
Use of dicyanamide ionic liquids for extraction of metal ions
S. Boudesocque,
A. Mohamadou,
L. Dupont*,
A. Martinez and
I. Déchamps
Université de Reims Champagne-Ardenne, Institut de Chimie Moléculaire de Reims (ICMR), CNRS UMR 7312, UFR des Sciences Exactes et Naturelles, Bâtiment 18 Europol'Agro, BP 1039, F-51687 Reims Cedex 2, France. E-mail: laurent.dupont@univ-reims.fr
First published on 31st October 2016
Abstract
Hydrophobic ionic liquids (ILs) were generated by association between tetraalkylammonium cations and dicyanamide (Dca−) anion. Extraction of Cu(II), Ni(II), Pb(II) and Cd(II) from water was then performed using these ILs at room temperature. The use of a coordinating Dca− anion allows high extraction yields for copper and cadmium to be obtained. The extraction mechanism of Cu(II) has been studied by the determination of the concentration of the organic cation released upon extraction and of the concentrations of counter ions co-extracted. Our results show that the extraction mechanism proceeds via a mixed process involving both cation exchange and ion-pairing, the proportions of which depend upon the nature of the anion and, more precisely, upon its position in the Hofmeister series. The coordination of Cu(II) in ionic liquid phase was followed by UV-vis and EPR spectroscopies.
Introduction
In recent years, industrial activities have grown rapidly, increasing the volume and toxicity of their residues, among which are liquid effluents containing heavy metals. The high degree of toxicity1 of these heavy metals makes them harmful for humans and the environment, leading to the promulgation of laws regulating the elimination of these residues. Therefore, the recovery of metals from liquid effluents is of great importance in the fields of wastewater and pollution reduction.2The separation and concentration of metals involve not only environmental but also economic gains, because of the continuous increase in the value of metals. The traditional processes for the elimination of metals from industrial aqueous effluents include chemical precipitation, coagulation, solvent extraction, electrolysis, membrane separation, ion-exchange and adsorption.3,4 Among these technologies, liquid–liquid extraction is one of the most efficient techniques to separate and concentrate metal ions from industrial wastewaters. The extracting agents are dissolved in an organic solvent (kerosene, toluene, etc.) used as diluent. The major drawback of this technique is the loss of organic diluent via volatilization, which has a detrimental impact on the environment and on human health. Consequently, “greener” extraction methods are being sought and the use of Ionic Liquids (ILs) as an alternative to traditional organic solvents could overcome this drawback.5
In the last decade, significant work has highlighted the potential of Room Temperature Ionic Liquids (RTILs) as substitutes for traditional solvents used in liquid–liquid extraction processes for the separation of metal ions.6–9 Ionic liquids made of imidazolium and pyridinium cations associated with fluorinated anions, have been extensively studied.6–9 However, conventional ionic liquids contain weak chelating moieties, and have a low capability to dissolve metal salts, mainly serving as diluents for molecular extractants.9–16 To overcome the use of molecular extractants, more recent developments have focused on the design of Task-Specific Ionic Liquid (TSIL)17–22 by the introduction of chelating functions on the organic cation or by the use of a strong coordinating anion associated with a hydrophobic cation.23–26 However, the development of “green” extraction processes with ILs suffers from a lack of knowledge about the mechanisms involved, especially in regard to the transfer mechanism of metal ions towards the IL phase. The ionic nature of these solvents can result in a variety of extraction mechanisms, including ion-pair extraction (IP) and cationic exchange (CE).27–29 This can be an advantage to implementing a selective extraction process but it can also generate harmful effects on the environment if the ionic liquid components are lost to the aqueous phase by an ion-exchange reaction.
The use of ionic liquids with substituted imidazolium cations and/or fluorinated anions may have serious disadvantages from an economic and environmental point of view. The replacement of these two components by more environmentally friendly ones might be required to generate ionic liquids of lower cost and with a smaller environmental impact. It is known that the use of quaternary ammonium or phosphonium cations with a long alkyl chain allows a hydrophobic ionic liquid with a non-fluorinated anion to be obtained.30–33 Recent research has shown that these quaternary ammonium ionic liquids may be used as a pure extracting phase, without using an organic diluent, if it is saturated with water in advance to reduce its viscosity.30
Here, we report the ability of a task-specific ionic liquid to remove heavy metals from water. The ionic liquids studied were generated by the association between a cationic tetraalkylammonium ion and the coordinating dicyanamide anion (Dca−![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (CN)2N−). In previous studies, we showed the ability of an ionic liquid with Dca− to extract Cu(II), Cd(II), Ni(II) and Pb(II) from water.33 The choice of the tetraalkyl-ammonium ion is justified by its availability, its limited cost, and its structural modularity, which allows the control of the hydrophobicity of the cation by varying the alkyl chain length bound to the ammonium group. The dicyanamide anion is the cheapest anion commonly used to generate ILs, with a moderate toxicity comparable to that of a TFA− [CF3SO2−] anion and less than that of the usual anions such as Tf2N− [(CF3SO2)2N−], PF6− or BF4−.34 The chelating ability of the Dca− anion for metal ions is well known.35,36 In this paper, firstly, we examine the extraction properties of dicyanamide ionic liquids towards a panel of metal ions of environmental concern. Secondly, we investigate the fundamental aspects which govern the partitioning and the extraction mechanism of Cu(II) in the biphasic system H2O/IL, such as the metal concentration, and the nature of the anion of the metal salt.
(CN)2N−). In previous studies, we showed the ability of an ionic liquid with Dca− to extract Cu(II), Cd(II), Ni(II) and Pb(II) from water.33 The choice of the tetraalkyl-ammonium ion is justified by its availability, its limited cost, and its structural modularity, which allows the control of the hydrophobicity of the cation by varying the alkyl chain length bound to the ammonium group. The dicyanamide anion is the cheapest anion commonly used to generate ILs, with a moderate toxicity comparable to that of a TFA− [CF3SO2−] anion and less than that of the usual anions such as Tf2N− [(CF3SO2)2N−], PF6− or BF4−.34 The chelating ability of the Dca− anion for metal ions is well known.35,36 In this paper, firstly, we examine the extraction properties of dicyanamide ionic liquids towards a panel of metal ions of environmental concern. Secondly, we investigate the fundamental aspects which govern the partitioning and the extraction mechanism of Cu(II) in the biphasic system H2O/IL, such as the metal concentration, and the nature of the anion of the metal salt.
Results and discussion
Extraction of Cu(II), Ni(II), Cd(II) and Pb(II) from aqueous solutions
The association of tetrahexyl(or tetraoctyl)ammonium bromide ([THN+][Br−] or [TON+][Br−]) with sodium dicyanamide ([Na+][Dca−]) or lithium bis(trifluoromethylsulfonyl)imide ([Li+][Tf2N−]) by anionic metathesis generates hydrophobic ILs (Fig. 1) with high yield (up to 75%) and a degree of purity up to 98.5%. At room temperature, these ILs ([THN+][Tf2N−], [TON+][Tf2N−], [THN+][Dca−] and [TON+][Dca−]) are yellowish viscous liquids, except for [THN+][Dca−], which is slightly brown. At room temperature, the four ionic liquids form two liquid phases in contact with water. The solubility in water (determined by NMR measurements (see Experimental section)) of [TON+][Dca−] and [THN+][Dca−] are 0.0043 and 0.04% (w/w), respectively.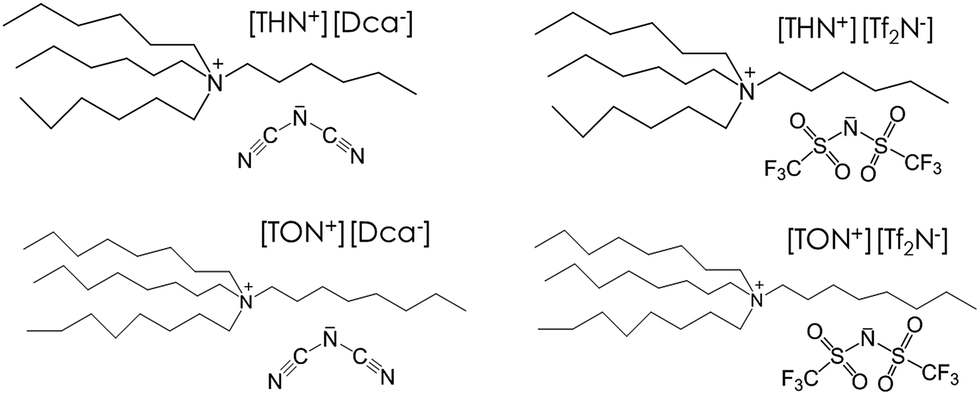 | ||
| Fig. 1 Structure of [tetraalkylN+][Dca−] and [tetraalkylN+][Tf2N−]. | ||
To examine the extraction properties of [THN(or TON)+][Dca−] towards Cu(II), Ni(II), Cd(II) and Pb(II), we determined the percentage of extraction (% E) from 0.1 mol dm−3 solutions of the nitrate salts of these metals at 295 K. For this purpose, the results obtained with [THN(or TON)+][Dca−] were compared to those obtained with [THN(or TON)+][Tf2N−] which can be considered as a conventional IL, since it is constituted with only weakly coordinating moieties. The extraction yields (% E) for each IL are depicted in Fig. 2.
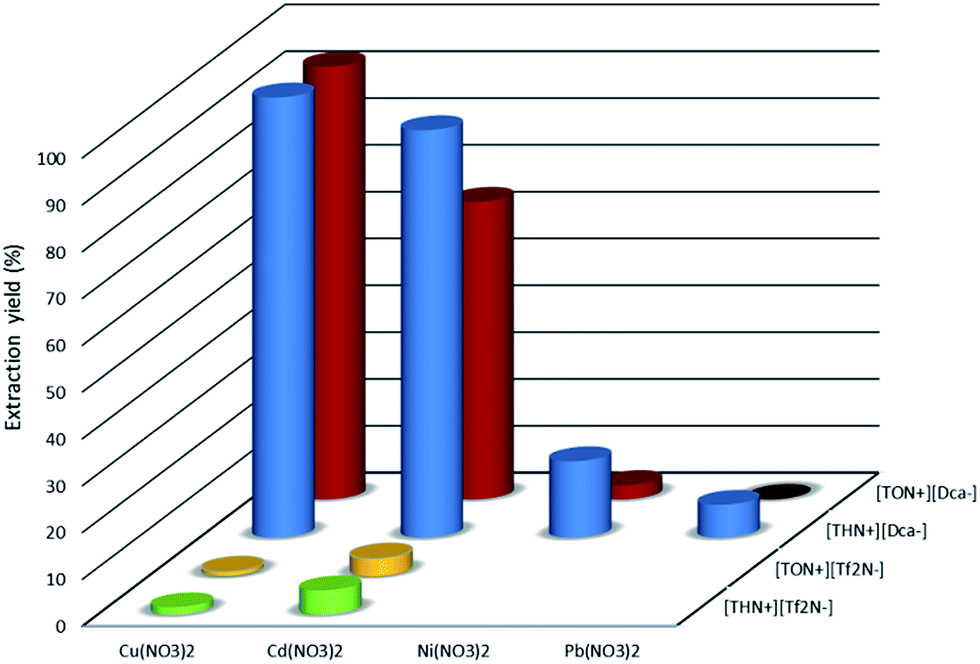 | ||
| Fig. 2 Extraction yields (%E) for aqueous nitrate salt with [THN(or TON)+][Dca−] and [THN(or TON)+][Tf2N−]; Cmetal = 0.1 mol dm−3; Vw/VIL = 1. | ||
For [THN(or TON)+][Tf2N−], the extraction yields are low with copper and cadmium and do not exceed 5%. No significant extraction is measured in the case of Ni(II) and Pb(II) in these experimental conditions.
The incorporation of Dca− as an anionic constituent of the tetraalkylammonium ionic liquid causes striking modifications in the extraction behavior of ionic liquids compared to generic ionic liquids. The Dca−-anion-based ionic liquids give an extraction yield of higher than 80% with copper nitrate, whereas only [THN+][Dca−] extracted cadmium salts with an extraction yield of up to 80%. The opposite trend is observed with Ni(II) and Pb(II). Indeed, the extraction yields of these two cations with [THN+][Dca−] and [TON+][Dca−] do not exceed 20%. No extraction is observed even for lead nitrate in the case of [TON+][Dca−].
The higher extraction properties of Dca−-anion-based ILs compared to generic ILs were already in evidence in our previous work33 and were ascribed to the ability of the Dca− anion to interact with metallic cations through the formation of metal complexes via their nitrogen atoms.35–37
Although there is no quantitative data available in the literature, such as the stability constants of metal–Dca complexes,38 it is reasonable to think that the higher extraction yields observed for Cu(II) and Cd(II) compared to those of Ni(II) and Pb(II) are related to their higher affinity for the Dca− anion, which favors their transfer from aqueous to ionic liquid phases.
Fig. 2 shows that the cation of the ionic liquid has a significant influence on the extraction of the metal cation. Indeed, the extraction yield is significantly higher with [THN+][Dca−] than with [TON+][Dca−]. This trend is related, on the one hand, to the more hydrophilic character of the tetrahexylammonium cation, which may favor the transfer of the metal in the ionic liquid phase via a cationic exchange process, and, on the other hand, to the fact that the concentration of the Dca− ion, in the ionic liquid phase, is higher in [THN+][Dca−] than in [TON+][Dca−]. Indeed, taking into account the density (0.91 and 0.87 of [THN+][Dca−] and [TON+][Dca−], respectively) and the molar mass of the two ionic liquids, the molar concentrations of [THN+][Dca−] and [TON+][Dca−] are estimated at 2.10 and 1.60 mol dm−3, respectively. Such a difference may explain the better extraction properties of [THN+][Dca−], which contains a higher concentration of extractant. The extraction mechanism of the metal cation will be discussed in a subsequent section.
In regard to the selective recovery of metal ions, the important differences in the extraction percentages for Cu2+ and Cd2+ versus Ni2+ and Pb2+ observed with [THN+][Dca−] and [TON+][Dca−] suggest that these two ILs could be used for the selective separation of these metal ions. The selective extraction of metals by commercial ionic liquids was also studied and requires a high HCl concentration.39 One of the advantages of ionic liquids with a Dca− ion is the avoidance of an acidic medium.
Furthermore, an appreciable difference in extraction yield is observed between Cu2+ and Cd2+ with [TON+][Dca−]. This could allow the selective separation of these two metals. It can be concluded from the above results that it is possible to tailor ILs for use as extractive agents for specific metal ion separations.
Fig. 3 compares the extraction of different copper salts in [THN+][Dca−] and [TON+][Dca−]. The results point to the significant influence of the counter-ion associated with the metal cation on the extraction efficiency of the ionic liquid. The extraction yield of copper increases in the following order: SO42− < Cl− < BF4− < NO3− < ClO4−.
 | ||
| Fig. 3 Extraction yields (% E) for aqueous copper salt with [THN(or TON)+][Dca−]; Cmetal = 0.1 mol dm−3; Vw/VIL = 1. | ||
In a previous work,33 we provided evidence that the anion of a metal salt may influence the transfer mechanism of the metal between aqueous and ionic liquid phases. To discuss the specific anion effect, we must refer to the extraction mechanism of the metal from an aqueous solution to an ionic liquid phase. It is commonly described that partitioning of a metal ion proceeds via ion exchange reactions or via “ion-pairing” extractions or via both of them.27–29
When a cation exchange occurs, the metal ion in the aqueous phase is exchanged with the cation of the ionic liquid. The metal ion is then replaced by the cation of the ionic liquid, which moves to the aqueous phase (when the anion exchange occurs, the anion of the IL is exchanged with the metal's counter-ion). In the case of the extraction by ion-pairing, also called neutral extraction, the metal cation and its counter-anion are co-extracted in the ionic liquid phase. The two mechanisms are depicted by the following two equations in the case of CuX2 extracted by [THN+][Dca−]:
Cationic exchange (CE)
| Cu2+(w) + 2THN+(IL) + nDca−(IL) ⇌ Cu(Dca)n(n−2)−(IL) + 2THN+(w) | (1) |
Ion-pairing (IP)
| Cu2+(w) + 2X−(w) + nDca−(IL) ⇌ Cu(Dca)n(n−2)−(IL) + 2X−(IL) | (2) |
These equilibria take into account the formation of copper–dicyanamide complexes in ionic liquid phases with no definite stoichiometry. As a first assumption, it is considered that the anion of the metal salt remains uncoordinated in the ionic liquid phase. This point will be discussed in the following section. In the case of cationic exchange, the release of organic cations to the aqueous phase leads to a gradual dissolution of the IL, which has a negative implication for the practical use of these ILs.
Consequently, the issue to be addressed is to determine the required conditions which will lead to extraction by ion-pairing and which will present a real environmental benefit.
In our previous work,33 we provided evidence that the extent of ion-pairing in the overall metal transfer is related to the strength of the association between the organic cation of the ionic liquid and the anion of the metal salt. The ability of the counter-anion to generate a hydrophobic association with the organic cation (tetraalkylN+) determines the ease with which the anion can be transferred from the aqueous phase to the IL phase and promote the extraction of a metal cation via an ion-pairing mechanism. The high affinity of BF4− NO3− and ClO4− for a trialkyl-betaine ammonium cation favors their co-extraction with Cu(II) to maintain the electroneutrality of the IL phase. This was corroborated by IR spectra of different ILs phases after tetrafluoroborate or perchlorate copper(II) extraction, showing the characteristic vibration of ClO4− or BF4− ions, respectively. It is reasonable to think that the same effects apply with [THN+][Dca−] and [TON+][Dca−]. Comparing the extraction of monovalent copper salt, the high hydrophilicity of Cl− compared to BF4− NO3− and ClO4− limits the transfer of metal into the ionic liquid phase and the efficiency of the extraction process, even if the extraction yield remains interesting. This is even more marked with copper sulfate. Due to the high hydrophilicity of the sulfate ion, copper sulfate is slightly extracted with [THN+][Dca−] and not at all with [TON+][Dca−]. To support this affirmation, it is interesting to note that the influence of the counter-anions of the metal on its partitioning between aqueous and ionic liquid phases is easily correlated to their solvation energy. Indeed, the transfer of an anion from aqueous to ionic liquid phase requires a dehydration process, which generates an energy cost.
The anion dehydration reaction constitutes a brake on the extraction process of the metallic cation by ion-pairing. This is particularly true for the sulfate anion, which has a high solvation energy estimated at −1080 kJ mol−1.40,41 The extraction of copper sulfate proceeds mainly by ion exchange and the extraction yield is poor because the solubility of the organic cation in the aqueous phase is limited. Once that limit is reached, ion exchange becomes less favourable and extraction ceases, since neutral extraction may not take place, which explains the low extraction efficiency measured for copper sulfate. We examine the extraction process in the following sections. Chloride ions, which have a solvation energy equal to −381 kJ mol−1, are transferable into liquid ionic phase, up to a certain limit, which explains the higher extraction efficiency than is the case with the sulfate ion. In this case, the extraction proceeds via a joint mechanism involving cation exchange reactions and extraction by ion-pair. When the solubility of the organic cation reaches its upper limit, neutral extraction remains the dominant mechanism. Tetrafluoroborate, nitrate and perchlorate ions have a weak energy of solvation equal to −274, −314 and −219 kJ mol−1, respectively,40 which is why they are more easily transferable into ionic liquid phase than chloride ions. Moreover, these anions may generate a hydrophobic association with the organic cations, more stable than the [THN+][Dca−] ion-pairs, reducing the solubility of organic cations in aqueous media and leading exclusively to an extraction mechanism of copper by ion-pairing. Under our experimental conditions, this results in a nearly quantitative extraction of copper specifically for nitrate, tetrafluoroborate and perchlorate salts.
A similar influence of the anion of the metal salt has been reported by Janssen et al.42 for the extraction of copper with protic trialkyammonium hexa(octa or deca)noate salts. Janssen et al.42 connect the influence of the anion to its relative position in the Hofmeister series. The Hofmeister effect has been studied for over a century, and is observed in a wide range of interfacial phenomena, including the denaturation of proteins, the behavior of colloids, and the structure of microemulsions, among other examples. The Hofmeister series, or lyotropic series, proposes a classification of ions as a function of their ability to decrease (salting out) or increase (salting in) the solubility of proteins in aqueous media. The Hofmeister series ranks inorganic anions and common organic anions in the following order (for a panel of usual anions):
| SO42− > HPO42− > CH3COO− > Cl− > NO3− > ClO4− |
The physical phenomena underlying the Hofmeister effect remain poorly understood, but it is admitted that small and/or polycharged anions are able to structure the water molecules around their hydration shell. This disturbs the natural hydrogen-bonded network of water, and decreases the concentration of free water. Moreover, they interact with the charges on the surface of the protein, thereby exposing hydrophobic patches on the protein surface, provoking their aggregation or precipitation. These anions are called kosmotropes or water structure stabilizers. They enhance the aggregation or precipitation of proteins. Chaotropic anions or “water structure breakers,” have an opposite effect to kosmotropes, thus enhancing the solubility of protein in water. Kosmotropic anions are located at the left-hand end of the Hofmeister series and chaotropic anions are on the right.43,44 Janssen et al. postulate that chaotropic anions are adsorbed in the ionic liquid phase, at the interface between the hydrophobic and hydrophilic domains of the ionic liquid, stabilizing its different domains and reducing the free energy of the system. This provides a thermodynamic driving force to transfer inorganic anions from the aqueous to the IL phase, thus enhancing the extraction of the metal by the ion-pairing process. The Hofmeister effect is relevant, but it is difficult to prove because of the difficulty of obtaining information on the distribution of anions in the ionic liquid phase. The contribution of the solvation energy of the anion to the transfer process of the metal salts is a direct and accurate explanation of the anion's influence on the metal distribution between the organic and aqueous phases. It is corroborated by earlier results which show that ClO4−, BF4− and to a lesser extent NO3− combine easily with hydrophobic cations to generate hydrophobic ionic liquids.45,46 However, since the location of anions on the Hofmeister series is highly correlated with the solvation energy of these latter, the striking influence of the anion of the copper on metal extraction may be perceived indirectly as a manifestation of the Hofmeister effect. In the metal extraction process, it is pertinent to think that the uncoordinated anions co-extracted with the metal cation are associated with THN+/TON+ to form an ionic liquid diluted in [THN+ (or TON+)][Dca−]. In this case, it is more convenient to rewrite eqn (2):
| Cu2+(w) + 2X−(w) + nTHN+Dca−(IL) ⇌ (n − 2)THN+Cu(Dca)n(n−2)−(IL) + 2THN+X−(IL) | (2b) |
The coordination of the anion in the ionic liquid phase will be further studied in the following section. To determine the extent of ion exchange in the overall extraction process, the concentration of organic cations transferred from the ionic liquid to the aqueous phase, associated with the transfer of metal salt in the opposite direction, should be evaluated. In order to do this, we determined the concentration of organic cations by 1H NMR measurements before and after copper extraction from chloride and nitrate salts in [THN+][Dca−] and [TON+][Dca−].
For [TON+][Dca−], the concentration of organic cations in aqueous chloride and nitrate copper solutions after extraction is lower than that in pure water, meaning that the anions of the metal can combine with the organic cations in the aqueous phase to form ion-pairs which migrate into the ionic liquid phase. With these two salts, the extraction follows an ion-pair mechanism (Table 1).
| E% | C mol dm−3 | % IP | |
|---|---|---|---|
| [THN+][Dca−] | — | 0.95 × 10−3 | — |
| [THN+][Dca−]/CuCl2 | 18 | 1.95 × 10−3 | 66 |
| [THN+][Dca−]/Cu(NO3)2 | 70 | 0.53 × 10−3 | 100 |
| [TON+][Dca−] | — | 0.081 × 10−3 | — |
| [TON+][Dca−]/CuCl2 | 15 | 0.049 × 10−3 | 100 |
| [TON+][Dca−]/Cu(NO3)2 | 71 | 0.052 × 10−3 | 100 |
With [THN+][Dca−], the results prove a difference in the extraction mode between Cu(NO3)2 and CuCl2. With Cu(NO3)2, the extraction proceeds mainly through ion-pairing. With CuCl2, the concentration of organic cations in the aqueous phase is higher than that in pure water, indicating that extraction also involves the transfer of metal cations through a cation exchange process. The occurrence of ion-exchange with CuCl2 in [THN+][Dca−] is related (i) to the higher solubility of THN+ compared to that of TON+, which promotes the extrasolubilisation of the organic cation in aqueous phase and (ii) to the more hydrophilic character of the chloride ion compared to the nitrate ion, which limits its transfer to the organic phase. The quantitative use of this data shows that the extraction process associates cation exchange and ion-pairing reactions to an extent of 33% and 66%, respectively. It would have been interesting to complete this study with CuSO4, but the very low extraction efficiency measured with this salt does not provide sufficiently accurate measurements. We must nevertheless remember that our analysis is valid only at this copper concentration. The influence of metal concentration will be examined in the following section.
To highlight the occurrence of the cation exchange mechanism in the overall extraction process of copper chloride in [THN+][Dca−], we compare the extraction efficiency of copper chloride in pure water and in saturated aqueous solution of tetrahexyl ammonium chloride. All the experimental conditions are otherwise identical ([CuCl2] = 0.033 mol dm−3, Vw/VIL = 2.6). The extraction efficiency of copper chloride decreases from 33% in water to 20% in a saturated solution of [THN+][Cl−]. This result shows that the release of organic cations in aqueous solution constitutes a driving force for the transfer of copper into the ionic liquid phase. The presence of tetrahexyl ammonium cations in aqueous solution limits the transfer of IL cations in aqueous solution and blocks the cation exchange process.47,48 In these conditions, only ion-pair extraction may occur, which explains the decrease in the extraction efficiency of copper chloride in the presence of [THN+][Cl−].
To complete our investigations, we measured the transfer of chloride and nitrate ions from sodium salt solutions, in both ionic liquids, by ion-chromatography to evaluate qualitatively the strength of the interaction between the organic cation of the ionic liquid and the anion of the metal salt. In [THN+][Dca−] no significant extraction could be measured from a 0.02 mol dm−3 NaCl solution, whereas in [TON+][Dca−] the extraction percentage is estimated at 7%. Sodium nitrate, at the same concentration, is extracted at a rate of 50% in [THN+][Dca−] and 38% in [TON+][Dca−]. It is interesting to note that extraction of sodium salt proceeds mainly by ion-pair. Indeed, the release of Dca− ions in aqueous solution, associated with the extraction of sodium salt, remains limited to only a few percent, indicating that the anionic exchange process does not take place. The difference in extraction yield between nitrate and chloride ions confirms the stronger interaction of nitrate ions with THN(or TON)+ cations, leading to the formation of more stable ion-pairs. This partly explains the higher copper extraction efficiency observed with the nitrate salt and the extraction by ion-pair. This corroborates our previous interpretation of the influence of the anion of the copper salt on the extraction process.
Influence of metal concentration
The effect of initial metal concentration in the aqueous solution on the efficiency of the extraction process for copper nitrate, chloride and sulfate is shown in Fig. 4. For copper nitrate, the extraction percentage of Cu2+ decreased as the metal ion concentration increased. This effect is significant but relatively limited, since the extraction efficiency fell by 25% when the metal concentration was in the range 0.01 to 0.1 mol dm−3. It is interesting to note that the decrease in the extraction efficiency does not reflect a saturation of the extraction capacity of the ionic liquid, since the concentration of extracted copper increases with the concentration of the aqueous solution. For example, for [THN+][Dca−] in contact with a 0.01 mol dm−3 aqueous copper solution, the concentration of metal extracted into ionic liquid phase is equal to 0.023 mol dm−3 whereas with a 0.1 mol dm−3 solution, the concentration of copper in the ionic liquid is 0.16 mol dm−3. | ||
| Fig. 4 Effect of the initial concentration of copper salt on the extraction yield. Vw/VIL = 2.6. | ||
For copper sulfate, the decrease in extraction yield with the increase in copper concentration in [THN+][Dca−] reflects a saturation of copper in the ionic liquid phase. Indeed, the concentration of extracted copper in ionic liquid phase remains nearly constant whatever the concentration of aqueous solution, and comprised between 3 and 4 × 10−3 mol dm−3. As we will see in the following section, sulfate ions are not co-extracted with copper. In this case, extraction proceeds only by cationic exchange and the copper extraction is concomitant with the release of THN+ cations in the aqueous phase. Considering a mean concentration of copper in the ionic liquid phase of 3.5 × 10−3 mol dm−3, the concentration of ammonium cation that should be transferred to aqueous solution in order to maintain the electroneutrality of the two phases should be equal to 4.5 × 10−3 mol dm−3. In the aqueous phase, the respective concentrations of THN+ and Dca− ions are related by the solubility product of [THN+][Dca−], so the release of THN+ in aqueous solution should lead to a transfer of dicyanamide anions from the aqueous to the ionic liquid phase. The solubility product of [THN+][Dca−] is a limiting factor for copper extraction.49,50
The key point about the mechanisms presented in eqn (1) and (2) is that ion exchange requires no transfer of anions from the aqueous phase, while the neutral extraction requires a ratio of 1 between the extraction rate of anions and that of Cu2+. The co-extraction of sulfate, nitrate and chloride ions associated with the extraction of copper was followed by ion-chromatography or ICP-OES. For copper sulfate, no data is reported in Fig. 5, since sulfate is not co-extracted with copper and the ratio is close to zero under our experimental conditions. This result contrasts with the observations of Janssen et al.,42 which determine a significant extraction efficiency for copper sulfate (more than 80%) as well as a significant co-extraction of sulfate anions (more than 50%), in protic trialkyammoniumocta(hexa or deca)noate compounds. Such a difference between the observations of Janssen et al. and our results addresses the question of the structuration of ionic liquids at the nanoscale in polar and nonpolar domains and the modifications induced by the extraction of metal salts and their impact on the extraction process. Indeed, Janssen et al.42 point out that the presence of metal salt in ionic liquid phase leads to water absorption, which induces a change in the structuration of ionic liquid into hydrophilic and hydrophobic domains. Further investigations of the structuration of [THN(or TON)+][Dca−] in the absence and presence of metals salt are needed to obtain a better understanding of the driving forces that operate in the extraction process.
 | ||
| Fig. 5 Ratio (A−/C+) between the extraction rate of anion and that of Cu(II) for the different salts used at different concentrations. Vw/VIL = 2.6. | ||
Such investigations are presently beyond the scope of this paper.
The ratios reported in Fig. 5, for nitrate and chloride ions, confirm the observations based on NMR experiments. With copper, nitrate ion-pairing is the dominant mechanism. Indeed the ratio values are close to 1. The furthest value from 1 is 0.88, which is obtained with [THN+][Dca−] and with a copper concentration of 0.01 mol dm−3, meaning that with these experimental conditions cationic exchange may occur to a small extent. For 0.02 mol dm−3, the ratio values are slightly above 1, meaning that anionic exchange with dicyanamide anions may take place. For copper chloride at 0.01 mol dm−3 the ratio is clearly below neutral extraction with [THN+][Dca−], indicating a significant extent of ion exchange, which confirms the NMR results described in the preceding section. As expected, ion exchange is less marked in the case of [TON+][Dca−], with ratio values closer to 1. The increase in copper concentration leads to an increase in the ratio (A−/C+), indicating that extraction by ion-pair is favored at high metal concentration. All these results are in agreement with those previously reported by Janssen et al.40
Effect of salt concentration
According to the equilibrium (2), the addition of an indifferent electrolyte may drive the neutral extraction process and should increase the extraction rate of the metal. The extraction in salt media is a credible path for the implementation of an extraction process, limiting the release of ionic species in aqueous solutions, showing also a real environmental benefit. This has already been experienced with hydrophilic ionic liquids for the selective separation of polluting metals.51 The addition of a background electrolyte is also an effective way to erase the influence of the anion of the metal salt in the extraction process. Fig. 6 depicts the extraction of copper under the influence of increasing concentrations of sodium chloride, nitrate and sulfate. The addition of Na2SO4 has no effect on the copper extraction efficiency, meaning that sulfate is too hydrophilic to be extracted with copper in the ionic liquid phase, even at high concentration. As expected, the addition of chloride or nitrate salt leads to an increase in extraction efficiency according to the equilibria (2). The higher influence of NaCl compared to NaNO3 above 1 mol dm−3 is unexpected since, in pure water, copper nitrate is more efficiently extracted than copper chloride. Indeed, the extraction yield of copper is 94% at 2 mol dm−3 of NaCl, whereas it is only 81% with NaNO3 at the same concentration. The lesser influence of sodium nitrate, especially at concentrations higher than 1 mol dm−3, can be ascribed to a specific interaction of nitrate with organic cations, which may form stable ion-pairs by anionic metathesis, leading to a partial exchange between nitrate and dicyanamide anions following the equilibrium:| [TON+][Dca−]IL + NO3−(w) ⇌ [TON+][NO3−]IL + Dca−(w) |
 | ||
| Fig. 6 Effect of salt concentration on copper extraction. Vw/VIL = 2.6; CCu = 0.1 mol dm−3. | ||
This hypothesis is confirmed by the biggest solubility of NaNO3 with regard to that of the NaCl in [THN+ (or TON+)] [Dca−]. The use of a high concentration of nitrate ions, while promoting copper extraction, may lead to a release of Dca− in the aqueous phase, which can alter the green characteristics of the process.
Our results show that the development of the use of ionic liquids to implement green liquid/liquid extraction processes needs to evaluate the extent of all the ion-exchange reactions involved in the extraction process, and therefore identify ways to minimize them. In particular, the use of an indifferent salt seems to be a relevant approach, but it is important to take into account the possibility of anion exchange reactions, and to use appropriate salt concentrations, allowing an increase in metal extraction without generating anion exchange.
The stoichiometry of extracted species in aqueous salt solutions was determined using the slope analysis method (Fig. 7). In the case of NaCl, the plots of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) DCu against logCNaCl are found to be linear, with a slope of 2.14 and a good correlation coefficient. This confirms the extraction of CuCl2 in ionic liquid phase. Although it is strongly suspected that, in NaNO3, Cu(NO3)2 remains the main extracted species, it cannot be deduced from the slope analysis of experimental data, for the reasons evoked earlier.
DCu against logCNaCl are found to be linear, with a slope of 2.14 and a good correlation coefficient. This confirms the extraction of CuCl2 in ionic liquid phase. Although it is strongly suspected that, in NaNO3, Cu(NO3)2 remains the main extracted species, it cannot be deduced from the slope analysis of experimental data, for the reasons evoked earlier.
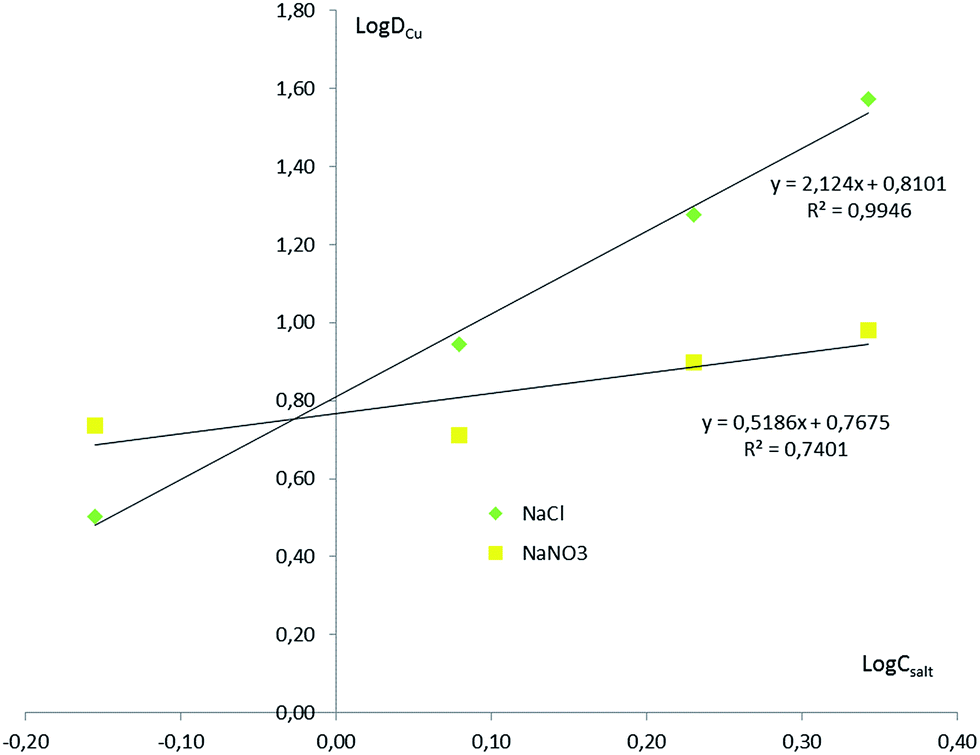 | ||
| Fig. 7 log–log plot of logDCu versus logCsalt. Vw/VIL = 2.6; CCu = 0.1 mol dm−3. | ||
As can be seen in Fig. 8, the extraction of nickel(II), which is very low in aqueous solution, can be enhanced by the addition of background salt more specifically. However, unlike the results obtained with Cu2+, no effect is observed with NaCl, while the addition of NaNO3 leads to an extraction yield of 60%. The different trends observed with Ni2+ and Cu2+, in the presence of an indifferent electrolyte, highlight that these metals may be recovered selectively from a mixture by the successive use of NaCl and NaNO3.
 | ||
| Fig. 8 Effect of background electrolyte on Ni2+ extraction in [THN+][Dca−]. Vw/VIL = 2.6; CNi = 0.1 mol dm−3. | ||
Structure and coordination mode of aqueous Cu(II) species
After examining the extraction mode, our investigations focused on the coordination mode of Cu(II) in the IL phase. To obtain insight into the nature of the copper species in the IL phase, UV-vis and RPE analyses were monitored in the [THN+][Dca−] phase after extraction. The UV-visible spectra of Cu(NO3)2 in ionic liquid and in aqueous phases are depicted in Fig. 9. For greater convenience, the visible and UV regions are presented separately. The UV-visible spectra show the same features as that previously reported for [BuGBOEt+][Dca−].33 In the visible region (Fig. 9a), one single d–d transition was observed, with a maximum wavelength shifted from 10 nm towards higher energies compared to copper nitrate in aqueous solution. The relatively broad shape of the band and the value of the molar extinction coefficient would be indicative of an octahedral geometry of the copper complex. The increase in molar absorbance in IL indicates that the Cu(II) complex is more distorted than the Cu(H2O)62+ species. In the UV region (Fig. 9b), the spectrum shows two supplementary transitions centered at 390 nm and 300 nm, respectively. The transition at 390 nm is observed with all the Cu(II) salts and is ascribed to an LMCT or a MLCT, characteristic of the coordination of Dca− anion to Cu(II). | ||
| Fig. 9 UV-vis spectra of IL phase after Cu(NO3)2 extraction (a) visible region and (b) UV region. | ||
With copper chloride, the maximum in the visible region is shifted towards a higher wavelength, underlining the implication of donor atoms of lower fields in the coordination sphere of the metal center. This seems to indicate that chloride ions are involved in the coordination of the copper center, likely giving a copper complex with mixed ligands with dicyanamide and chloride anions, Cu(Dca−)n(Cl−)m. The transition in the UV region is equally shifted from 390 to 423 nm. As can be seen in Fig. 10, the shift is gradual with an increase in aqueous copper concentration, meaning that the rate of chloride coordination is progressive and highly correlated with the ratio of anions co-extracted with the metal.
 | ||
| Fig. 10 Shift of the maximum wavelength, in the visible region, as a function of CuCl2 concentration. | ||
The EPR spectra of ionic liquid phases were recorded after extraction of aqueous solutions of CuCl2, Cu(NO3)2, Cu(BF4)2 and Cu(ClO4)2 (Fig. 11). The corresponding anisotropic parameters are listed in Table 2.
 | ||
| Fig. 11 EPR spectra of IL phases after Cu(II) extraction. | ||
| [Cu2+] mol dm−3 | A− | g | A (10−4 cm−1) |
|---|---|---|---|
| 0.1 | NO3− | g⊥ = 2.075 | A⊥ = 20 |
| g∥ = 2.345 | A∥ = 140 | ||
| 0.1 | BF4− | g⊥ = 2.075 | A⊥ = 20 |
| g∥ = 2.345 | A∥ = 140 | ||
| 0.1 | ClO4− | g⊥ = 2.075 | A⊥ = 20 |
| g∥ = 2.345 | A∥ = 140 | ||
| 0.1 | Cl− | g⊥ = 2.070 | A⊥ = 20 |
| g∥ = 2.330 | A∥ = 125 | ||
| 0.025 | Cl− | g⊥ = 2.075 | A⊥ = 20 |
| g∥ = 2.340 | A∥ = 135 |
The EPR spectra are anisotropic and characteristic of an axially symmetric monomeric copper(II) complex. The values of g∥, higher than those of g⊥, are in agreement with a dx2−y2 ground state and the allowed transitions ΔMS = 1, characteristic of a D4h symmetry.52 The parallel and perpendicular features are typical of a Cu2+ ion in an axial field with a tetragonal distortion by elongation along the axial direction. The hyperfine coupling constant A∥ is also a good indicator of the distortion from geometrical coordination in copper(II) complexes.
The EPR spectra of Cu(NO3)2, Cu(BF4)2 and Cu(ClO4)2 exhibit the same features, with similar g⊥, g∥ and A⊥ and A∥ values (A∥ = 140 × 10−4 cm−1), meaning that the coordination of Cu2+ ions in the three ionic liquids phases is similar. It is reasonable to believe that for the three salts, the anion is not in the coordination sphere of copper in the ionic liquid phase. The EPR parameters of copper extracted from a CuCl2 solution at 0.025 mol dm−3 are close to those determined for Cu(NO3)2, Cu(BF4)2 and Cu(ClO4)2, meaning that at this concentration, the coordination of Cl− in ionic liquid phase is relatively weak. The weaker A∥ value observed for the 0.1 mol dm−3 CuCl2 solution, compared to the other solutions indicates a high implication of Cl− in the coordination of copper in relation to a higher co-extraction percentage of this anion. This result corroborates the observation made from UV-vis spectra.
Experimental
Chemicals and reagents
All chemicals and reagents used in this study were used as received, without further purification. Sodium nitrate (99%) sodium chloride (99%), sodium dicyanamide (99%), and bis(trifluoromethane)sulfonylimide lithium salt were obtained from Sigma-Aldrich (Diegem, Belgium). Tetrahexyl(or tetraoctyl)ammonium bromide (98%) and tetrahexylammonium chloride were purchased from Acros Organics (Geel, Belgium). The metal salts had a purity of 99% or better and were purchased from Acros Organics (Geel, Belgium) or Sigma-Aldrich (Diegem, Belgium).The stocks of aqueous metal solutions were prepared by dissolving the corresponding salt of the respective metals (analytical grade purchased from Sigma-Aldrich-Fluka Chemical) in double-distilled and deionized water.
The tetrahexyl(or octyl)ammonium dicyanamide salt [THN(or TON)+][Dca−] were synthesized by metathesis between the corresponding ammonium bromide salt and sodium dicyanamide in aqueous media. In a flask, 15 g of tetrahexyl(or octyl)ammonium bromide was added to 200 mL of water. The suspension was heated to 70 °C, and 7.9 g (or 6.3 g) of sodium dicyanamide, (2.5 eq.) were slowly added to the suspension. The mixture was stirred at 70 °C for one hour, then slowly cooled at ambient temperature and then stirred for 72 h. To ensure complete metathesis, every 24 h 150 mL of aqueous solution were extracted from the reaction medium and replaced by a NaDca solution at the same initial concentration (40 or 31.5 g dm−3). This operation was repeated twice. This protocol allows the ionic liquid to be obtained with a purity higher than 98.5% and avoids the use of expensive silver reagent.33 The obtained tetraalkylammonium dicyanamide ionic liquid was recovered with ethyl acetate (3 × 40 mL). The organic layer was washed firstly with dilute NaDca solution (0.03 mol dm−3) and secondly with pure water, dried over MgSO4, and filtered. The filtrate was evaporated to give a pale yellow liquid.
The tetrahexyl(or octyl)ammonium bis(trifluoromethanesulfonyl)imide [THN(or TON)+][Tf2N−] were obtained by the procedure described above.
The purity of ionic liquids was checked by elemental analysis or 1H NMR. The absence of bromide ions was checked by ion chromatography.
Physical measurements
Elemental analyses (C, H, and N) were carried out on a Perkin-Elmer 2400 C, H, N element analyzer in our University. The UV-visible spectra of metal solutions were recorded using a Carry-5000 Varian spectrophotometer. The metal analyses were performed by UV-vis spectroscopy using EDTA for Cu(II) and Ni(II) and by ICP-OES, Thermo Fisher ICAP Series for Cd(II) and Pb(II).The 1H NMR spectrum was recorded in CDCl3 at room temperature with a Bruker AC 600 spectrometer. Chemical shifts (in ppm) for 1H NMR spectra were referenced to residual protic solvent peaks. The concentrations of organic cations in aqueous phase were determined by 1H NMR with an internal reference.
The concentrations of NO3− and Cl− ions in aqueous solution before and after extraction were determined using a Metrohm ion-HPLC with a conductivity detector.
The anisotropic X-band EPR spectra (9.431 GHz) of the frozen solutions were recorded at 150 K (Bruker ER4111VT variable temperature unit) using a Bruker ESP 300e spectrophotometer equipped with a Bruker E035M gaussmeter and a HP 5350B microwave frequency counter. The g values were controlled with diphenylpicrylhydrazyl (g = 2.0037). The EPR spectra were simulated using XSophe software version 1.1.4 developed by the Center for Magnetic Resonance and the Department of Mathematics of the University of Queensland, Brisbane, Australia, for Bruker Biospin GMBH.53 The software is a linewidth model with an angular dependence of g and a Simplex optimisation method with the copper element in natural abundance.
Extraction of Cu(II), Ni(II), Cd(II) and Pb(II) from aqueous solutions
The metal ion distribution ratios were determined by mixing definite volumes of IL and aqueous phase. The mixture was shaken during 24 h to reach equilibrium and then centrifuged (2000 × g, 5 min). The phases were then separated for analysis. The aqueous phase composition was analyzed by spectrophotometry UV-vis or by ICP-OES. The metal ion concentration in the IL phase was deduced from the difference between the initial concentration of metal ions in the aqueous phase and the concentration of metal ions before and after extraction.The efficiency of the extraction process was evaluated by calculation of the extraction percentage (% E) using the following equation:

The metal extraction percentages (% E) were determined at 298 K. The concentration of metal solutions lies in the range 10−3 to 0.1 mol dm−3. The experiments were made in triplicate to ensure the reproducibility of the assay, and the mean values of extraction yields were considered, for each system studied.
The concentrations of organic cations in aqueous phase were determined from extraction experiments using the following procedure: 3.5 mL of aqueous metal salt was recovered from the metal extraction experiment, involving 4 mL of aqueous metal solution in contact with 1 g (1.13 mL) of ionic liquids during 24 hours. NaOH 1 M was added to the solution to precipitate the residual metal. The solution was then centrifuged, to eliminate the precipitate, and lyophilized. The lyophilisat was recovered with 600 μL of CDCl3. We add also 50 μL of a solution of methyl pyridine (dissolved in CDCl3) as internal reference.
Conclusions
The high level of extraction observed for Cu(II) and Cd(II) in the present study demonstrates the possibility of designing, in a simple way, ILs able to extract metallic cations efficiently by using a common coordinating anion such as dicyanamide. The ILs are composed of inexpensive moieties and the extraction does not require the use of a chelating agent, which is a valuable advantage for process design. In some cases, extraction can be improved by the use of simple salts such as NaCl or NaNO3. The rational use of background salts can even improve the selectivity of the extraction process. Our study has focused on the fundamental aspects which govern the metal ion partitioning into IL solvents. Our main result highlights the influence of the anion of the metal salt on the extraction efficiency and on the extraction mode of the metals. The effect of the anion is highly related to its hydrophilic/hydrophobic character. A hydrophobic anion will be more easily co-extracted with the metals than a hydrophilic one, and will favor the transfer of metal salt into the ionic liquid phase via ion-pairing. These results confirm what has been previously evidenced in our earlier survey and in the study of Jansen et al., indicating that these phenomena are general and applicable to many systems. They clearly contribute to establishing a correlation between the influence of the anion on the extraction process and its relative position in the Hofmeister series, as proposed by Jansen et al. This study completes the knowledge already gained in this area. Further experiments will concern the reversibility of the extraction process and the recyclability of the ionic liquids.Notes and references
- E. Lichtfouse, J. Schwarzbauer and D. Robert, Environmental chemistry. Green chemistry and pollutants in ecosystems, Springer, Berlin, 2005, p. 780 Search PubMed.
- R. A. Kumbasar, Hydrometallurgy, 2009, 95, 290 CrossRef CAS.
- J. F. Blais, S. Dufresne and G. Mercier, Rev. Sci. Eau, 1999, 12(4), 687 CAS.
- F. Fu and Q. Wang, J. Environ. Manage., 2011, 92, 407 CrossRef CAS PubMed.
- G. J. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765 RSC.
- H. Zhao and S. V. Malhotra, Aldrichimica Acta, 2002, 35, 75 CrossRef CAS.
- A. P. De los Rıos, F. J. Hernandez-Fernandez, L. J. Lozano, S. Sanchez, J. I. Moreno and C. Godinez, J. Chem. Eng. Data, 2010, 55, 605 CrossRef.
- R. Germani, M. V. Mancini, G. Savelli and N. Spreti, Tetrahedron Lett., 2007, 48, 1767 CrossRef CAS.
- N. Papaiconomou, J. M. Lee, J. Salminen, M. Von Stosch and J. M. Prausnitz, Ind. Eng. Chem. Res., 2008, 47, 5080 CrossRef CAS.
- H. Zhao, S. Xia and P. Ma, J. Chem. Technol. Biotechnol., 2005, 80, 1089 CrossRef CAS.
- A. E. Visser, R. P. Swatloski, S. T. Griffin, D. H. Hartman and R. D. Rogers, Sep. Sci. Technol., 2001, 36, 804 CrossRef.
- S. Dai, Y. H. Ju and C. E. Barnes, J. Chem. Soc., Dalton Trans., 1999, 1201 RSC.
- A. Sengupta, P. K. Mohapatra, M. Iqbal, J. Huskens and W. Verboom, Dalton Trans., 2012, 41, 6970 RSC.
- R. Lertlapwasin, N. Bhawawet, A. Imyin and S. Fuangswasdi, Sep. Purif. Technol., 2010, 72, 70 CrossRef CAS.
- K. Shimojo and M. Goto, Anal. Chim., 2009, 76, 5039 Search PubMed.
- L. Y. Yuan, J. Peng, L. Xu, M. L. Zhai, J. Q. Li and G. S. Wei, J. Phys. Chem. B, 2009, 113, 8948 CrossRef CAS PubMed.
- J. D. Holbrey, A. E. Visser, S. K. Spear, W. M. Reichert, R. P. Swatloski and R. D. Roger, Green Chem., 2003, 5, 129 RSC.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis Jr and R. D. Rogers, Chem. Commun., 2001, 135 RSC.
- J. M. Reyna Gonzales, A. A. Torriero, A. I. Siriwardana, I. M. Burgar and A. M. Bond, Anal. Chem., 2010, 82, 7691 CrossRef PubMed.
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis Jr and R. D. Rogers, Environ. Sci. Technol., 2002, 36, 2523 CrossRef CAS PubMed.
- J. R. Harjani, T. Friscic, L. R. Mac Gillivray and R. D. Singer, Dalton Trans., 2008, 4595 RSC.
- J. H. Olivier, F. Camerel, J. Selb, P. Retailleau and R. Ziessel, Chem. Commun., 2009, 1133 RSC.
- M. P. Jensen, J. Neuefeind, J. V. Beitz, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2003, 123, 15469 Search PubMed.
- K. Kidani, N. Hirayama and H. Imura, Anal. Sci., 2008, 24, 1251 CrossRef CAS PubMed.
- H. Mehdi, K. Binnemans, K. Van Hecke, L. Van Meervelt and P. Nockemann, Chem. Commun., 2010, 46, 234 RSC.
- M. P. Jensen, J. A. Dzielawa, P. Rickert and M. L. Dietz, J. Am. Chem. Soc., 2002, 124, 10664 CrossRef CAS PubMed.
- M. L. Dietz and J. A. Dzielawa, Chem. Commun., 2001, 2124 RSC.
- M. L. Dietz, J. A. Dzielawa, I. Laszak, B. A. Young and M. P. Jensen, Green Chem., 2003, 6, 682 RSC.
- M. L. Dietz and D. C. Stepinski, Green Chem., 2005, 7, 747 RSC.
- A. Root and K. Binnemans, Phys. Chem. Chem. Phys., 2016, 18, 16039 RSC.
- D. Parmentier, T. Van der Hoogerstraete, D. Banerjee, Y. A. Valia, S. J. Metz, K. Binnemans and M. C. Kroon, Dalton Trans., 2016, 45, 9661 RSC.
- V. Egorov, D. I. Djigailo, D. S. Momotenko, D. V. Chernyshow, I. I. Torochesnikova, S. V. Smirnova and I. V. Pletner, Talanta, 2010, 80, 1177 CrossRef CAS PubMed.
- A. Messadi, A. Mohamadou, S. Boudesocque, L. Dupont and E. Guillon, Sep. Purif. Technol., 2013, 107, 172 CrossRef CAS.
- S. Viboud, N. Papaiconomou, A. Cortesia, G. Chatelb, M. Drayeb and D. Fontvieille, J. Hazard. Mater., 2016, 215–216, 40 Search PubMed.
- S. Martın, M. Gotzone Barandika, J. I. Ruiz de Larramendi, R. Corte, M. Font-Bardia, L. Lezama, Z. E. Serrna and X. Solans, Inorg. Chem., 2001, 40, 3687 CrossRef.
- A. Mohamadou, G. A. van Albada, H. Kooijman, B. Wieczorek, A. L. Spek and J. Reedijk, New J. Chem., 2003, 27, 983 RSC.
- M. J. Deng, I. W. Sun, P. Y. Chen, J. K. Chang and W. T. Tsai, Electrochim. Acta, 2008, 53, 5812 CrossRef CAS.
- K. J. Powell, Academic software Mini SC database version 5.3, 1999 Search PubMed.
- A. P. De Los Rios, F. J. Hernández-Fernández, F. J. Alguacil, L. J. Lozano, A. Ginestá, I. García-Díaz, S. Sánchez-Segado, F. A. López and C. Godínez, Sep. Purif. Technol., 2012, 97, 350 CrossRef.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995 RSC.
- S. L. Garvey, Talanta, 2012, 95, 25 CrossRef CAS PubMed.
- C. H. C. Janssen, N. A. Macias-Ruvalcaba, M. Aguilar-Martinez and M. N. Kobrak, Sep. Purif. Technol., 2016, 168, 275 CrossRef CAS.
- D. W. Smith, J. Chem. Educ., 1977, 54, 540 CrossRef CAS.
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358 RSC.
- Y. De Gaetano, A. Mohamadou, S. Boudesocque, J. Hubert, R. Plantier-Royon and L. Dupont, J. Mol. Liq., 2015, 207, 60 CrossRef CAS.
- A. Messadi, A. Mohamadou, S. Boudesocque, L. Dupont, P. Fricoteaux, A. Nguyen-Van-Nhien and M. Courty, J. Mol. Liq., 2013, 184, 68 CrossRef CAS.
- S. L. Garvey, Sep. Purif. Technol., 2014, 123, 145 CrossRef CAS.
- S. L. I. Toh, Solvent Extr. Ion Exch., 2006, 24, 33 CrossRef CAS.
- Y. Watanabe, J. Chem. Eng. Data, 2014, 59, 696 CrossRef CAS.
- T. Hamamoto, J. Phys. Chem. B, 2015, 119, 6317 CrossRef CAS PubMed.
- D. Depuydt, L. Liu, C. Glorieux, W. Dehaen and K. Binnemans, Chem. Commun., 2015, 14183 RSC.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143 CrossRef CAS.
- M. Griffin, A. Muys, C. Noble, D. Wang, C. Eldershaw, K. E. Gates, K. Burrage and G. R. Hanson, Mol. Phys. Rep., 1999, 26, 60 CAS.
| This journal is © The Royal Society of Chemistry 2016 |