
NH2-functionalized carbon-coated Fe3O4 core–shell nanoparticles for in situ preparation of robust polyimide composite films with high dielectric constant, low dielectric loss, and high breakdown strength†
Xinliang Fanga,
Shanfeng Wang*b,
Yanxiao Lia,
Xiaoyun Liua,
Xinxin Li*a,
Shaoliang Lina,
Zhong-Kai Cuic and
Qixin Zhuang*a
aKey Laboratory of Advanced Polymer Materials of Shanghai, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: qxzhuang@ecust.edu.cn
bDepartment of Materials Science and Engineering, Institute of Biomedical Engineering, The University of Tennessee, Knoxville, TN 37996, USA
cDepartment of Chemistry, Université de Montréal, C.P. 6128, Succ. Centre Ville, H3C 3J7, Montréal, Québec, Canada
First published on 3rd November 2016
Abstract
Novel Fe3O4/polyimide (PI) composite films with extraordinary dielectric properties were prepared via in situ polymerization of PI in the presence of 4,4′-diaminodiphenyl ether (ODA), pyromellitic dianhydride (PMDA) and compatible amine-functionalized carbon-coated Fe3O4 nanoparticles (Fe3O4@C–NH2). Fe3O4 nanoparticles prepared via solvothermal reaction were coated with crosslinked glucose and then surface-grafted via carbodiimide coupling. The core–shell structure of Fe3O4@C–NH2 was thereafter obtained with amine functionalities on the carbonized-glucose shell. Homogeneous dispersion of Fe3O4@C–NH2 and strong interfacial covalent bonding on the shell surface contributed to high dielectric constants (ε) and extremely low dielectric losses (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) in the composite films. The highest dielectric constant of the composite films was 58.6 at 1 kHz when the Fe3O4@C–NH2 composition was 1.13 vol%, as well as a low dielectric loss of 0.0091 and a high breakdown strength of 126.5 ± 11.3 MV m−1 were observed without considerable sacrifice of the thermal stability and mechanical properties.
δ) in the composite films. The highest dielectric constant of the composite films was 58.6 at 1 kHz when the Fe3O4@C–NH2 composition was 1.13 vol%, as well as a low dielectric loss of 0.0091 and a high breakdown strength of 126.5 ± 11.3 MV m−1 were observed without considerable sacrifice of the thermal stability and mechanical properties.
Introduction
Polymer nanocomposites have attracted much attention in recent years because of their many advantages such as flexibility, ease of processing, and tunable properties.1–4 Polymer nanocomposites with a high dielectric constant (ε) and ease of processing are promising dielectric materials for embedded capacitors, considering their flexibility and compatibility with organic printed circuit boards.5 Polyvinylidene fluoride (PVDF) is one of the most studied polymer matrices for being incorporated with carbon nanotubes (CNT),6,7 BaTiO3,8 or a porous graphene sandwich9 to improve its dielectric properties because of its intrinsically high dielectric constant of ∼12. The dielectric constants of these composites did rise with increasing of the filler composition; however, the dielectric loss increased as well by adding conductive fillers as their direct contact induces a high leakage current. The dielectric constants of radial ZnO/PVDF composites were reported to be ∼65 at 1 kHz, and the dielectric losses were ∼0.5,10 which were too high for practical applications. Besides PVDF, poly(p-phenylene benzobisoxazole),11–16 epoxy,17–19 polyurethane,20,21 polyethersulfone,22 polyarylene ether nitriles,23 poly(methyl methacrylate),24 and other polymers25–27 were also reported as polymer matrices for fabricating dielectrics.Polyimide (PI) exhibits great potential as a high-performance dielectric material on the basis of its heat resistance and outstanding mechanical properties.28 The dielectric constants of most aromatic PI films are between 2.8 and 5.0, while the dielectric loss values vary from 0.001 to 0.03. Because the low dielectric loss of PI is ideal for practical applications in high temperature environments, researchers committed to improve its dielectric constant. Previous efforts on achieving high-ε PI-based composites primarily focused on those filled with high-ε ferroelectric ceramics, such as nano-TiO2,29 Al2O3,30 BaTiO3,31,32 and CaCu3–Ti4O12.33,34 For these composites, ε was barely over 50 even when the filler volume fraction was increased to 50% or higher. In addition, the composite films were limited because of reduced processability and dramatic compromise in mechanical properties.
Another strategy was to introduce conductive fillers, including CNT (ε = 31.3)35 and graphene (ε = 36.9)36 into the PI matrix. Although the mechanical properties were satisfactory, the dielectric constants of the resulting composites were only ∼35.0, much lower than those of ferroelectric ceramic/PI composites. Fe3O4 spherical nanoparticles were promising candidates because of their unique magnetic response and large surface area.37–41 Haldar et al. reported ε values of ∼2000 and ∼500 in Fe3O4/polyaniline composites and polypyrrole-modified poly(N-vinyl carbazole)–Fe3O4 nanocomposites, respectively.42,43 Therefore, a high dielectric constant and a low dielectric loss are expected in Fe3O4/PI composites at the percolation threshold when modified Fe3O4 nanoparticles are well dispersed in the PI matrix. This material design strategy was applied in this study to achieve the goals in dielectric properties with a new preparative method of functionalized Fe3O4 nanoparticles.
Although Fe3O4 nanoparticles are highly competent as conductive fillers, two limitations have been identified in their practical applications: (1) Fe3O4 nanoparticles readily aggregate because of their large specific surface area and high surface free energy, making nanoscale dispersion of Fe3O4 nanoparticles in a polymer matrix extremely challenging;44 (2) Fe3O4 nanoparticles can be easily oxidized in the absence of a protective shell. To overcome these barriers, a strategy has been proposed to prepare core-shell-structured carbon-coated Fe3O4 nanoparticles (Fe3O4@C) via hydrothermal carbonization.45 The carbon layer can effectively protect Fe3O4 nanoparticles from being oxidized and the outermost polysaccharide layer holds a variety of functional groups, such as carboxyl and hydroxyl groups, on the surface.46,47 Fe3O4@C was further decorated with p-phenylene diamine (PPD) in the presence of N,N′-dicyclohexyl carbodiimide (DCC) catalyst to introduce amine functionality (Fe3O4@C–NH2), so as to improve the reactivity and dispersion of the nanoparticles.36 The functionalized core–shell nanoparticles were homogeneously distributed throughout the polymer matrix via in situ polymerization of PI, and many appealing properties (high dielectric constant, low dielectric loss, and high breakdown strength) of the Fe3O4@C–NH2/PI composites were achieved as expected while the thermal stability and tensile strength were also as excellent as the PI matrix with scarce sacrifice.
Experimental section
Materials
Reagents including FeCl3·6H2O, ethylene glycol, sodium acetate, polyethylene glycol, DCC, HNO3, DMF and ethanol were supplied by Alfa Aesar (Massachusetts, USA). Glucose was purchased from Aldrich Chemical Company (Milwaukee, USA). PPD and 4,4′-diaminodiphenyl ether (ODA) were obtained from Aladdin (Shanghai, China). Pyromellitic dianhydride (PMDA) was provided by J & K (Beijing, China). DMAc was dried with calcium hydride (CaH2) prior to distillation. Other reagents were of high purity and used as received.Synthesis of Fe3O4 nanoparticles48
1.35 g FeCl3·6H2O (5 mmol) was dissolved in 40 mL ethylene glycol, followed by addition of 3.6 g sodium acetate (NaAc) and 1.0 g polyethylene glycol (PEG, Mw = 20000 g mol−1). The solution was under vigorously magnetically stirring for 30 min, and sealed in a 100 mL Teflon-lined stainless-steel autoclave reactor. The autoclave reactor remained at 200 °C for 8 h, and cooled down to room temperature. The black powder was washed thoroughly with ethanol and dried at 60 °C for 6 h.
Synthesis of Fe3O4@C core–shell nanoparticles49
Fe3O4 nanoparticles were soaked in 0.1 M HNO3 solution for 5 min, and separated the Fe3O4 nanoparticles from the solution with a magnet followed by extensive wash with deionized water. The treated Fe3O4 nanoparticles (2 out of 5 mmol) were dispersed in 60 mL water under vigorous stirring for 30 min with assistance of 7.2 g glucose (40 mmol). The solution was transferred to a 100 mL Teflon-lined stainless-steel autoclave reactor. The autoclave reactor remained at 180 °C for 6 h, and cooled down to room temperature. The black powder was washed thoroughly with ethanol and dried at 60 °C for 6 h.Preparation of Fe3O4@C–NH2 nanoparticles
0.05 g Fe3O4@C was dispersed in 20 mL DMAc with ultrasonication, followed by addition of 0.05 g DCC. After 4 h, 0.05 g PPD was added and the dispersion was stirred for 24 h under nitrogen protection at room temperature before filtration and thorough wash with DMF and ethanol. Fe3O4@C–NH2 was obtained and dried in a vacuum oven at 60 °C for 24 h.Preparation of Fe3O4@C–NH2/PI films via in situ polymerization of PI
10.05 mg Fe3O4@C–NH2 was dispersed in 20 mL anhydrous DMAc with ultrasonication for 30 min, and a stable dispersion was obtained. 1.00 g 4,4′-diaminodiphenyl ether (ODA, 5 mmol) was added and dissolved with magnetic stirring at room temperature under nitrogen protection, followed by addition of 1.09 g pyromellitic dianhydride (PMDA, 5 mmol) with 4 equal batches in quantity within 4 h. After PDMA was completely dissolved, the dispersion was stirred for 24 h and poured into a glass Petri dish, followed by drying for 48 h and heat treatment at 100, 200, and 300 °C for 2 h, respectively, and then at 400 °C for 5 min. Fe3O4@C–NH2/PI composite films containing 0.13 vol%, 0.27 vol%, 0.55 vol%, 0.83 vol%, 1.13 vol%, and 1.40 vol% of Fe3O4@C–NH2 were obtained. Fe3O4/PI films were prepared following the same procedure.Characterization
Fourier transform infrared spectroscopy (FTIR) spectra were acquired on Nicolet Magna-IR 550 spectrometer using pressed KBr pellets. X-ray diffraction (XRD) was performed on a D/MAX 2550 VB/PC rotating anode X-ray multi-crystal diffractometer equipped with Ni-filtered CuKα radiation and operated at 60 mA and 40 kV. The morphologies of Fe3O4, Fe3O4@C and Fe3O4@C–NH2 nanoparticles were examined using field-emission scanning electron microscopy (SEM, FESEM, Hitachi S-4800) and high-resolution transmission electron microscopy (TEM, HRTEM, JEOL JEM-2100). The fractured cross sections of both PI and Fe3O4@C–NH2/PI composite films were examined using the SEM and a Fe3O4@C–NH2/PI composite film with a thickness of 60–70 nm was prepared through ultrathin section and observed using the TEM. Thermogravimetric analysis (TGA) was performed at a heating rate of 10 °C min−1 under nitrogen stream. The dielectric constants were measured on a CONCEPT 40 broadband dielectric spectrometer (Novocontrol Technologies GmbH & Co. KG, Germany). The breakdown strength was evaluated by a DC dielectric strength tester with a sphere–sphere stainless electrode (DH, Shanghai Lanpotronics Co., China) at room temperature. Mechanical properties were investigated with a Rheometric Scientific DMTA5 under tensile mode, with results averaged over more than 5 replicates.Results and discussion
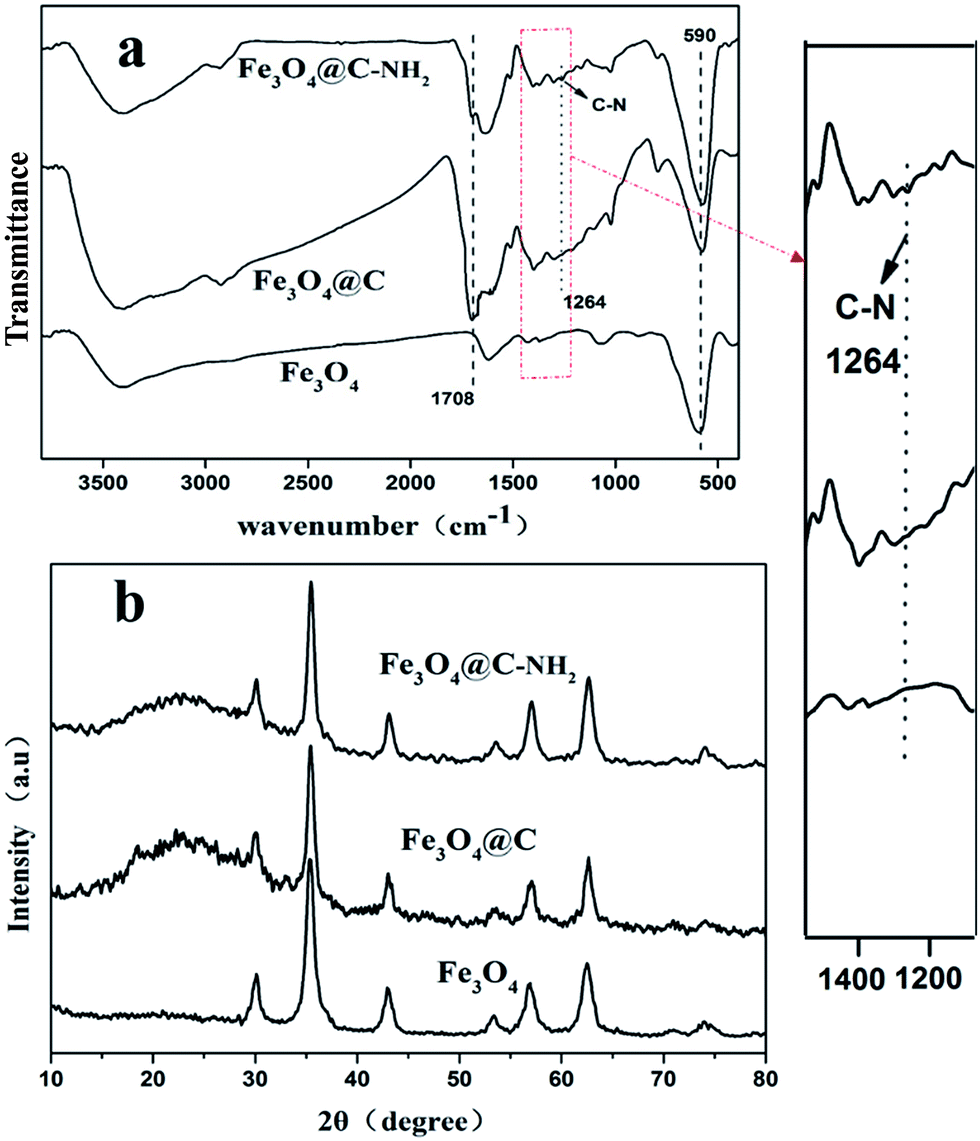
The Fe3O4 or Fe3O4@C–NH2 volume composition (f) in the Fe3O4/PI and Fe3O4@C–NH2/PI composites was defined as Vp/(Vp + Vm) × 100%, where Vp and Vm are the volumes of Fe3O4 or Fe3O4@C–NH2 and PI used for composite preparation, respectively. Vp and Vm were calculated from their weights and the densities, which are 5.18 and 1.40 g cm−1 for Fe3O4 and PI, respectively. Because the carbon shell (4 nm) of the Fe3O4@C–NH2 was much thinner than the diameter of the Fe3O4 (40 nm), we substituted the density of Fe3O4@C–NH2 with the density of Fe3O4 in calculating f for Fe3O4@C–NH2 in the composites. Thus the weight compositions of the nanoparticles of 1, 2, 3, 4, and 5 wt% were converted to f values of 0.27, 0.55, 0.83, 1.13, and 1.40 vol%.Fe3O4, Fe3O4@C, and Fe3O4@C–NH2 nanoparticles were characterized using FTIR, as shown in Fig. 1a. The absorption band at 3410 cm−1 indicated the presence of residual hydroxyl groups, and the bands at 1708 and 1635 cm−1 were associated with stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC, respectively. This result confirmed successful carbonization of glucose during the hydrothermal carbonization and the presence of considerable residual hydrophilic functionalities resulting from the incomplete carbonization of glucose.50,51 The bands in 1000–1300 cm−1 were attributed to stretching vibrations of C–O and bending vibrations of O–H. Compared to Fe3O4@C, Fe3O4@C–NH2 showed weaker stretching vibrations of CO and emergence of stretching vibrations of C–N at 1264 cm−1 from the amide in Fe3O4@C–NH2, indicating successful amidation between Fe3O4@C and PPD. The band at 590 cm−1 corresponding to stretching vibrations of Fe–O,50 confirmed the presence of Fe3O4 in the composites.
O and CC, respectively. This result confirmed successful carbonization of glucose during the hydrothermal carbonization and the presence of considerable residual hydrophilic functionalities resulting from the incomplete carbonization of glucose.50,51 The bands in 1000–1300 cm−1 were attributed to stretching vibrations of C–O and bending vibrations of O–H. Compared to Fe3O4@C, Fe3O4@C–NH2 showed weaker stretching vibrations of CO and emergence of stretching vibrations of C–N at 1264 cm−1 from the amide in Fe3O4@C–NH2, indicating successful amidation between Fe3O4@C and PPD. The band at 590 cm−1 corresponding to stretching vibrations of Fe–O,50 confirmed the presence of Fe3O4 in the composites.
 | ||
| Fig. 1 (a) FTIR spectra and (b) XRD profiles of Fe3O4, Fe3O4@C, and Fe3O4@C–NH2 nanoparticles. | ||
From the XRD profiles presented in Fig. 1b, characteristic diffraction peaks at 2θ of 30.05°, 35.50°, 43.19°, 57.11°, and 62.67° were well indexed to the cubic spinel structured magnetite (JCPDS file No. 19-0629),52 indicating that the crystalline phase of Fe3O4 remained unchanged during the coating and the amidation processes. After the coating process, a relatively broad halo (17.13–29.14°) centered at 2θ of 22.51° was identified in the profiles of Fe3O4@C and Fe3O4@C–NH2 samples, indicating that the carbon matrix was amorphous.53
The morphology of as-synthesized nanoparticles was further captured by using SEM and TEM. As shown in Fig. 2a and b, a virtually spherical shape was observed for the as-synthesized Fe3O4 nanoparticles with a mean diameter of ∼40 nm. Based on the SEM images of Fe3O4@C and Fe3O4@C–NH2 nanoparticles in Fig. 2c and e, the mean diameters of Fe3O4@C and Fe3O4@C–NH2 nanoparticles were estimated to be ∼40 nm as well, suggesting that the shape and size remained unchanged after the coating and the amidation processes. In contrast to Fig. 2b, d and f presented a core–shell structure, in which black Fe3O4 nanoparticles were encapsulated by a 4 nm thick gray carbon shell (see the high magnification images). The uniform amorphous carbonaceous coating was most likely generated by the carbonization of glucose coating on the Fe3O4 nanoparticles during the hydrothermal carbonization.50 Dispersion of Fe3O4, Fe3O4@C, and Fe3O4@C–NH2 in N,N-dimethylacetamide (DMAc) at the concentration of 3 mg mL−1 was presented in Fig. 2g–i, respectively. The comparison between Fe3O4 and Fe3O4@C showed that the carbon layer with hydrophilic functionalities was of vital importance in protecting the Fe3O4 nanoparticles from oxidation and promoting the dispersion of the nanoparticles in DMAc. After amidation, the dispersion of Fe3O4@C–NH2 in DMAc was further improved.
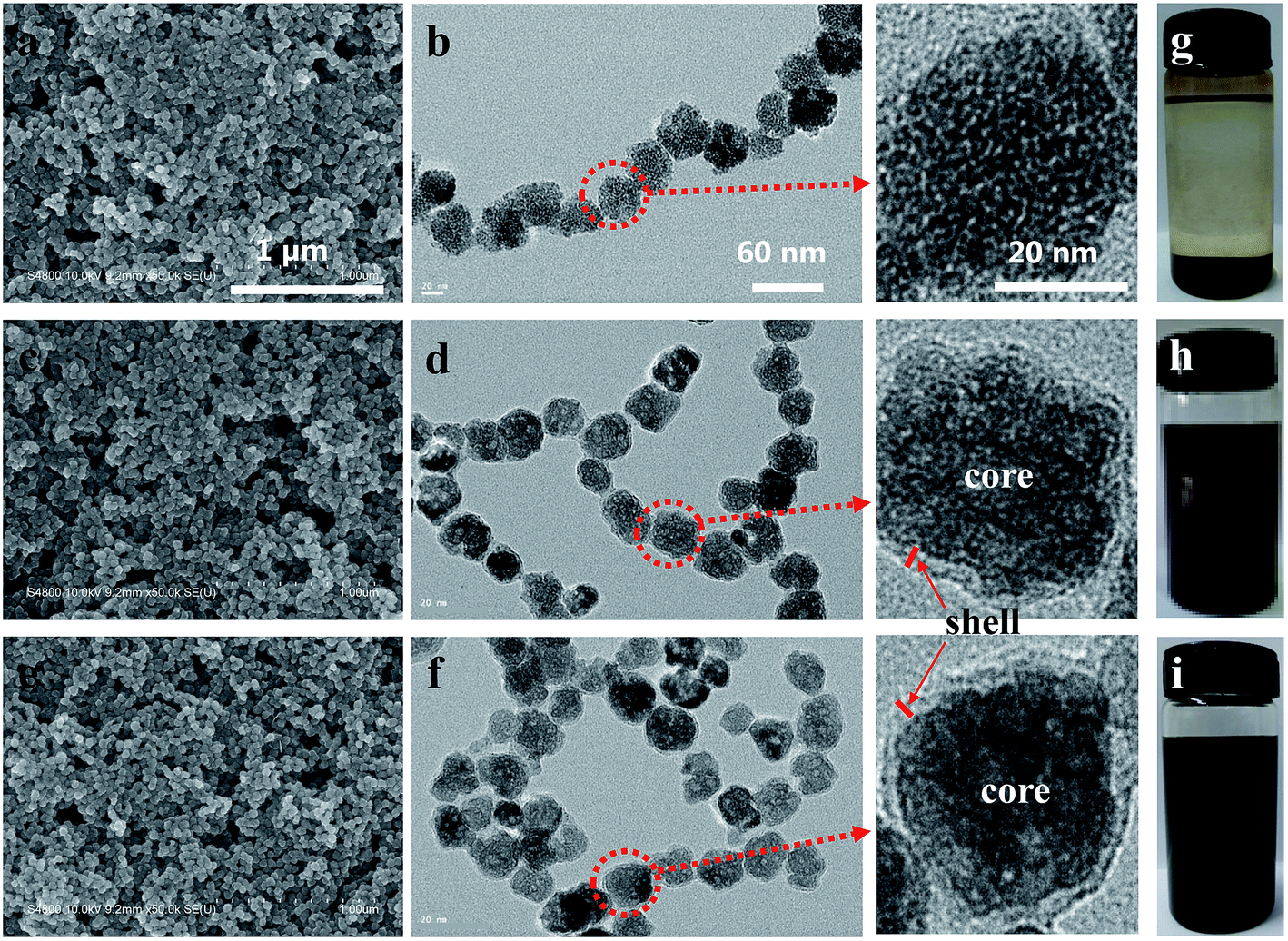 | ||
| Fig. 2 SEM (a, c, e) and TEM (b, d, f) images, and dispersion in DMAc at 3 mg mL−1 (g, h, i) of the nanoparticles of Fe3O4 (a, b, g), Fe3O4@C (c, d, h), and Fe3O4@C–NH2 (e, f, i) with high magnification TEM images of individual nanoparticles. Scale bars of 1 μm, 60 nm, and 20 nm in the images in the top row are applicable for the images in their respective columns. | ||
The dispersion of Fe3O4@C–NH2 in the PI matrix was a pivotal factor in controlling the physical properties and performance of the composites. SEM images of the fractured surface of the neat PI and the Fe3O4@C–NH2/PI (f = 1.13 vol%) composite film at the same magnification were compared and further supported this key point. The smooth fractured cross section of the neat PI film (Fig. 3a) was altered dramatically upon the introduction of the nanoparticles; the fractured cross-section of the Fe3O4@C–NH2/PI composite films was rather rough and ridged (Fig. 3b). As shown in Fig. 3c, the Fe3O4@C–NH2 nanoparticles were uniformly dispersed without observable aggregation, leading to the expectation of high dielectric constants and low dielectric losses in the composites. However, the diameters of the Fe3O4@C–NH2 nanoparticles shown in Fig. 3c were from 5 to 45 nm. The discrepancy between the data and the values obtained from SEM and TEM images in Fig. 2 was as a result of that the cross-section film thickness of nanocomposites (60–70 nm) was very close to the nanoparticle diameter (∼40 nm).
 | ||
| Fig. 3 SEM images of the fractured cross sections of (a) the neat PI (×40000) and (b) the 1.13 vol% Fe3O4@C–NH2/PI (×40000) composite films; (c) TEM image of the 1.13 vol% Fe3O4@C–NH2/PI composite film. Scale bars of 1 μm, 1 μm, and 50 nm are present in the images of (a), (b), and (c), respectively. | ||
Thermal stability is one of the important properties for PI-based nanocomposites used as high-performance engineering plastics. As shown in TGA (Fig. 4a), the onset thermal decomposition temperatures at 5% mass loss of neat PI and Fe3O4@C–NH2/PI nanocomposites with f values of 0.27 vol%, 0.55 vol%, 0.83 vol%, 1.13 vol%, and 1.40 vol% were 591.5, 563.1, 557.2, 549.5, 522.5, and 504.8 °C, respectively. The thermal stability of the nanocomposites only decreased slightly upon increasing the nanoparticle composition. Compared to neat PI and nanocomposites containing 0.27–1.13 vol% Fe3O4@C–NH2, the onset decomposition temperature of Fe3O4@C–NH2/PI (f = 1.40 vol%) nanocomposite was much lower, primarily attributed to agglomeration of the Fe3O4@C–NH2 nanoparticles at compositions higher than the percolation threshold. In contrast, as shown in Fig. 4b, the onset thermal decomposition temperatures of the Fe3O4/PI nanocomposites greatly decreased from 591.5 to 428.8 °C with increasing f from 0% (neat PI) to 1.40 vol%, which were much lower than those of the Fe3O4@C–NH2/PI nanocomposites because of the poor dispersion of Fe3O4 in PI. The agglomeration of the Fe3O4 nanoparticles in PI hindered the amidization process of PI and that the larger interface gap between the thermal conductive Fe3O4 aggregates and the PI matrix might serve as diffusion path for heat and degradation products.
 | ||
| Fig. 4 TGA diagrams (a, b) and stress–strain curves (c, d) of the neat PI film and the Fe3O4@C–NH2/PI (a, c) and Fe3O4/PI (b, d) composite films with various nanoparticle volume compositions. | ||
Representative stress–strain curves of the neat PI and the Fe3O4@C–NH2/PI composites are illustrated in Fig. 4c. The tensile strength and the elongation at break of Fe3O4@C–NH2/PI composite films were lower than those of the neat PI films. With increasing f from 0 (neat PI) to 1.40 vol%, the tensile strength gradually decreased from 108 ± 5.8 to 85 ± 9.3 MPa. Meanwhile, tensile modulus decreased from 1.2 ± 0.2 to 0.9 ± 0.1 GPa and elongation at break decreased from 30.1 ± 10.6% to 19.8 ± 10.3%, showing similar trends with increasing the composition of Fe3O4@C–NH2. Despite these slight decreases due to interrupted integrity of PI matrix introduced by aggregation of Fe3O4@C–NH2 nanoparticles, these nanocomposite films still exhibited excellent mechanical properties relative to other polymer candidates (∼73 MPa).54,55 Compared to the Fe3O4@C–NH2/PI composites, the tensile strength of Fe3O4/PI composites (Fig. 4d) decreased more significantly from 108 ± 5.8 to 76 ± 9.6 MPa, mainly due to the poor dispersion of Fe3O4 in the matrix.
The dielectric properties of Fe3O4@C–NH2/PI and Fe3O4/PI nanocomposite films with various nanoparticle compositions were plotted versus frequency in Fig. 5. Relative to Fe3O4/PI nanocomposite films, superior dielectric properties were found in Fe3O4@C–NH2/PI nanocomposite films and this might attribute to better dispersion of Fe3O4@C–NH2 in PI with assistance of the compatible core–shell structure of the nanoparticles and the subsequent amidation. As shown in Fig. 5a, the dielectric constant of Fe3O4@C–NH2/PI composite films decreased with increasing the frequency from 10 Hz to 1 × 106 Hz and more strikingly the addition of Fe3O4@C–NH2 significantly promoted the dielectric constant of PI. A low percolation threshold was identified in the Fe3O4@C–NH2/PI composite films. Furthermore, the dielectric loss of the Fe3O4@C–NH2/PI nanocomposite films was extremely low (Fig. 5b). The dielectric constant of Fe3O4/PI composite films presented in Fig. 5c also decreased with increasing the frequency from 10 Hz to 1 MHz, but it only mildly increased with increasing the composition of Fe3O4. The dielectric constants of the Fe3O4@C–NH2/PI composite films were prominently higher than those of the Fe3O4/PI composite films. No percolation threshold was observed upon adding Fe3O4 nanoparticles into PI, primarily due to the poor dispersion of Fe3O4 in PI. The dielectric loss of Fe3O4/PI composite films (Fig. 5d) decreased when the frequency increased to 1 × 106 Hz and the values were also extremely low. Taking these two parameters into consideration, the above observations indicated that Fe3O4 nanoparticles could not effectively improve the dielectric properties of PI while incorporation of Fe3O4@C–NH2 into PI was a very effective approach in promoting the dielectric properties of the PI matrix.
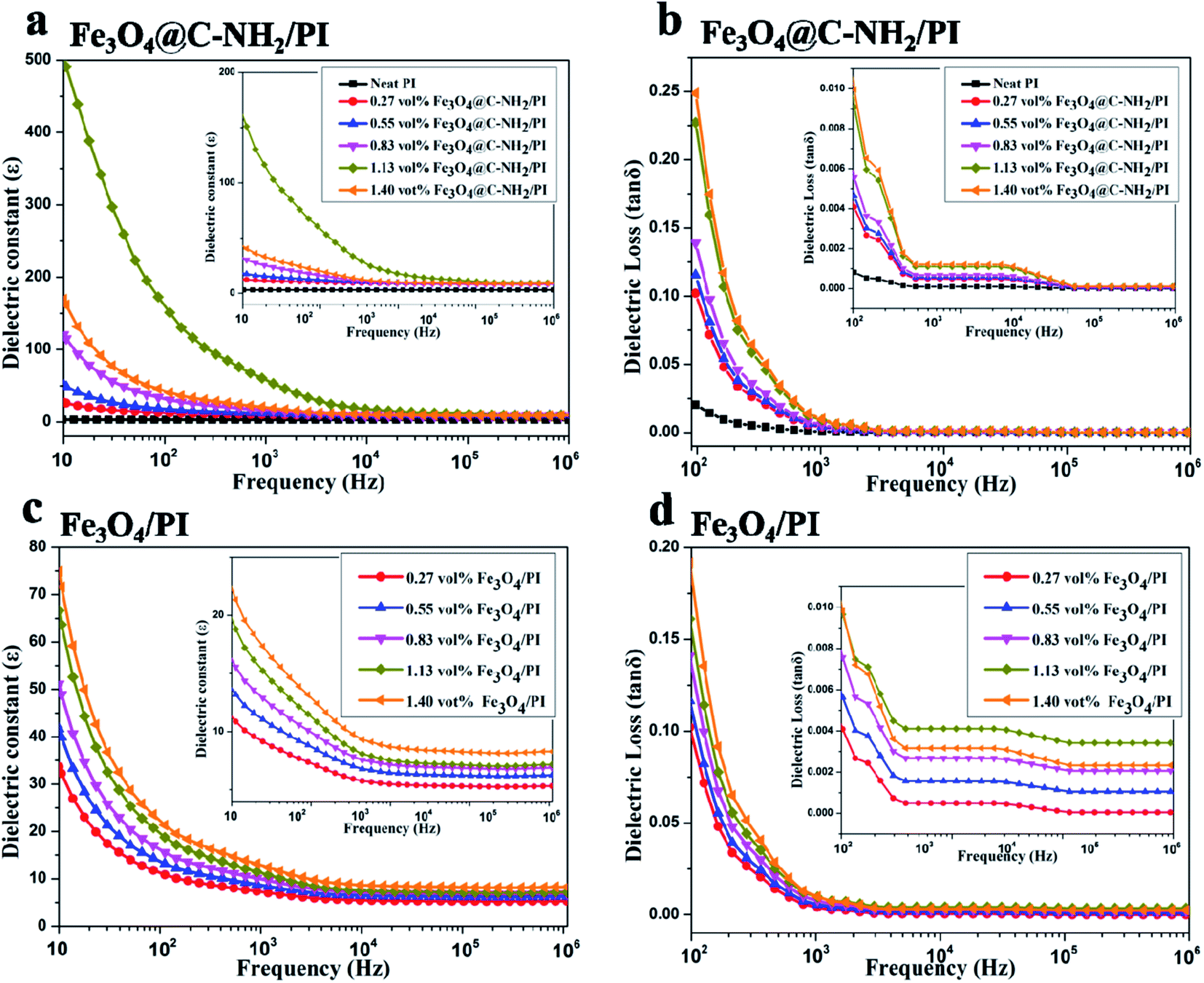 | ||
| Fig. 5 Room-temperature dielectric constant (a, c) and dielectric loss (b, d) as functions of frequency in the Fe3O4@C–NH2/PI (a, b) and Fe3O4/PI (c, d) composite films with various nanoparticle volume compositions. The insets are the enlarged areas in the high frequency range. | ||
The dielectric constant and loss of Fe3O4@C–NH2/PI and Fe3O4/PI composites at the frequency of 1 kHz are plotted as functions of f in Fig. 6a and b, respectively. The dielectric constant increased dramatically when the Fe3O4@C–NH2 composition approached and exceeded the percolation threshold, as predicted by the percolation theory.56 The enhancement of the dielectric constant was primarily attributed to the formation of microcapacitor network.57 The maximum of the dielectric constant for the Fe3O4@C–NH2/PI nanocomposite was 58.6 at f = 1.13 vol%, significantly higher than that of the neat PI (∼3.0) by a factor of ∼20, indicating that homogeneous dispersion of the Fe3O4@C–NH2 nanoparticles in the matrix.58 The dielectric loss of the Fe3O4@C–NH2/PI composites was between 0.0027 and 0.25, a few orders of magnitude lower than those of other conventional percolative composites.10,59 Although adding Fe3O4 nanoparticles directly into PI only mildly increased its dielectric constant by a factor of ∼4, the dielectric losses of Fe3O4/PI composites were also extremely low with a maximum value of 0.0089 for the Fe3O4 composition of 1.40 vol% in the studied range (Fig. 6b). The above results indicated that the Fe3O4@C–NH2/PI composite films were promising polymer-based dielectric materials with a high dielectric constant and an extremely low dielectric loss.
 | ||
| Fig. 6 Dielectric constant and dielectric loss at 1 kHz as functions of the volume fraction of Fe3O4@C–NH2 (a) and Fe3O4 (b) nanoparticles. Inset in (a): linear fitting (line) of dielectric constant vs. fc − f in a double-natural-logarithmic plot. | ||
The percolation threshold can be calculated using the following equation:
| ε ∼ (fc − f)−s for f < fc | (1) |
The dielectric constants of the neat PI and the Fe3O4@C–NH2/PI (f = 1.13 vol%) composite were plotted as a function of temperature, shown in Fig. 7a. At elevated temperatures, the dielectric constant of the neat PI remained constant at ∼3. However, temperature showed a remarkable impact on the dielectric constant of the composite. The dielectric constant of the composite increased with increasing temperature from 20 to 250 °C. This increment can be explained by the increased conductivity of semi-conducting Fe3O4 particles and thermal expansion of PI,10,61 as well as polarization of permanent dipoles in the polymer at the molecular level (Scheme 1).
 | ||
| Fig. 7 (a) Temperature dependence of dielectric constants of the neat PI (open circles) and the 1.13 vol% Fe3O4@C–NH2/PI composite (solid symbols) at different frequencies. (b) Breakdown strengths of the Fe3O4@C–NH2/PI (solid circles) and Fe3O4/PI (open circles) composite films as functions of Fe3O4@C–NH2 or Fe3O4 composition. | ||
 | ||
| Scheme 1 Synthesis of Fe3O4@C–NH2/PI composite films. | ||
High dielectric constant and high breakdown strength were equally important for dielectric materials because they are decisive factors in governing the operating electric field of the dielectric materials.62 The breakdown strengths of the neat PI, Fe3O4@C–NH2/PI, and Fe3O4/PI composite films were measured and plotted in Fig. 7b as functions of Fe3O4@C–NH2 or Fe3O4 compositions at room temperature. The breakdown strength of the neat PI film was as high as 286.5 ± 32.6 MV m−1. Addition of a small quantity of Fe3O4@C–NH2 resulted in a significant decrease in breakdown strength to 192.8 ± 23.3 MV m−1 in Fe3O4@C–NH2/PI (f = 0.13 vol%) composite films. However, as the Fe3O4@C–NH2 composition further increased, the breakdown strength of the composite film only decreased mildly, similar to the findings in CNT/PI and graphene/PI composites.35,36 The breakdown strength of the Fe3O4@C–NH2/PI (f = 1.13 vol%) composite film remained at a high level of 126.5 ± 11.3 MV m−1. The open symbols in Fig. 7b showed that adding Fe3O4 nanoparticles directly into PI resulted in a significant decrease in the breakdown strength from the value for the neat PI to 169.4 ± 27.5 MV m−1 for the Fe3O4/PI (f = 0.13 vol%) composite film. The breakdown strengths of the Fe3O4/PI composites were always lower than those of Fe3O4@C–NH2/PI at the same f values. In addition, as the Fe3O4 composition further increased, the breakdown strength decreased faster for the Fe3O4/PI composite films than that of the Fe3O4@C–NH2/PI composite films, showing that Fe3O4@C–NH2 nanoparticles could better maintain the breakdown strength of the composites than Fe3O4 nanoparticles.
Conclusion
In summary, we synthesized NH2-functionalized Fe3O4@C and in situ polymerized Fe3O4@C–NH2/PI composite films to enhance dielectric properties. The dielectric properties of the Fe3O4@C–NH2/PI composite films were optimized by varying the nanoparticle composition. At Fe3O4@C–NH2 composition of 1.13 vol%, the dielectric constant of the nanocomposite films at 1 kHz was promoted to 58.6, higher than that of the neat PI (∼3.0) by a factor of ∼20. An extremely low dielectric loss of 0.0091 and a high breakdown strength of 126.5 ± 11.3 MV m−1 were also identified. This finding could be explained by the homogeneous dispersion of Fe3O4@C–NH2 in the PI matrix and the strong interfacial covalent bonding between Fe3O4@C–NH2 nanoparticles and the PI matrix. The Fe3O4@C–NH2/PI nanocomposite films witnessed satisfactory thermal stability as well as mechanical properties for multifunctional applications in extreme operating environment, suggesting that they are potential high-performance candidates for dielectric applications in capacitors.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51573045) and the International Collaboration Research Program of Science and Technology Commission of Shanghai (16520722000).Notes and references
- X. Zhang, W. Chen, J. Wang, Y. Shen, L. Gu, Y. Lin and C.-W. Nan, Nanoscale, 2014, 6, 6701–6709 RSC.
- Q. Li, L. Chen, M. R. Gadinski, S. Zhang, G. Zhang, H. Li, A. Haque, L. Q. Chen, T. Jackson and Q. Wang, Nature, 2015, 523, 576–579 CrossRef CAS PubMed.
- S. Zhang, N. Zhang, C. Huang, K. Ren and Q. M. Zhang, Adv. Mater., 2005, 17, 1897–1901 CrossRef CAS.
- M. Zhang, Y. Li, Z. Su and G. Wei, Polym. Chem., 2015, 6, 6107–6124 RSC.
- W. Jillek and W. K. C. Yung, Int. J. Adv. Manuf. Tech., 2005, 25, 350–360 CrossRef.
- S. Yu, W. Zheng, W. Yu, Y. Zhang, Q. Jiang and Z. Zhao, Macromolecules, 2009, 42, 8870–8874 CrossRef CAS.
- L. L. Sun, B. Li, Y. Zhao, G. Mitchell and W. H. Zhong, Nanotechnology, 2010, 21, 305702 CrossRef CAS PubMed.
- K. Yang, X. Huang, Y. Huang, L. Xie and P. Jiang, Chem. Mater., 2013, 25, 2327–2338 CrossRef CAS.
- L. Chu, Q. Xue, J. Sun, F. Xia, W. Xing, D. Xia and M. Dong, Compos. Sci. Technol., 2013, 86, 70–75 CrossRef CAS.
- G. Wang, Y. Deng, Y. Xiang and L. Guo, Adv. Funct. Mater., 2008, 18, 2584–2592 CrossRef CAS.
- S. Wang, Y. Chen, Q. Zhuang, X. Li, P. Wu and Z. Han, Macromol. Chem. Phys., 2006, 207, 2336–2342 CrossRef CAS.
- S. Wang, P. Wu and Z. Han, Polymer, 2001, 42, 217–226 CrossRef CAS.
- Z. Xie, Q. Zhuang, Q. Wang, X. Liu, Y. Chen and Z. Han, Polymer, 2011, 52, 5271–5276 CrossRef CAS.
- S. Wang, P. Guo, P. Wu and Z. Han, Macromolecules, 2004, 37, 3815–3822 CrossRef CAS.
- Q. Zhuang, X. Liu, Q. Wang, X. Liu, J. Zhou and Z. Han, J. Mater. Chem., 2012, 22, 12381–12388 RSC.
- Y. Chen, Q. Zhuang, X. Liu, J. Liu, S. Lin and Z. Han, Nanotechnology, 2013, 24, 245702 CrossRef PubMed.
- R. A. Pethrick, E. A. Hollins, I. McEwan, A. J. MacKinnon, D. Hayward, L. A. Cannon, S. D. Jenkins and P. T. McGrail, Macromolecules, 1996, 29, 5208–5214 CrossRef CAS.
- C. Min, D. Yu, J. Cao, G. Wang and L. Feng, Carbon, 2013, 55, 116–125 CrossRef CAS.
- D. Lairez, J. R. Emery, D. Durand and R. A. Pethrick, Macromolecules, 1992, 25, 7208–7210 CrossRef CAS.
- C. Wu, X. Huang, X. Wu, L. Xie, K. Yang and P. Jiang, Nanoscale, 2013, 5, 3847–3855 RSC.
- F. Galantini, S. Bianchi, V. Castelvetro and G. Gallone, Smart Mater. Struct., 2013, 22, 055025 CrossRef.
- F. Wang, D. Zhou and Y. Hu, Phys. Status Solidi A, 2009, 206, 2632–2636 CrossRef CAS.
- X. Huang, Z. Pu, L. Tong, Z. Wang and X. Liu, J. Mater. Sci.: Mater. Electron., 2012, 23, 2089–2097 CrossRef CAS.
- M. Li, X. Huang, C. Wu, H. Xu, P. Jiang and T. Tanaka, J. Mater. Chem., 2012, 22, 23477–23484 RSC.
- J. Y. Kim, J. Lee, W. H. Lee, I. N. Kholmanov, J. W. Suk, T. Kim, Y. Hao, H. Chou, D. Akinwande and R. S. Ruoff, ACS Nano, 2014, 8, 269–274 CrossRef CAS PubMed.
- L. J. Romasanta, M. Hernandez, M. A. Lopez-Manchado and R. Verdejo, Nanoscale Res. Lett., 2011, 6, 508 CrossRef PubMed.
- N. Ning, X. Bai, D. Yang, L. Zhang, Y. Lu, T. Nishi and M. Tian, RSC. Adv., 2014, 4, 4543–4551 RSC.
- K. Kriechbaum, D. A. Cerrón-Infantes, B. Stöger and M. M. Unterlass, Macromolecules, 2015, 48, 8773–8780 CrossRef CAS.
- X. Liu, J. Yin, Y. Kong, M. Chen, Y. Feng, Z. Wu, B. Su and Q. Lei, Thin Solid Films, 2013, 544, 54–58 CrossRef CAS.
- A. Alias, Z. Ahmad and A. B. Ismail, Mater. Sci. Eng., B, 2011, 176, 799–804 CrossRef CAS.
- K. Abe, D. Nagao, A. Watanabe and M. Konno, Polym. Int., 2013, 62, 141–145 CrossRef CAS.
- E. Hamciuc, C. Hamciuc, I. Bacosca, M. Cristea and L. Okrasa, Polym. Compos., 2011, 32, 846–855 CrossRef CAS.
- Z. M. Dang, T. Zhou, S. H. Yao, J. K. Yuan, J. W. Zha, H. T. Song, J. Y. Li, Q. Chen, W. T. Yang and J. Bai, Adv. Mater., 2009, 21, 2077–2082 CrossRef CAS.
- Q. Chi, J. Sun, C. Zhang, G. Liu, J. Lin, Y. Wang, X. Wang and Q. Lei, J. Mater. Chem. C, 2014, 2, 172–177 RSC.
- Y. Chen, B. Lin, X. Zhang, J. Wang, C. Lai, Y. Sun, Y. Liu and H. Yang, J. Mater. Chem. A, 2014, 2, 14118–14126 CAS.
- X. Fang, X. Liu, Z.-K. Cui, J. Qian, J. Pan, X. Li and Q. Zhuang, J. Mater. Chem. A, 2015, 3, 10005–10012 CAS.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- X. Xu, C. Deng, M. Gao, W. Yu, P. Yang and X. Zhang, Adv. Mater., 2006, 18, 3289–3293 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- K. Mori, Y. Kondo, S. Morimoto and H. Yamashita, J. Phys. Chem. C, 2008, 112, 397–404 CAS.
- A. Zhu, L. Yuan and S. Dai, J. Phys. Chem. C, 2008, 112, 5432–5438 CAS.
- I. Haldar, M. Biswas and A. Nayak, Polym.-Plast. Technol. Eng., 2014, 53, 1317–1326 CrossRef CAS.
- I. Haldar, M. Biswas and A. Nayak, Synth. Met., 2011, 161, 1400–1407 CrossRef CAS.
- G. Wang, X. Zhang, A. Skallberg, Y. Liu, Z. Hu, X. Mei and K. Uvdal, Nanoscale, 2014, 6, 2953–2963 RSC.
- T. Yao, T. Cui, H. Wang, L. Xu, F. Cui and J. Wu, Nanoscale, 2014, 6, 7666–7674 RSC.
- Z. Wang, H. Guo, Y. Yu and N. He, J. Magn. Magn. Mater., 2006, 302, 397–404 CrossRef CAS.
- Z. Zhang, H. Duan, S. Li and Y. Lin, Langmuir, 2010, 26, 6676–6680 CrossRef CAS PubMed.
- H. Wang, L. Sun, Y. Li, X. Fei, M. Sun, C. Zhang, Y. Li and Q. Yang, Langmuir, 2011, 27, 11609–11615 CrossRef CAS PubMed.
- D. Qi, H. Zhang, J. Tang, C. Deng and X. Zhang, J. Phys. Chem. C, 2010, 114, 9221–9226 CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- Q. Peng, Y. Dong and Y. Li, Angew. Chem., Int. Ed., 2003, 42, 3027–3030 CrossRef CAS PubMed.
- X. Yu, J. Wan, Y. Shan, K. Chen and X. Han, Chem. Mater., 2009, 21, 4892–4898 CrossRef CAS.
- R. Demir Cakan, M. M. Titirici, M. Antonietti, G. Cui, J. Maier and Y. S. Hu, Chem. Commun., 2008, 3759–3761 RSC.
- L. Liu, B. Liang, W. Wang and Q. Lei, J. Compos. Mater., 2006, 40, 2175–2183 CrossRef CAS.
- H. Cai, F. Yan, Q. Xue and W. Liu, Polym. Test., 2003, 22, 875–882 CrossRef CAS.
- Y. Chen, S. Zhang, X. Liu, Q. Pei, J. Qian, Q. Zhuang and Z. Han, Macromolecules, 2015, 48, 365–372 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- W. Zhang, W. He and X. Jing, J. Phys. Chem. B, 2010, 114, 10368–10373 CrossRef CAS PubMed.
- Y. Shen, Y. H. Lin and C. W. Nan, Adv. Funct. Mater., 2007, 17, 2405–2410 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Z. M. Dang, J. B. Wu, L. Z. Fan and C. W. Nan, Chem. Phys. Lett., 2003, 376, 389–394 CrossRef CAS.
- M. Molberg, D. Crespy, P. Rupper, F. Nüesch, J.-A. E. Månson, C. Löwe and D. M. Opris, Adv. Funct. Mater., 2010, 20, 3280–3291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21249b |
| This journal is © The Royal Society of Chemistry 2016 |