
Effect of electronic coupling on the electrocatalytic performance of platinum metal†
Yan Fengab,
Caixia Wanga,
Feng Yea,
Hui Liu*ac and
Jun Yang*ac
aState Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: liuhui@ipe.ac.cn; jyang@ipe.ac.cn; Fax: +86-10-8254 4915; Tel: +86-10-8254 4917
bUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
cCenter for Mesoscience, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
First published on 19th August 2016
Abstract
Engineering the electronic coupling in platinum-based heterogeneous nanomaterials is an effective way to enhance their activity in electrocatalysis. Herein, we study the relationship between the electronic coupling and electrocatalytic activity of Pt metal through a core–shell construction. We prepare core–shell Ag–Pt, Au–Pt, and hollow Pt nanoparticles with comparable sizes and morphologies, and tailor the electron density around the Pt atoms via the differences in electronegativities between the core and shell components. Specifically, core–shell Ag–Pt nanoparticles are active for the methanol oxidation reaction due to the presence of electron donation from Ag to Pt, while core–shell Au–Pt nanoparticles exhibit superior activity for the oxygen reduction reaction due to the electron withdrawing effect from Pt by Au. The bimetallic core–shell particles therefore have the feasibility of tuning a Pt surface for two very different structure sensitive catalytic reactions, and further enhancement in electrocatalysis might be achieved by optimizing both the core size and shell thickness in the core–shell materials.
1. Introduction
Platinum (Pt)-based nanomaterials with heterogeneous structures would be particularly useful for the electrocatalytic applications, e.g. methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR).1–7 In principle, the adjacent components with different electron affinities in the heterogeneous nanostructures would interact with the Pt metal via electronic coupling through the interfaces between the different domains, inducing changes in the electron density around the Pt atoms, which could tune the electrocatalytic behavior of the Pt metal by altering the adsorption of reactants on the catalyst surface. For an instance, the heterodimers consisting of Pt particles and Au clusters exhibit superior stability for catalyzing ORR.8 The electronic interaction between Au and Pt would modify the Pt electronic structure toward a lower Pt surface energy, or lower-lying Pt d-band state, which is helpful to increase the oxidation potential of Pt electrocatalysts against dissolution under potential cycling regimes.However, the effect of electronic coupling on the electrocatalytic property of Pt is inconsistent in literature. Typically, Yang and Ying demonstrated that the heterogeneous Ag2S–Pt nanocomposites have poor catalytic activity for ORR due to the presence of strong electron donation effect from Ag2S to the Pt domains, which leads to the decrease of the 5d vacancies in Pt, and results in too weak adsorption of oxygen on the Pt surfaces.9 On the other hand, Sun and coworkers reported that owing to the charge transfer from Fe to Pt, the Pt nanoparticles in the Pt–Fe3O4 heterogeneous structures show a 20-fold increase in mass activity for ORR compared with the single component Pt particles.10 Considering the great importance of electronic coupling in catalytic applications, the efforts to figure out the effect of electronic coupling on the Pt activity in electrocatalysis would be undoubtedly significant.
Herein, we demonstrate the tuning of electronic coupling through a core–shell construction. We hypothesize that the heterogeneous growth of a thin Pt shell on a metal core with different electronegativity may tailor the electronic coupling effect in catalysis for enhancing the diversity in the control of Pt catalytic properties. We will demonstrate this with the design and synthesis of bimetallic core–shell Ag– or Au–Pt nanoparticles with comparable sizes and morphologies, and will also show that the catalytic activity of Pt shells may be varied from MOR to ORR by only altering their internal cores (Ag or Au). In specific, in comparison with their counterparts (pure hollow Pt nanostructures with comparable sizes), the core–shell Ag–Pt nanoparticles are active for MOR due to presence of electronic donation effect from the Ag core to the Pt shell, while the core–shell Au–Pt nanoparticles leverages on the electronic withdrawing effect from the Au core to Pt shell to increase the ORR activity of the Pt surface. The study in this work not only clarifies the relationship between electronic coupling and electrocatalysis of Pt metal, but also offers a vivid example to tune the material properties by means of a structural tailoring.
2. Experimental
2.1 General materials
Gold(III) chloride trihydrate (HAuCl4·3H2O, ACS reagent, ≥49.0% Au basis), silver nitrate (AgNO3, 99%), potassium tetrachloroplatinate(II) (K2PtCl4, 98%), acetic acid (CH3COOH, 98%), sodium borohydride (NaBH4, 98%), tri-sodium citrate dihydrate (C6H5Na3O7·2H2O, ≥99%), oleylamine (70%, technical grade), tetra-n-octylammonium bromide (TOAB, 98%), aqueous HClO4 solution (70%, ACS reagent), and Nafion 117 solution (5% in a mixture of lower aliphatic alcohols and water) from Sigma-Aldrich, methanol (99%), ethanol (99.5%), and toluene (99.5%) from Beijing Chemical Works, and Vulcan XC-72 carbon powders with BET surface area of 250 m2 g−1 and average particle size of 40–50 nm (XC-72C) from Cabot, were used as received. All glassware and Teflon-coated magnetic stirrers were cleaned with aqua regia, followed by copious rinsing with de-ionized water before drying.2.2 Synthesis of core–shell Au–Pt nanoparticle
A one-pot approach was used for the synthesis of bimetallic Au–Pt nanoparticles with a core–shell construction.11 Typically, 0.2 mmol of HAuCl4·3H2O and 0.1 mmol of K2PtCl4 were dissolved in 20 mL of oleylamine placed in a three-necked flask equipped with a condenser and stir bar. The solution was heated to 195 °C under flowing N2 and kept at this condition for 2 h for the reduction of Au3+ and Pt2+ ions by oleylamine, which also serves as the capping agent. After reaction, the core–shell Au–Pt nanoparticles were precipitated with methanol, collected by centrifugation, washed with methanol, and then re-dispersed in 20 mL of toluene.2.3 Synthesis of core–shell Ag–Pt nanoparticle
The phase transfer technique and seed-mediated growth was combined for the synthesis of bimetallic Ag–Pt nanoparticles with core–shell structures. For the preparation of Ag seeds using phase transfer approach, 8 mL of 100 mM aqueous tri-sodium citrate solution was added to 200 mL of 1 mM aqueous AgNO3 solution. Under vigorous stirring, 6 mL of 100 mM aqueous NaBH4 solution was introduced dropwise to obtain Ag hydrosol, in which sodium citrate serves as the stabilizer. The molar ratio of NaBH4 to the Ag+ ions was kept above 1.5 to ensure the reduction of the Ag salts to their zerovalent state. After reaction, the Ag hydrosol was mixed with 200 mL of 1 mM ethanolic solution of TOAB and the mixture was stirred for 2 min. Then, 50 mL of oleylamine was added and stirring was continued for another 3 minutes. The Ag nanoparticles were extracted into the oleylamine layer rapidly, leaving behind a colorless aqueous solution. Subsequently, 0.1 mmol of K2PtCl4 was added to the Ag colloidal solution in oleylamine, and the mixture was brought to 180 °C, and kept at this temperature for 3 h under flowing N2 for the reduction of the Pt metal precursor. After the reactions, the core–shell Ag–Pt nanoparticles were also purified by precipitation with methanol, centrifugation, and washing with methanol, and re-dispersed in 20 mL of toluene.2.4 Synthesis of hollow Pt nanoparticles supported on carbon substrates
To benchmark the core–shell nanoparticles in electrocatalysis, pure Pt nanoparticles with hollow interiors supported on carbon substrates (hPtNPs/C) were also prepared using the strategies we established before.12 In detail, 0.2 mmol of AgNO3 was added to 20 mL of oleylamine. The solution was heated to 165 °C and kept this temperature under flowing N2 for 1 h to reduce the Ag+ ions using oleylamine. Then 0.1 mmol of K2PtCl4 was added swiftly, and the mixture was continuously heated at 165 °C for 2 h. After reaction, the core–shell Ag–Pt nanoparticles were precipitated with methanol and re-dispersed into 20 mL of toluene. Subsequently, a calculated amount of carbon powder was added to the toluene solution of core–shell Ag–Pt nanoparticles. After aging the mixture for 24 h under stirring, the core–shell Ag–Pt nanoparticles supported on carbon substrates (20% Pt on carbon support) were collected by centrifugation, which were then re-dispersed in 20 mL of acetic acid by ultrasonication and refluxed for 3 h at 120 °C to remove the oleylamine from the particle surface. Afterwards, the carbon-supported core–shell Ag–Pt nanoparticles were collected by centrifugation, washed thrice with water, and then dried at room temperature in vacuum. Finally, the core–shell Ag–Pt particles supported on carbon substrates were agitated with 20 mL of saturated NaCl solution for 24 h under to remove the Ag component from the core region. The resulting hPtNPs/C were then collected by centrifugation, washed thrice with water, and then dried at room temperature in vacuum.2.5 Particle characterization
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were performed on the JEOL JEM-2100 electron microscope operating at 200 kV. An energy dispersive X-ray spectroscopy (EDX) analyzer attached to the TEM operating in the scanning transmission electron microscopy (STEM) mode was used to determine the chemical compositions as well as their distribution in the nanoparticles. X-ray photoelectron spectroscopy (XPS) was conducted on a VG ESCALAB MKII spectrometer. Samples for XPS analyses were obtained by concentrating the toluene solution of nanoparticles to 0.5 mL using flowing N2. Then 10 mL of methanol was added to precipitate the nanoparticles, which were recovered by centrifugation, washed twice with methanol, and dried at room temperature in vacuum.2.6 Electrochemical measurements
Electrochemical measurements were performed using a glass-carbon electrode (Pine Research Instrumentation) connected to a Bio-logic VMP3 (with EC-lab software version 9.56) potentiostat. A leak-free Ag/AgCl (SCE, saturated with KCl) and a Pt mesh (1 × 1 cm2) attached to a Pt wire were used as the reference and counter electrodes, respectively. For the evaluation of the catalytic activity for MOR and ORR, the core–shell Ag–Pt and Au–Pt nanoparticles were loaded on the Vulcan XC-72 carbon substrates with a mass ratio of 20% (Pt base) from their toluene solutions, followed by refluxing in acetic acid at 120 °C for 3 h to remove the surface coatings (oleylamine). Then 5 mg of carbon-supported nanoparticles (core–shell Ag–Pt, core–shell Au–Pt, or hollow Pt) was dispersed into 1 mL of ethanol containing 0.05 mL of Nafion solution to form an ink, which was dispensed onto the 5 mm glassy-carbon electrode to produce a working electrode with a nominal catalyst loading of 25 μg cm−2 (Pt basis). The cyclic voltammograms of core–shell Ag–Pt and Au–Pt nanoparticles in argon-purged HClO4 (0.1 M) were used to calculate the electrochemically active surface areas (ECSAs) of Pt. The catalyst performance for MOR was also determined by cyclic voltammetry in an electrolyte (1 M methanol in 0.1 M perchloric acid) at a potential window of −0.2 V to 1 V with a scanning rate of 20 mV s−1. The catalyst performance in room temperature ORR was evaluated in 0.1 M HClO4 electrolyte solution using a glassy carbon rotating disk electrode (RDE) at a rotation rate of 1600 rpm. Negative-going linear sweep voltammograms were recorded from 0.8 to 0 V at a scanning rate of 20 mV s−1 in the presence of bubbling ultra-pure oxygen to maintain a saturated oxygen atmosphere near the working electrode. The current densities for each catalyst (core–shell Ag–Pt, core–shell Au–Pt, and hollow Pt) were normalized by its ECSA to obtain the specific activities.3. Results and discussion
In the design of this study, the thin Pt shells with an Ag, Au, or a hollow interior at the core region are comparable in size and morphology. The rigorous experimental protocols adopted here also allow the comparison in electrocatalysis to be made under controlled surface conditions, and therefore a consistent basis for investigating the effect of electronic coupling on the electrocatalytic performance of Pt metal is established. The only difference among the samples is the existence of an Ag core, Au core, or a hollow interior underneath the Pt shells.3.1 Bimetallic core–shell Au–Pt, Ag–Pt, and hollow structured Pt nanoparticles
The core–shell Au–Pt nanoparticles were synthesized by co-reducing Au and Pt metal precursors in oleylamine at elevated temperature.11 Although the Au and Pt metal salts were heated simultaneously, the difference in their reduction kinetics results in the preferential formation of Au nanoparticles, which seed the subsequent deposition of Pt metal. Fig. 1a and b show the TEM and HRTEM images of the as-prepared core–shell Au–Pt nanoparticles, respectively, which indicate that the core–shell particles are spherical. The average diameter of the core–shell Au–Pt nanoparticles is ca. 14.6 nm, as calculated from the insert of Fig. 1a for the histogram, which shows the size distribution. The distributions of Au and Pt element in an arbitrary single particle, which were analyzed by EDX under the high-angle annular dark-field scanning TEM (HAADF-STEM) mode, were used to confirm the core–shell structure. Nanoscale element mappings (Fig. 1c–f) reveal that the Au in the bimetallic nanoparticles is concentrated in the core region, while the Pt signal is distributed throughout the entire particle, clearly indicating the formation of Au–Pt core–shell structure. The mapping analyses are in accordance to the element profile analyses of an arbitrary single Au–Pt particle (Fig. 1g), which also supports that the bimetallic Au–Pt nanoparticles thus obtained have core–shell constructions. | ||
| Fig. 1 TEM image (a), HRTEM image (b), elemental mapping analyses (c–f), and EDX-based element profiles (g) of bimetallic core–shell Au–Pt nanoparticles as-prepared by co-reducing Au and Pt metal precursors in oleylamine at elevated temperature. Insert in (a) is the histogram to show the size distribution of core–shell Au–Pt particles. | ||
Although the core–shell Ag–Pt nanoparticles could also be prepared by co-reducing the corresponding metal precursors in oleylamine,11 the Ag seeds formed this way usually have multiply twinned nature, which has strong influence on the stability of the core–shell structure, and would promote the inside-out diffusion of Ag in core–shell nanoparticles.13,14 Hence, in this study, we combine the phase transfer technique with the seed-mediated growth for the synthesis of bimetallic Ag–Pt nanoparticles with stable core–shell geometries. The Ag seeds with single crystal phase were firstly produced in aqueous phase by NaBH4 reduction of AgNO3, and they were then transferred into oleylamine using an ethanol mediated approach.15,16 Fig. S1 in ESI† for the TEM image shows that the Ag particles are spherical and monodispersed with an average diameter of ca. 11.4 nm. The HRTEM image illustrates the uniform lattice orientations in these nanoparticles, confirming that these Ag particles are of highly single crystallinity (ESI Fig. S1b†).
The single crystal Ag nanoparticles were then used as seeds for the formation of bimetallic core–shell Ag–Pt nanoparticles. At elevated temperature, the Pt metal reduced from its precursors by oleylamine nucleated preferentially on the surface of existing Ag nanoparticles. After reaction, the core–shell products were purified and re-dispersed in toluene. Fig. 2a and b exhibit the TEM and HRTEM images of the bimetallic core–shell Ag–Pt products as-prepared by the seed-mediated growth method, which contain well dispersed nanoparticles with an average size of ca. 14.8 nm (insert of Fig. 2a). Again, the core–shell structure in bimetallic Ag–Pt nanoparticles was verified by the distribution of Ag and Pt in the final nanoparticles. As displayed by Fig. 2c–f, the nanoscale element mappings of a single particle (Fig. 2c) illustrate that Pt is distributed throughout the entire particle (Fig. 2e and f), while the Ag was concentrated in the core region (Fig. 2d and f). The EDX-based line scanning analysis (Fig. 2g) of the same single particle also supports the formation of bimetallic Ag–Pt nanoparticles with Ag and Pt residing in the core and shell regions, respectively.
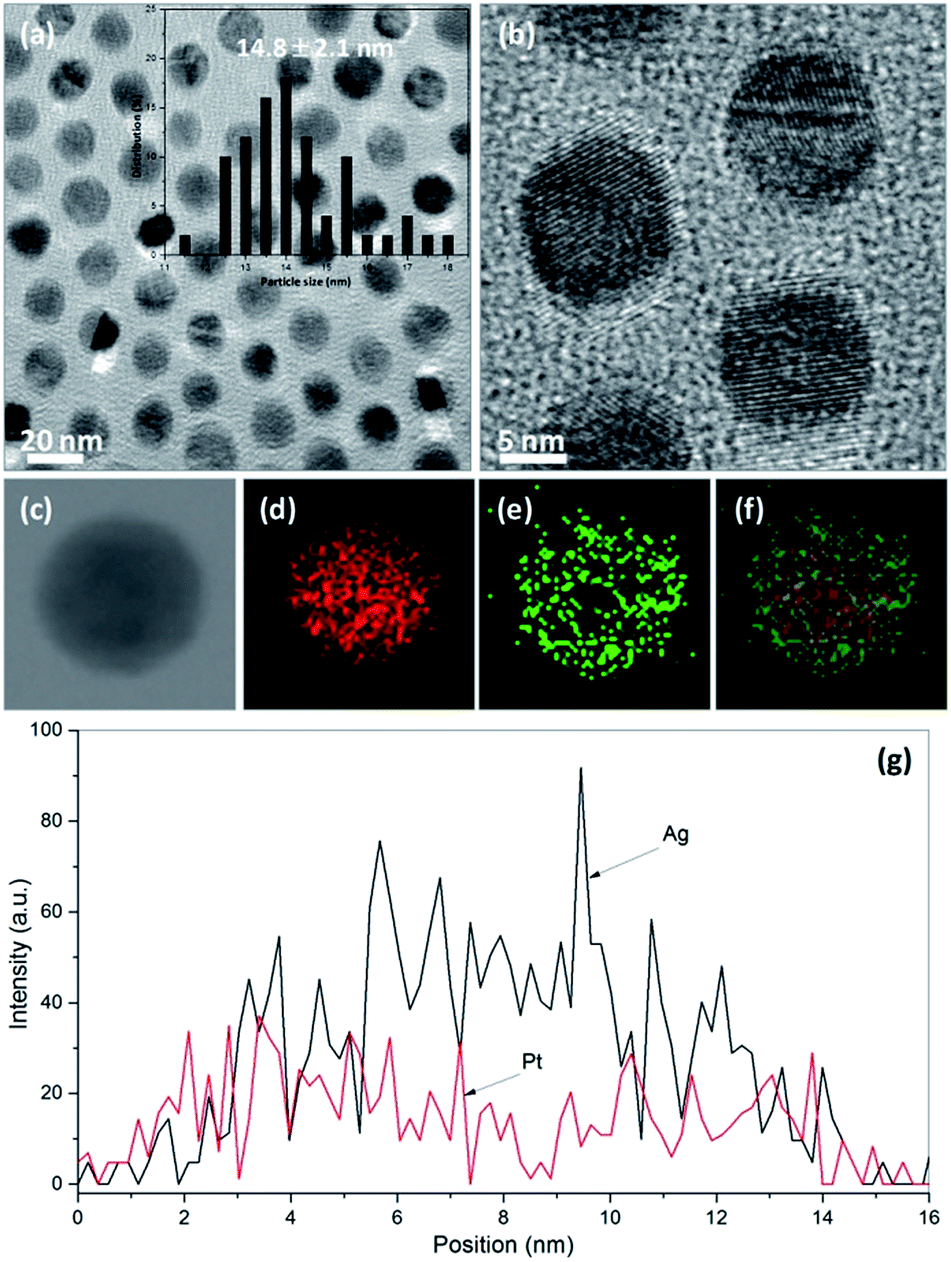 | ||
| Fig. 2 TEM image (a), HRTEM image (b), elemental mapping analyses (c–f), and EDX-based element profiles (g) of bimetallic core–shell Ag–Pt nanoparticles as-prepared by seed-mediated growth method in oleylamine at elevated temperature. Insert in (a) is the histogram to show the size distribution of core–shell Ag–Pt particles. | ||
Both core–shell Ag–Pt and core–shell Au–Pt nanoparticles were loaded on Vulcan carbon substrates and tested for electrocatalytic activity for the MOR and ORR at room temperature. As shown by ESI Fig. S2a and b† for the representative TEM images, the core–shell Ag–Pt and Au–Pt nanoparticles could be dispersed very well on the carbon substrates by conventional means. The loading of the core–shell particles on carbon supports was fixed at 20 wt% of Pt. To benchmark the activity of core–shell Ag– or Au–Pt nanoparticles as electrocatalysts for MOR and ORR, pure hollow Pt nanostructures were also supported on same Vulcan carbon substrates with mass ratio of 20%, labeled as hPtNPs/C, using the approaches we developed previously.12 As described in experimental section, core–shell Ag–Pt nanoparticles were firstly prepared in oleylamine by successively reducing the AgNO3 and K2PtCl4 in oleylamine at elevated temperature, which were then loaded on carbon substrates, followed by agitating with saturated NaCl solution to eliminate the Ag component from the core region. The TEM image of the as-prepared hPtNPs/C and the histogram to show the size distribution of hollow Pt particles were presented in ESI Fig. S2c and d,† respectively, which suggest the well dispersion of hollow structured Pt nanoparticles with an average diameter of ca. 14.2 nm on the carbon substrates.
3.2 Electronic coupling in core–shell nanoparticles
An important feature of core–shell materials affecting the electrocatalytic property is the electronic coupling effect between the core and shell metals. The differences in the electronegativities of Ag or Au core and Pt shell (1.83 for Ag, 2.54 for Au, and 2.28 for Pt, respectively) imply that electrons may be donated to or withdrawn from Pt in core–shell Ag– or Au–Pt nanoparticles. The XPS analyses were therefore used to determine the electron transfer in core–shell particles. As exhibited by Fig. 3a, compared with those of pure hollow Pt nanostructures (no electronic coupling in them), the binding energies of the Pt 4f7/2 and 4f5/2 peaks for core–shell Ag–Pt nanoparticles are shifted appreciably to lower values, while shifted to higher values for core–shell Au–Pt nanoparticles. This is indication of electron donation from Ag to Pt in core–shell Ag–Pt nanoparticles and electron withdrawing from Pt by Au in core–shell Au–Pt nanoparticles. The electron-donation or withdrawing effect imposed on Pt shells by Ag or Au cores could result in a substantial increase or decrease in the local electron density around the Pt sites, which would definitely affect the adsorption of reactants on the Pt atoms and thus alter their catalytic properties. | ||
| Fig. 3 4f XPS spectra of Pt in core–shell Ag–Pt, Au–Pt, and pure hollow Pt nanoparticles (a); room-temperature CO stripping tests over the core–shell Ag–Pt, Au–Pt, and pure hollow Pt nanoparticles in 0.1 M HClO4 electrolyte (b). | ||
The effect of the electronic coupling on the chemisorption of reactants on the Pt surface could be confirmed by the CO stripping tests. Fig. 3b shows the CO stripping voltammograms of hollow Pt nanostructures, core–shell Ag–Pt, and core–shell Au–Pt nanoparticles after holding the working electrode at −0.15 V for 30 min in CO saturated 0.1 M HClO4 electrolyte. The CO stripping peak shifts to a lower potential for the core–shell Ag–Pt nanoparticles, while shifts to a higher value for the core–shell Au–Pt nanoparticles as compared to the pure hollow Pt nanostructures, suggesting a more facile or difficult removal of CO from the Pt shells in core–shell Ag–Pt or core–shell Au–Pt nanoparticles. The different behavior of CO removal for core–shell Ag– and Au–Pt nanoparticles relative to hollow Pt nanostructures is related to the nature of the Pt–CO bond. The chemisorption of CO on Pt surface involves the donation of lone pair electrons from the filled carbon σ orbital of CO to the empty 5d-orbital of Pt, which is compensated by the back donation of electrons from the Pt dπ to the π* orbitals of CO.17,18 Therefore, a high or low local density of electrons around Pt is associated with weak or strong CO chemisorption.
3.3 Electrocatalytic performance of hollow and core–shell nanoparticles
We use cyclic voltammetry to determine the electrochemically active surface areas (ECSAs) of hollow Pt, core–shell Ag–Pt and Au–Pt nanoparticles. As shown by Fig. 4a. The specific ECSAs calculated by integrating the charge associated with the hydrogen adsorption/desorption potential region after double-layer correction, are 51.3 m2 gPt−1 for hollow Pt, 36.2 m2 gPt−1 for core–shell Ag–Pt, and 31.5 m2 gPt−1 for core–shell Au–Pt nanoparticles, respectively. The hollow interiors may account for the higher ECSAs of hollow structured Pt nanoparticles. | ||
| Fig. 4 Cyclic voltammograms of pure hollow Pt nanostructures, core–shell Ag–Pt, and core–shell Au–Pt nanoparticles in argon-purged HClO4 electrolyte (0.1 M) at a scan rate of 50 mV s−1 (a); cyclic voltammograms of pure hollow Pt nanostructures, core–shell Ag–Pt, and core–shell Au–Pt nanoparticles in argon-purged HClO4 (0.1 M) with methanol (1 M) at a scan rate of 20 mV s−1 (b); ORR polarization curves recorded in an O2-saturated HClO4 solution (0.1 M) at a sweep rate of 20 mV s−1 and a rotating speed of 1600 rpm for the pure hollow Pt nanostructures, core–shell Ag–Pt, and core–shell Au–Pt nanoparticles (c). | ||
The voltammograms of MOR were obtained in the potential window of 0–1 V at a sweeping rate of 20 mV s−1 (Fig. 4b). The current densities in Fig. 4b were normalized by the ECSAs of Pt. As displayed, the comparison in current densities manifests that for these three samples, the core–shell Ag–Pt nanoparticles have the highest specific activities for methanol oxidation. As summarized in ESI Table S1,† the current densities in forward san for core–shell Ag–Pt, Au–Pt, and hollow Pt nanoparticles are 43.7, 28.7, and 32.8 mA cm−2, respectively. The superior activity of core–shell Ag–Pt nanoparticles for MOR could be attributed to the stronger electron donation effect from Ag to Pt in core–shell Ag–Pt nanoparticles. Analogous to the electronic coupling in the bimetallic Pt–Sn system,17 the electron transfer from Ag to Pt in core–shell Ag–Pt nanoparticles leads to the weaker chemisorption of CO, an intermediate product in the MOR and catalyst poison, and promote the MOR.
We also performed the evaluation of core–shell Ag–Pt, Au–Pt, and hollow Pt nanoparticles in catalyzing ORR by using a glass carbon rotating disk electrode (RDE). In contrast to their poor activity for MOR, the core–shell Au–Pt nanoparticles exhibit the highest activity for ORR in comparison with their core–shell Ag–Pt and hollow Pt counterparts. Polarization curves for the ORR over these three samples were presented in Fig. 4c, and ESI Table S2† summarized the ORR activities of these three samples. The half-wave potentials for core–shell Au–Pt nanoparticles are 650 mV, 40 mV and 80 mV higher than that of hollow Pt (610 mV) and core–shell Ag–Pt nanoparticles (570 mV), respectively. The kinetic current density at half-wave potential of core–shell Au–Pt nanoparticles is also the highest (14.7, 7.71, and 12.6 mA cm−2 for core–shell Au–Pt, Ag–Pt, and hollow Pt nanoparticles, respectively). The electron withdrawing effect from Pt by Au in core–shell Au–Pt nanoparticles may account for their good catalytic activity for ORR, and this is in accordance to the studies reported by Watanabe and co-workers,19 in which the alloying with Fe, Ni or Co was used to enhance the electrocatalytic activity of Pt for ORR. Fe, Ni or Co has more 5d vacancies than Pt and could withdraw electrons from the latter. This withdrawing effect induces an increase in 5d vacancies in Pt, resulting in improved oxygen adsorption to favor the oxygen reduction. In addition, the poor activity of core–shell Ag–Pt nanoparticles for ORR is also well consistent with the results documented in literature.20 The strong electron donation effect from Ag to Pt in core–shell Ag–Pt nanoparticles leads to a decrease of the 5d vacancies in Pt. Thus, the adsorption of oxygen on the Pt surfaces is too weak for the oxygen dissociation reaction, accounting for their poor activity for ORR.
The long-term stabilities of core–shell Ag–Pt, Au–Pt, and pure hollow Pt nanoparticles in MOR and ORR were illustrated by the chronoamperograms. As evinced by ESI Fig. S3,† the slow decay rates of core–shell Ag–Pt nanoparticles for MOR and core–shell Au–Pt nanoparticles for ORR support the electron donation to Pt and withdrawing from Pt shells would improve their electrocatalytic performance for MOR and ORR, respectively.
4. Conclusions
In summary, we have demonstrated the tuning of electronic coupling of Pt shells through a core–shell construction. We prepared core–shell Ag–Pt, Au–Pt, and hollow Pt nanoparticles with comparable sizes and morphologies. The differences in electronegativities between the core and shell components were used to tailor the electron density around the Pt atoms. Compared with the pure hollow Pt nanostructures (no electronic coupling in them), we found that electron donation from Ag to Pt in core–shell Ag–Pt nanoparticles is essential for their high activity for MOR, while the electron withdrawing effect from Pt by Au in core–shell Au–Pt nanoparticles accounts for their enhanced ORR catalytic activity. The investigations in this work clarify the relationship between electronic coupling and electrocatalysis of Pt metal. In addition, by optimizing both the core size and shell thickness in the core–shell materials, further enhancement in activity for MOR and ORR might be expected.Acknowledgements
Financial support from National Natural Science Foundation of China (No. 21376247, 21476246, 21406231, 21506225, 21573240) and Center for Mesoscience, Institute of Process Engineering, Chinese Academy of Sciences (COM2015A001) is gratefully acknowledged.Notes and references
- P. D. Cozzoli, T. Pellegrino and L. Manna, Chem. Soc. Rev., 2006, 35, 1195 RSC.
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasché and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 3273 CrossRef CAS.
- Y. Feng, H. Liu, P. Wang, F. Ye, Q. Tan and J. Yang, Sci. Rep., 2014, 4, 6204 CrossRef CAS PubMed.
- J. Yang and H. Liu, Metal-based composite nanomaterials, Springer-Verlag, New York, 2014 Search PubMed.
- H. Liu, Y. Feng, D. Chen, C. Li, P. Cui and J. Yang, J. Mater. Chem. A, 2015, 3, 3182 CAS.
- J. Qu, Y. Ye, D. Chen, Y. Feng, Q. Yao, H. Liu, J. Xie and J. Yang, Adv. Colloid Interface Sci., 2016, 230, 29 CrossRef CAS PubMed.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220 CrossRef CAS PubMed.
- J. Yang and J. Y. Ying, Angew. Chem., Int. Ed., 2011, 50, 4637 CrossRef CAS PubMed.
- C. Wang, H. Daimon and S. Sun, Nano Lett., 2009, 9, 1493 CrossRef CAS PubMed.
- P. Hou, H. Liu, J. Li and J. Yang, CrystEngComm, 2015, 17, 1826 RSC.
- H. Liu, F. Ye and J. Yang, Ind. Eng. Chem. Res., 2014, 53, 5925 CrossRef CAS.
- H. Liu, J. Qu, Y. Chen, J. Li, F. Ye, J. Y. Lee and J. Yang, J. Am. Chem. Soc., 2012, 134, 11602 CrossRef CAS PubMed.
- P. Hou, P. Cui, H. Liu, J. Li and J. Yang, Nano Res., 2015, 8, 512 CrossRef CAS.
- J. Yang, J. Y. Lee, T. C. Deivaraj and H.-P. Too, J. Colloid Interface Sci., 2004, 277, 95 CrossRef CAS PubMed.
- J. Yang, J. Y. Lee and J. Y. Ying, Chem. Soc. Rev., 2011, 40, 1672 RSC.
- A. K. Shukla, A. S. Aricò, K. M. El-Khatib, H. Kim, P. L. Antonucci and V. Antonucci, Appl. Surf. Sci., 1999, 137, 20 CrossRef CAS.
- A. Nilsson, L. G. M. Pettersson and J. K. Nørskov, Chemical bonds at surfaces and interfaces, Elsevier B. V., 2008 Search PubMed.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750 CrossRef CAS.
- Z. Peng, J. Wu and H. Yang, Chem. Mater., 2010, 22, 1098 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and HRTEM images of the Ag seed particles, TEM images and chronoamperograms of carbon-supported core–shell and hollow nanoparticles, tables to summarize the electrochemical measurements in this study. See DOI: 10.1039/c6ra16626a |
| This journal is © The Royal Society of Chemistry 2016 |