chemical Co., Ltd., China. Benzotrifluoride (99%) was purchased from J&K Scientific Ltd. All materials and chemicals were used without further purification.2.2 Radiation induced graft polymerization
Quantitative FMA-C8 and 0.5 g PES were dissolved in 20 mL DMAc at room temperature, and then a determined amount of 4-methoxyphenol, as the inhibitor, was mixed in the solution. The homogeneous solution was transferred to a 50 mL glass tube, bubbled with nitrogen for 20 min before it was sealed and then irradiated by a 60Co γ-ray source at a 0.3 kGy h−1 dose rate at room temperature.After a definite time period, the tube was removed from the irradiation source. The product (including the graft polymer PES-g-PFMA-C8 and homopolymer PFMA-C8) was very slowly precipitated into a vessel of methanol with stirring. Then the precipitate was extracted by F113 in a Soxhlet extractor for 72 h to thoroughly remove the homopolymer, and F113 was replaced every day during the extraction process. Finally, the PES-g-PFMA-C8 copolymer was dried at 60 °C in a vacuum oven until a constant weight was obtained.
2.3 Membrane preparation
The pristine PES membrane was prepared via the NIPS method, where a casting solution was prepared by mixing PES (15 wt%), LiCl (3 wt%), PVP (10 wt%) and DMAc (72 wt%) under stirring at 60 °C for about 48 h. After standing for 24 h at room temperature to any remove bubbles, the casting solution was cast onto a polyester nonwoven fabric by a steel scraper where the clearance was 0.15 mm. The obtained nascent membrane was then immersed in deionized water at 21 °C after exposure in air for 15 s. The membrane was rinsed with deionized water for 24 h to remove the residual solvent and additive agent. Finally, the prepared PES membrane was dried at room temperature.The PES membrane was then modified by the spraying method according to the following procedure. 0.1 g PES-g-PFMA-C8 was mixed with 50 g benzotrifluoride at 60 °C for 48 h to form a mixture. After that, the mixture was dispersed for 1 h with the aid of ultrasonication. The obtained dispersion was sprayed onto the PES membrane (6 cm × 10 cm) thrice by a spray gun (jet nozzle diameter = 0.8 mm, W-101, Anest Iwata Co., Japan). Each time, the 4 s spraying operation was followed by a 5 min time interval. During the interval, the sprayed membrane was left standing naturally. The spraying distance, pressure and temperature were kept at 20 cm, 0.2 MPa and room temperature, respectively. Finally, the sprayed membrane was naturally dried at room temperature for 24 h to ensure that benzotrifluoride evaporated completely. In this work, two types of modified PES membranes were fabricated by using different degrees of grafting (DG) of PES-g-PFMA-C8, as listed in Table 1.
| Membrane | PES-g-PFMA-C8 | ||
|---|---|---|---|
| Content (wt%) | DG (%) | Polymerization reaction condition | |
| PES-L | 0.2 | 14.5 | Absorbed dose 5.1 kGy, monomer concentration 3.5 vol%, inhibitor concentration 5 mmol L−1 |
| PES-H | 0.2 | 20.3 | Absorbed dose 5.1 kGy, monomer concentration 5 vol%, inhibitor concentration 1.5 mmol L−1 |
2.4 Characterization of the polymers
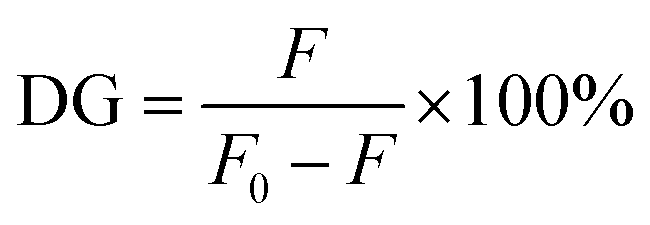 | (1) |
2.5 Characterization of the membranes
2.6 Vacuum membrane distillation experiments
VMD was conducted using the laboratory setup shown in our previous work.28 The effective membrane area was 15.00 cm2. The system piping, membrane module and storage tanks were all thoroughly insulated to minimize heat loss to the environment. A hot saline solution (3.5 wt% NaCl) was pumped to the feed side of the membrane by a digital peristaltic pump (WT600-2J, Baoding Longer Co., Ltd., China) at a constant flow rate of 1.0 L min−1. The feed volume was approximately 2.5 L and the feed temperature was monitored at the feed tank. The other side of the module was connected to a diaphragm vacuum pump (V410, Chemvak, Germany) to draw out the water vapor penetrating the membrane under the vacuum degree of 92 kPa. The water vapor could be condensed in the condenser filling with cold water and collected in a glass flask. The temperature of the cooling water was maintained at 10 ± 0.5 °C.Membrane flux was measured gravimetrically by determining the quantity of water collected in the glass flask every 30 minutes. The flux, J (kg m−2 h−1), was calculated using the following equation:
 | (2) |
The conductivity of the feed and the condensate was measured to represent the salt concentration. An electric conductivity monitor (DDS-307A, Shanghai Precision Scientific Instrument Co. Ltd., China) was used to monitor the conductivity of the feed and permeate. Salt rejection, R (%), was calculated using the following equation:
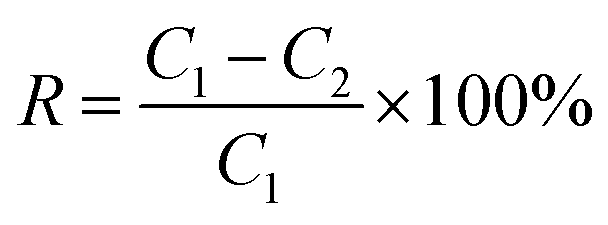 | (3) |
3 Results and discussion
3.1 Synthesis of PES-g-PFMA-C8
Fig. 1 shows the effects of the above mentioned parameters on the DG of PES-g-PFMA-C8. With respect to the effect of absorbed dose (Fig. 1a), at the critical absorbed dose of 5.1 kGy, the highest DG (18.4%) was observed. The DG increased when the absorbed dose was below this critical value, which is due to the fact that more active sites could be generated at a higher absorbed dose and these active sites act as the grafting initiator in polymerization.30 Subsequently, the DG decreased from 18.4% to 5.9% as the absorbed dose increased from 5.1 kGy to 12 kGy. A similar phenomenon was found in the simultaneous grafting of N-vinylpyrrolidone onto cotton-cellulose, where the DG started to decrease when the absorbed dose was beyond 5 kGy.31 Actually, scission of the grafted chains occurs under excessive irradiation, which is a usual phenomenon in simultaneous irradiation induced graft polymerization systems.32 In general, the radiation resistance of grafted side chains is lower than that of the bulk polymer, especially those containing aromatic rings (such as PES).32,33 Therefore, scission of the grafted side chains occurs under excessive irradiation condition, which might result in a decrease in DG.
 | ||
| Fig. 1 Effects of the reaction conditions on the DG of PES-g-PFMA-C8: (a) effect of absorbed dose (monomer concentration: 5 vol%, inhibitor concentration: 5 mmol L−1); (b) effect of monomer concentration (absorbed dose: 5.1 kGy, inhibitor concentration: 5 mmol L−1); and (c) effect of inhibitor concentration (absorbed dose: 5.1 kGy, monomer concentration: 5 vol%). | ||
The effect of monomer concentration on DG is shown in Fig. 1b. Clearly, the DG increased linearly from 7.1% to 30.7% with an increase in monomer concentration up to 8 vol%, and then started to decrease. This means that there was also a critical value of monomer concentration.30 In the simultaneous grafting system, graft polymerization and homopolymerization are a pair of competing reactions.13 The free radical concentration of FMA-C8 increased as the monomer concentration of FMA-C8 increased, which improved both the copolymerization and homopolymerization reaction rates. It is considered that the copolymerization reaction is dominant when the monomer concentration is below the critical value, which results in an increase in DG. However, once the monomer concentration exceeds the critical value, the free radicals of FMA-C8 contributing to copolymerization reaction become saturated. Meanwhile, the homopolymerization of FMA-C8 and the bimolecular termination between FMA-C8 are accelerated.20 As a result, the value of DG tends to level off.
In simultaneous irradiation systems, the addition of inhibitor is an essential step to suppress the side reaction of homopolymerization, especially for highly reactive monomers such as methyl acrylate and fluorinated methyl acrylate. Fig. 1c demonstrates the effect of inhibitor (4-methoxyphenol) concentration on DG. As the inhibitor concentration increases from 0.5 mmol L−1 to 1.5 mmol L−1, the DG increased from 14.9% to 20.3%. This indicates that the suppression effect on the side reaction of homopolymerization in the simultaneous irradiation system is enhanced by the addition of an appropriate amount of inhibitor. However, excessive inhibitor results in a decrease in DG. The DG decreased continuously from 20.3% to 9.9% with the increase in inhibitor concentration from 1.5 mmol L−1 to 10 mmol L−1, which means that excessive inhibitor also inhibits the graft copolymerization reaction. Therefore, the inhibitor concentration should be optimized in order to obtain a copolymer with a desirable DG.32
 | ||
| Fig. 2 FTIR spectra of pristine PES and PES-g-PFMA-C8 with different DGs. | ||
In order to further confirm the elemental composition of PES and PES-g-PFMA-C8, XPS characterization was performed. The XPS wide-scan spectra of PES and PES-g-PFMA-C8 are shown in Fig. 3a. Three elements including C, S and O were found in pristine PES and the F element appeared in PES-g-PFMA-C8, which obviously confirms the presence of PFMA-C8 graft chains after grafting. Moreover, the amount of F element increased rapidly with the DG (Fig. 3b). As listed in Table 2, for the pristine PES, the most abundant element is C, followed by O and S. After grafting, the contents of the C, O, S elements all decreased, whereas the content of F element increased obviously. When the DG was increased to 30.7%, the content of F (44.87%) was higher than that of C (41.77%), which made F become the main element of the copolymer. The F/C ratio increased from 0.18 to 1.07 when the DG increased from 5.9% to 30.7%. A high F/C ratio can hold great significance for the hydrophobic property of materials, which is essential for MD application.34
 | ||
| Fig. 3 (a) XPS wide-scan spectra of pristine PES and PES-g-PFMA-C8 with different DGs; (b) XPS F 1s spectra of PES-g-PFMA-C8 with different DGs. | ||
| Sample | Element mass concentration | ||||
|---|---|---|---|---|---|
| C (%) | O (%) | S (%) | F (%) | F/C | |
| PES | 65.51 | 21.22 | 13.27 | 0 | 0 |
| PES-g-PFMA-C8 (DG = 5.9%) | 60.14 | 18.14 | 11.01 | 10.71 | 0.18 |
| PES-g-PFMA-C8 (DG = 14.5%) | 57.35 | 17.70 | 3.01 | 21.94 | 0.38 |
| PES-g-PFMA-C8 (DG = 20.3%) | 47.45 | 14.25 | 2.46 | 35.84 | 0.76 |
| PES-g-PFMA-C8 (DG = 30.7%) | 41.77 | 9.15 | 4.21 | 44.87 | 1.07 |
In addition, Fig. 4 shows the corresponding C 1s spectra for PES and PES-g-PFMA-C8 with different DGs after curve fitting. As shown in Fig. 4a, five peaks can be assigned to the different types of C found in the backbone of PES, including C–C (284.8 eV), C–O (286.3 eV) and three other peaks (290.6, 291.7 and 293.1 eV) due to the π–π* shake-up satellites on the aromatic benzene rings of PES. The binding energies of all components were in good accordance with those reported previously.35 In the spectra of PES-g-PFMA-C8 with a DG of 5.9%, the C 1s core-level spectra could also be fitted into five peak components, namely C–C (284.8 eV), C–O (286.3 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (290.4 eV), CF2 (291.5 eV) and CF3 (293.0 eV). Influenced by the aromatic benzene rings of PES, the binding energies of the latter three were not highly in accordance with the results of former literature.36 In contrast, PES-g-PFMA-C8 with a high DG (e.g. 30.7%) was different from that with a low DG (e.g. 5.9%). For one thing, the influence of the aromatic benzene rings became weak at a high DG and the binding energies of CO, CF2 and CF3 returned to the normal levels of 288.9 eV, 291.4 eV and 293.5 eV, respectively. For another, the intensities of the CO, CF2 and CF3 peaks, especially for the CF2 peak, increased drastically in comparison with PES-g-PFMA-C8 with a low DG (e.g. 5.9%). Nevertheless, these three peaks are attributed to the CO, CF2 and CF3 bonds in the PFMA-C8 graft chains, which demonstrate the successful grafting of FMA-C8 onto PES.
O (290.4 eV), CF2 (291.5 eV) and CF3 (293.0 eV). Influenced by the aromatic benzene rings of PES, the binding energies of the latter three were not highly in accordance with the results of former literature.36 In contrast, PES-g-PFMA-C8 with a high DG (e.g. 30.7%) was different from that with a low DG (e.g. 5.9%). For one thing, the influence of the aromatic benzene rings became weak at a high DG and the binding energies of CO, CF2 and CF3 returned to the normal levels of 288.9 eV, 291.4 eV and 293.5 eV, respectively. For another, the intensities of the CO, CF2 and CF3 peaks, especially for the CF2 peak, increased drastically in comparison with PES-g-PFMA-C8 with a low DG (e.g. 5.9%). Nevertheless, these three peaks are attributed to the CO, CF2 and CF3 bonds in the PFMA-C8 graft chains, which demonstrate the successful grafting of FMA-C8 onto PES.
 | ||
| Fig. 4 XPS C 1s spectra of (a) pristine PES, (b) PES-g-PFMA-C8 with a DG of 5.9% and (c) PES-g-PFMA-C8 with a DG of 30.7%. | ||
The thermal stability of pristine PES and PES-g-PFMA-C8 with different DGs was evaluated by TGA. As presented in Fig. 5, pristine PES clearly displays a single-step degradation with an initial decomposition temperature of 473 °C, which is in accordance with the reported results.37 However, a two-step thermal decomposition was clearly observed in the TGA curves of PES-g-PFMA-C8 with different DGs. The first degradation started at 280 °C, which was ascribed to the decomposition of PFMA-C8 graft chains. The second degradation started at 473 °C, which conformed to the diagram of pristine PES. By comparing PES-g-PFMA-C8 with different DGs, the weight loss ratio in the first degradation stage increased as the DG increased. After grafting, the thermal stability decreased a bit, whereas the weight of residue decreased with an increase in DG due to the relatively low thermal stability of the PFMA-C8 chain. Nevertheless, it is considered that these differences have no effect on their application in MD, because the normal operating temperatures of MD are below 90 °C.
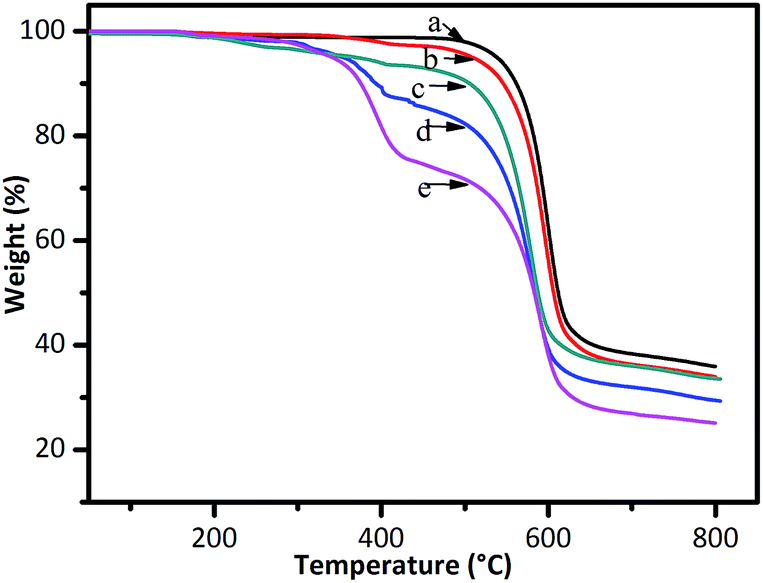 | ||
| Fig. 5 TGA curves of (a) pristine PES and PES-g-PFMA-C8 with DGs of (b) 5.9%, (c) 14.5%, (d) 20.3% and (e) 30.7%. | ||
3.2 Membrane characterization
As mentioned before, the properties of graft copolymers could be controlled by the DG. Mainly considering the solubility and hydrophobicity of the PES-g-PFMA-C8 copolymer, two moderate DGs of 14.5% and 20.3% were chosen for PES membrane modification in this work. The chemical feature in the near-surface region of the membranes was determined by XPS (as shown in Fig. 6). There was a significant difference in the composition of the membrane surface between the pristine PES membrane and the modified PES membranes. Specifically, no fluorine signal was detected in the pristine PES membrane, whereas it was clearly observed in both PES-L and PES-H membranes. The F 1s peak at 688.1 eV of the sprayed PES membranes demonstrates that PES-g-PFMA-C8 was successfully coated onto the PES base membrane surfaces. Also, the intensity of the F 1s peak of the PES-H membrane is slightly higher than that of the PES-L membrane due to the higher DG of PES-g-PFMA-C8. | ||
| Fig. 6 XPS wide-scan spectra of PES, PES-L and PES-H membranes. | ||
The perfluoroalkyl groups of the sprayed PES-g-PFMA-C8 endowed the modified PES membrane surfaces with distinct hydrophobic character. As listed in Table 3, the modified PES membranes exhibit obviously enhanced hydrophobicity. Advancing CA and receding CA both increased remarkably after the spraying modification. However, the differences between the advancing CA and receding CA of the sprayed membranes were lower than that of the pristine PES membrane, which indicate that the sprayed membranes have stronger resistance to wetting.38 Moreover, the static CA evolution with drop age of the membranes is compared in Fig. S1.† The static CA of the pristine PES membrane decreased clearly within 30 s, whereas the static CA of the sprayed membranes showed no obvious decline. This result also demonstrates that it is more difficult to wet the sprayed membranes. Surface roughness was also evaluated herein, and the Ra values are listed in Table 3 and the 3D images shown in Fig. S2.† Actually, very little hydrophobic copolymer was sprayed on the PES membrane due to the very dilute dispersion used, which resulted in a slight increase in the Ra value after spraying. Hence, the synergetic effect of surface chemistry and surface roughness increased the membrane surface hydrophobicity.39 Meanwhile, the maximum pore size decreased obviously by more than 34% after the spraying modification. The increased surface hydrophobicity and reduced maximum pore size together contribute to the increase in LEP of the membrane.40
| Sample | Maximum pore size (nm) | Mean pore size (nm) | θa (°) | θr (°) | Bulk porosity (%) | LEP (kPa) | Ra (nm) |
|---|---|---|---|---|---|---|---|
| PES | 68.92 | 9.97 | 82.3 ± 3.3 | 54.5 ± 2.7 | 85.8 | 295 | 2.38 |
| PES-L | 45.24 | 9.05 | 102.4 ± 2.8 | 80.1 ± 3.1 | 84.2 | 340 | 5.50 |
| PES-H | 41.10 | 8.68 | 114.3 ± 1.6 | 94.6 ± 2.9 | 83.1 | 410 | 5.67 |
SEM images of the membranes before and after spraying were taken to reveal the changes of surface and cross-sectional morphology (Fig. 7). The top surface layer is very critical for membranes used for the MD process, since it governs the MD performance.12 The pristine PES membrane presents a highly porous surface, as shown in Fig. 7(a-1). After spraying modification, the membrane surface structure becomes a little dense. As shown in Fig. 7(b-1) and (c-1), some of the surface pores are covered, which slightly decreases the membrane's surface mean pore size. The mean pore size decreased from 9.97 nm for the PES membrane to 8.68 nm for the PES-H membrane (as shown in Table 3). In addition, it could be seen clearly that the F element distributions on the modified membrane surfaces are uniform (Fig. 8a and b), which indicates even distributions of sprayed layer. Spraying modification does not significantly alter the cross-section structure of the membranes, as shown in Fig. 7. Besides, from the high magnification images of the membrane cross-section shown in Fig. 7(a-4)–(c-4), it is difficult to obtain the exact thicknesses of the sprayed layer, which might be due to the following two points. Firstly, the almost same backbone structure of PES and PES-g-PFMA-C8 might endow them with inherently fine interaction, which could enhance the anchoring strength of PES-g-PFMA-C8 on the PES membrane. Also, considering the quite low concentration of the spraying dispersion, the sprayed layer should be ultrathin. Therefore, it is expected that the ultrathin sprayed layer, homogeneous hydrophobization of the membrane surface, and reduced maximum pore size would be beneficial for MD desalination performance and stability.
 | ||
| Fig. 7 SEM observations of PES membrane (a-1) surface image (80k×), (a-2) cross-sectional image (600×), (a-3) cross-sectional image (5k×), (a-4) cross-sectional image (50k×); PES-L membrane (b-1) surface image (80k×), (b-2) cross-sectional image (600×), (b-3) cross-sectional image (5k×), (b-4) cross-sectional image (50k×); and PES-H membrane (c-1) surface image (80k×), (c-2) cross-sectional image (600×), (c-3) cross-sectional image (5k×), and (c-4) cross-sectional image (50k×). | ||
 | ||
| Fig. 8 SEM-EDS characterization of the fluorine element distribution of (a) PES-L membrane surface and (b) PES-H membrane surface. | ||
3.3 Membrane VMD performance
The membranes before and after modification were tested by the VMD process to evaluate their desalination performance using 3.5 wt% NaCl as the feed. As shown in Fig. 9a, the VMD fluxes increase exponentially for all the membranes with an increase in feed temperature because of the exponential increase in feed vapor pressure with temperature.41 Whereas, under the same operating temperature, the flux of the modified membrane is slightly lower than that of the pristine PES membrane, with the following flux order: PES > PES-L > PES-H. For example, the fluxes of the PES, PES-L and PES-H membranes at the feed temperature of 70 °C were 58.69, 52.67, 49.75 kg m−2 h−1, respectively. The differences in the pore structure parameters (such as pore size and porosity) are mainly responsible for this order. As given in Table 3, both the decrease in mean pore size and bulk porosity are consistent with this order. The lower porosity and pore size together reduced the effective evaporation area at the pore mouths, thus decreasing the membrane flux.42 As for the salt rejection, it can be observed from Fig. 9b that all the membranes hold a salt rejection higher than 99.9%, especially for the modified membranes whose salt rejections all exceed 99.95%. The similar high salt rejections of the three membranes are ascribed to their high LEP values, as listed in the Table 3. The sprayed PES-g-PFMA-C8 layer made membrane denser and more hydrophobic, which resulted in a lower permeation flux and higher salt rejection in the VMD desalination process. | ||
| Fig. 9 VMD performance of PES, PES-L and PES-H membranes: (a) flux, (b) rejection. (Feed solution: 3.5 wt% NaCl solution; feed flow rate: 1.0 L min−1; and vacuum pressure: 92 kPa). | ||
3.4 Desalination stability
Membrane wetting is always a great challenge for the long-term desalination operation of MD, and the long-term desalination stability of a membrane is a key issue for its practical application in MD. For membranes used for MD, improved hydrophobicity and reduced maximum pore size are both considered as effective methods to enhance their desalination stability.43 Although the membranes before and after modification in this work all could be used for MD desalination (Fig. 9), it is expected that their desalination stability would be different. An 80 h VMD treatment of a 3.5 wt% NaCl solution was operated to test the desalination stability of the three membranes. In the present work, the membranes were tested for 8 days (10 h per day), where the feed temperature was set at 70 °C. During the test, permeates were returned to the feed tank in order to maintain a constant salt concentration in the feed. Fig. 10 shows the flux and salt rejection of all the membranes versus time. As shown in Fig. 10a, the order of the initial flux of the tested membranes is PES (56.32 kg m−2 h−1) > PES-L (52.32 kg m−2 h−1) > PES-H (49.88 kg m−2 h−1), which is consistent with the results in Fig. 9a. However, the fluxes of the three membranes are almost identical at the end of the test. Compared with the modified membranes, the PES membrane had a relatively large fluctuation in the 80 h VMD process, and these changes were in the range of 45.50–58.99 kg m−2 h−1. The large fluctuation might be suffered from the fouling and pore wetting caused by NaCl crystallites due to the poor hydrophobicity and large maximum pore size of the PES membrane.46 As for the salt rejection, the PES membrane suffered a sudden decline in salt rejection after the 52nd hour, whereas such decline began at the 56th hour for the PES-L membrane. However, the rejection of the PES-H membrane was quite stable, which always maintained a level higher than 99.98%. The deteriorated salt rejections on the PES and PES-L membranes may have resulted from the partial pore wetting caused by NaCl crystallites.47 As given in Table 3, the maximum pore size follows the order: PES > PES-L > PES-H, however the hydrophobicity order is reversed. Decreased hydrophobicity and increased maximum pore size both lowered the membrane's resistance to such pore wetting. Therefore, surface modification by spraying PES-g-PFMA-C8 polymer with a higher DG (i.e. 20.3%) on the PES membrane improved the membrane's VMD desalination stability. | ||
| Fig. 10 VMD performance stability of PES, PES-L and PES-H membranes: (a) flux, (b) rejection. (Feed solution: 3.5 wt% NaCl solution; feed temperature: 70 °C; feed flow rate: 1.0 L min−1; and vacuum pressure: 92 kPa). | ||
Table 4 shows a comparison of the MD desalination performance in this study and previous literature.4,17–19,40,44,45 The MD performance of the PES-H membrane is comparable to that of the reported flat-sheet MD membranes. In combination with the results of Fig. 10, this comparison confirms that the PES-based membrane fabricated in this study has distinct advantages on MD performance and long-term stability, which make it promising for application in seawater desalination.
| Membrane | Operation mode | Feed solution | Process operating property | Flux (kg m−2 h−1) | Salt rejection (%) | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Tf (°C) | Tp (°C) | Vacuum degree (kPa) | ||||||
| Modified PES | DCMD | 2 wt% NaCl | 50 | 20 | — | 8.1 | 99.70% | 17 |
| Modified PES | DCMD | 4 wt% NaCl | 61.5 | 18.3 | — | 24.5 | 99.97% | 18 |
| Modified PES | VMD | 7000 ppm NaCl | 65 | — | 72 | 15.2 | 98% | 19 |
| Modified PVDF | DCMD | 3.5 wt% NaCl | 70 | 25 | — | 28 | ∼100% | 4 |
| PVDF | DCMD | 3.5 wt% NaCl | 60 | 20 | — | ∼9 | 99.99% | 40 |
| PVDF | DCMD | 1 M NaCl | 60 | 20 | — | 33 | ∼100% | 44 |
| PVDF | VMD | Pure water | 25 | — | 99 | 10.8 | — | 45 |
| PES-H | VMD | 3.5 wt% NaCl | 60 | — | 92 | 36.5 | 99.98% | This study |
4 Conclusions
The PES-g-PFMA-C8 copolymer was synthesized in a homogenous system by γ-ray simultaneous irradiation. The existence of PFMA-C8 graft chains in the copolymer was confirmed by FTIR and XPS spectroscopy. The kinetics of the radiation-induced graft polymerization was investigated to control the DG of the copolymer. In this work, the obtained DGs were in the range of 5.9% to 30.7%. Then, PES-g-PFMA-C8 copolymers with moderate DG of 14.5% and 20.3% were each used to modify a hydrophilic PES base membrane by the spraying method. The spraying of PES-g-PFMA-C8 resulted in an increase in hydrophobicity and reduction of the maximum pore size, which endowed the membrane with improved resistance to pore wetting, especially when a high DG (e.g. 20.3%) of PES-g-PFMA-C8 was used. An 80 h long-term VMD desalination test at 70 °C using a 3.5 wt% NaCl solution as the feed showed that a steady flux of about 50.5 kg m−2 h−1 and high salt rejection of over 99.98% were obtained by the PES-H membrane. Whereas, the pristine PES membrane suffered from obvious pore wetting during the long-term VMD test. It can be concluded that the PES-g-PFMA-C8 polymer synthesized in this work could effectively improve the hydrophobicity and MD stability of membranes by a simple spraying method. Also, the modified PES membrane exhibits a high and comparable VMD desalination performance.Acknowledgements
The authors would like to thank the support from National Natural Science Funds of China (21106153), Solar Energy Initiative of the Chinese Academy of Sciences (KGCX2-YW-380) and the National High-Tech R&D Program of China (2012AA021202, 2014AA021005).References
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Marinas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- D. Hou, J. Wang, X. Sun, Z. Ji and Z. Luan, J. Membr. Sci., 2012, 405–406, 185–200 CrossRef CAS.
- B. Zhang, L. Liu, S. Xie, F. Shen, H. Yan, H. Wu, Y. Wan, M. Yu, H. Ma, L. Li and J. Li, RSC Adv., 2014, 4, 16561–16566 RSC.
- A. Razmjou, E. Arifin, G. Dong, J. Mansouri and V. Chen, J. Membr. Sci., 2012, 415–416, 850–863 CrossRef CAS.
- M. M. Teoh and T.-S. Chung, Sep. Purif. Technol., 2009, 66, 229–236 CrossRef CAS.
- M. Khayet, T. Matsuura, J. I. Mengual and M. Qtaishat, Desalination, 2006, 192, 105–111 CrossRef CAS.
- K. K. S. Baoan Li, J. Membr. Sci., 2005, 257, 60–75 CrossRef.
- H. Wang, S. Ding, H. Zhu, F. Wang, Y. Guo, H. Zhang and J. Chen, Sep. Purif. Technol., 2014, 126, 82–94 CrossRef CAS.
- M. Khayet, Adv. Colloid Interface Sci., 2011, 164, 56–88 CrossRef CAS PubMed.
- F. Liu, N. A. Hashim, Y. Liu, M. R. M. Abed and K. Li, J. Membr. Sci., 2011, 375, 1–27 CrossRef CAS.
- Y. Kong, X. Lin, Y. Wu, J. Chen and J. Xu, J. Appl. Polym. Sci., 1992, 46, 191–199 CrossRef CAS.
- M. Essalhi and M. Khayet, J. Membr. Sci., 2012, 417–418, 163–173 CrossRef CAS.
- B. Deng, J. Li, Z. Hou, S. Yao, L. Shi, G. Liang and K. Sheng, Radiat. Phys. Chem., 2008, 77, 898–906 CrossRef CAS.
- D. E. Suk, T. Matsuura, H. B. Park and Y. M. Lee, Desalination, 2010, 261, 300–312 CrossRef CAS.
- D. E. Suk, T. Matsuura, H. B. Park and Y. M. Lee, J. Membr. Sci., 2006, 277, 177–185 CrossRef CAS.
- D. E. Suk, G. Chowdhury and T. Matsuura, Macromolecules, 2002, 35, 3017–3021 CrossRef CAS.
- A. Rastegarpanah and H. R. Mortaheb, Desalination, 2016, 377, 99–107 CrossRef CAS.
- X. Wei, B. Zhao, X.-M. Li, Z. Wang, B.-Q. He, T. He and B. Jiang, J. Membr. Sci., 2012, 407–408, 164–175 CrossRef CAS.
- H. Abdallah, A. F. Moustafa, A. A. AlAnezi and H. E. M. El-Sayed, Desalination, 2014, 346, 30–36 CrossRef CAS.
- A. Bhattacharya and B. N. Misra, Prog. Polym. Sci., 2004, 29, 767–814 CrossRef CAS.
- H. Liu, M. Yu, H. Ma, Z. Wang, L. Li and J. Li, Radiat. Phys. Chem., 2012, 94, 129–132 CrossRef.
- R. Mazzei, E. Smolko, D. Tadey and L. Gizzi, Nucl. Instrum. Methods Phys. Res., Sect. B, 2000, 170, 419–426 CrossRef CAS.
- D. J. T. Hill, J. H. O'Donnell, M. C. S. Perera and P. J. Pomery, Int. J. Radiat. Appl. Instrum., Part C, 1992, 40, 127–138 CAS.
- Q. Qin, Z. Hou, X. Lu, X. Bian, L. Chen, L. Shen and S. Wang, J. Membr. Sci., 2013, 427, 303–310 CrossRef CAS.
- R. W. Betty and W. H. Rapson, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9(7), 1901–1919 CrossRef CAS.
- W. Geng, T. Nakajima, H. Takanashi and A. Ohki, Fuel, 2007, 86, 715–721 CrossRef CAS.
- L. Li, B. Deng, Y. Ji, Y. Yu, L. Xie, J. Li and X. Lu, J. Membr. Sci., 2010, 346, 113–120 CrossRef CAS.
- L. Liu, F. Shen, X. Chen, J. Luo, Y. Su, H. Wu and Y. Wan, J. Membr. Sci., 2016, 499, 544–554 CrossRef CAS.
- X. Yang, B. Deng, Z. Liu, L. Shi, X. Bian, M. Yu, L. Li, J. Li and X. Lu, J. Membr. Sci., 2010, 362, 298–305 CrossRef CAS.
- C. Xu, W. Huang, Y. Zhou, D. Yan, S. Chen and H. Huang, Radiat. Phys. Chem., 2012, 81, 426–431 CrossRef CAS.
- E. Takács, H. Mirzadeh, L. Wojnárovits, J. Borsa, M. Mirzataheri and N. Benke, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 217–220 CrossRef.
- M. M. Nasef and E.-S. A. Hegazy, Prog. Polym. Sci., 2004, 29, 499–561 CrossRef CAS.
- J. Farhataziz and M. A. J. Rodgers, Radiation chemistry: principles and applications, VCH Publishers, New York, 1987 Search PubMed.
- B. Deng, R. Cai, Y. Yu, H. Jiang, C. Wang, J. Li, L. Li, M. Yu, J. Li, L. Xie, Q. Huang and C. Fan, Adv. Mater., 2010, 22, 5473–5477 CrossRef CAS PubMed.
- B. Kaeselev, P. Kingshott and G. Jonsson, Desalination, 2002, 146, 265–271 CrossRef CAS.
- M. M. Koji Honda, H. Otsuka and A. Takahara, Macromolecules, 2005, 38, 5699–5705 CrossRef.
- A. Rahimpour, S. S. Madaeni and S. Mehdipour-Ataei, J. Membr. Sci., 2008, 311, 349–359 CrossRef CAS.
- M. Khayeta, C. Y. Feng and T. Matsuura, J. Membr. Sci., 2003, 213, 159–180 CrossRef.
- F. L. Huang, Q. Q. Wang, Q. F. Wei, W. D. Gao, H. Y. Shou and S. D. Jiang, eXPRESS Polym. Lett., 2010, 4, 551–558 CrossRef CAS.
- J. Zhang, Z. Song, B. Li, Q. Wang and S. Wang, Desalination, 2013, 324, 1–9 CrossRef CAS.
- J. I. Mengual, M. Khayet and M. P. Godino, Int. J. Heat Mass Transfer, 2004, 47, 865–875 CrossRef CAS.
- S. Bonyadi and T.-S. Chung, J. Membr. Sci., 2009, 331, 66–74 CrossRef CAS.
- L. D. Tijing, Y. C. Woo, J.-S. Choi, S. Lee, S.-H. Kimd and H. K. Shon, J. Membr. Sci., 2015, 475, 215–244 CrossRef CAS.
- S. Nejati, C. Boo, C. O. Osuji and M. Elimelech, J. Membr. Sci., 2015, 492, 355–363 CrossRef CAS.
- M. K. T. Matsuura, Ind. Eng. Chem. Res., 2001, 40, 5710–5718 CrossRef.
- M. Gryta, J. Membr. Sci., 2005, 265, 153–159 CrossRef CAS.
- P. Wang, M. M. Teoh and T.-S. Chung, Water Res., 2011, 45, 5489–5500 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22193a |
| This journal is © The Royal Society of Chemistry 2016 |