
Encapsulation of unmodified Gibbsite via conventional emulsion polymerisation using charged co-oligomers†
Olessya P. Loikoa,
Anne B. Spoelstraa,
Alexander M. van Herkab,
Jan Meuldijka and
Johan P. A. Heuts*a
aDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: j.p.a.heuts@tue.nl
bInstitute of Chemical and Engineering Sciences, 1 Pesek Road, Jurong Island 627833, Singapore
First published on 17th August 2016
Abstract
Gibbsite platelets were successfully encapsulated via starved-feed conventional emulsion polymerisation using anionic co-oligomers without the need for any surface modification of the platelets. Charged co-oligomers, consisting of butyl acrylate and acrylic acid units, were synthesized using atom transfer radical polymerisation (ATRP) and used as stabilisers for the initial Gibbsite platelets and the formed latex particles. Optimisation of co-oligomer concentration resulted in efficient encapsulation where every latex particle contained a Gibbsite platelet. Cryo-TEM characterisation showed the Gibbsite platelet completely covered with a polymer layer and this morphology was not affected by the investigated co-oligomer composition or chain length.
Introduction
Encapsulation of inorganic particles, in particular various types of clay, has been widely studied over the past decades because of the improvement of various properties of the final product.1–3 Polymer encapsulation of clay via (mini)emulsion polymerisation usually requires modification of the surface via a so-called hydrophobization process to make it more compatible with polymer.4–7 This is usually done using a relatively complicated and time-consuming chemical surface modification.8–10Most of the attempts to encapsulate covalently modified clay via (mini)emulsion polymerisation led to armored,7,11–14 dumbbell or peanut-shaped15,16 latex particles. Several studies with unmodified synthetic LAPONITE® RD and natural sodium montmorillonite using (mini)emulsion polymerisation resulted either in armored latex particles17,18 or led to (partial) coagulation.13 Therefore, it still remains challenging to encapsulate every single clay platelet.
In previous work,19 successful encapsulation of unmodified natural sodium montmorillonite was achieved via conventional emulsion polymerisation, but a fairly “exotic” mixed surfactant system (Triton X-100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sodium dodecylbenzene sulfonate (SDBS) = 6:1 w/w, at half its critical micelle concentration (CMC)) was required. For the current work, we tried first to encapsulate unmodified Gibbsite platelets using SDBS, but this strategy resulted in a very low encapsulation efficiency (see ESI†) and was therefore abandoned.
:sodium dodecylbenzene sulfonate (SDBS) = 6:1 w/w, at half its critical micelle concentration (CMC)) was required. For the current work, we tried first to encapsulate unmodified Gibbsite platelets using SDBS, but this strategy resulted in a very low encapsulation efficiency (see ESI†) and was therefore abandoned.
We previously also reported the successful encapsulation of unmodified synthetic Gibbsite platelets using charged co-oligomers containing a trithiocarbonate group, which were chain extended using a starved feed emulsion polymerization.9 This strategy was originally developed by Hawkett and co-workers and was successfully applied to encapsulate titanium dioxide,20 pigments,21 graphene oxide22 and multi-walled carbon nanotubes.23 In other work24 we tried a similar approach, but now using atom transfer radical polymerization (ATRP) instead of the earlier reported reversible addition fragmentation chain transfer (RAFT) for the chain extension. Instead of encapsulated Gibbsite platelets, this approach resulted in a “muffin-like” morphology, where the polymer seems to have grown from only a single side of the Gibbsite platelet.
Reviewing the abovementioned studies we came to the conclusion that synthetic short charged co-oligomers act as usual surfactants for both the platelets and the polymer particles in addition to providing hydrophobic regions around hydrophilic platelets. Since this combination of properties is what is required for encapsulation we focus in this paper on the use of charged co-oligomers in a conventional starved-feed emulsion polymerisation for the encapsulation of unmodified Gibbsite platelets (Scheme 1). Briefly, anionic co-oligomers, consisting of butyl acrylate and acrylic acid units, were synthesised via ATRP to obtain relatively narrow molar mass distributions. They were then adsorbed on the surface of the positively charged Gibbsite platelets and acted as stabilisers. Polymer encapsulation of platelets was conducted under starved-feed conventional emulsion polymerisation conditions by feeding monomer mixture, consisting of MMA and BA. The resulting morphology was analysed with cryo-TEM. Several parameters, such as co-oligomer concentration, composition and chain length were investigated with a focus on the resulting particle colloidal stability and morphology.
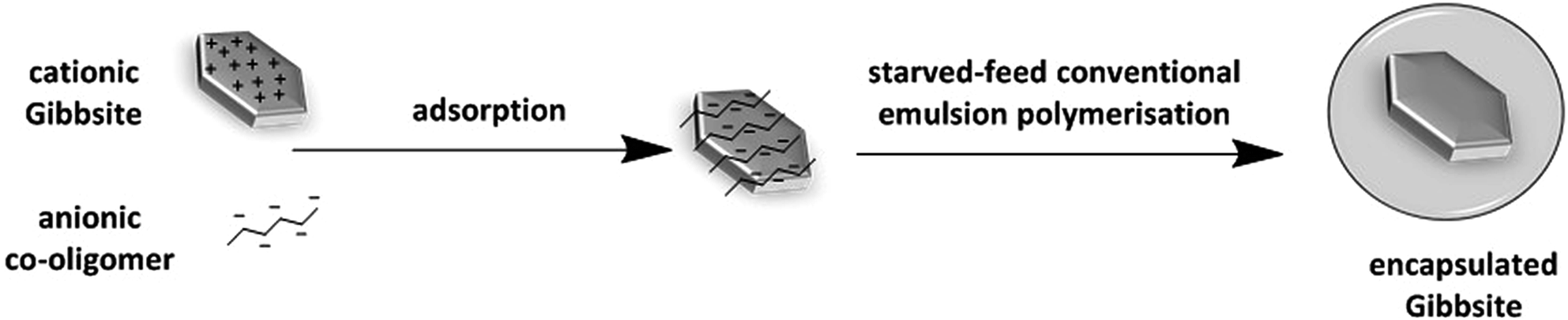 | ||
| Scheme 1 Schematic representation of the synthesis of polymer–Gibbsite nanocomposites by aqueous starved feed conventional emulsion polymerisation using charged co-oligomers as stabilisers. | ||
This approach is industrially attractive due to a very simple polymerisation process, which is based on the use of conventional emulsion polymerisation and no complicated or time-consuming surface modification procedures of the initial Gibbsite platelets is required.
Experimental
Materials
The monomers butyl acrylate (BA, Aldrich, 99%), methyl methacrylate (MMA, Aldrich, 99%) and tert-butyl acrylate (tBA, Aldrich, 99%) were purified by passing them through a column of inhibitor removal agent. CuBr (99.999%, Aldrich), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%, Aldrich), trifluoroacetic acid (TFA, 99%, Aldrich), hydrochloric acid (32%, Merck), dichloromethane (DCM), dimethylformamide (DMF), methanol, tetrahydrofuran (THF), aluminum isopropoxide (98%, Acros), aluminum sec-butoxide (95%, Fluka), and 2,2′-azobis[2-methyl-N-(2-hydroxyethyl) propionamide] (VA-086, Wako Chemicals GmbH) were used as received.Characterisation
Molar mass distributions, the number average molar mass (Mn) and polydispersity index (Đ) of the anionic co-oligomer and the composite latexes were measured by size exclusion chromatography (SEC) using a Waters SEC set-up equipped with a Waters model 510 pump, a model 410 differential refractometer and a 2487 dual λ absorbance detector (operated at λ = 254 nm). A set of two mixed bed columns (Mixed-C, Polymer Laboratories, 30 cm, 40 °C) was used. Tetrahydrofuran was used as the eluent, and the system was calibrated using polystyrene standards (range = 580–7.5 × 106 g mol−1). Data analysis was performed using the software Empower Pro version 2 from the Waters Corporation.1H NMR spectra were recorded on a Varian 400 MHz spectrometer using chloroform-d and water-d2 as solvents. Analysis of the spectra was done using the software MestReNova 9.0.0-12821 from Mestrelab Research S.L.
The particle size distribution and zeta potential (ζ) were determined at 23 °C using a Malvern Zetasizer Nano ZS instrument. The ζ potential was calculated from the electrophoretic mobility (μ) using the Smoluchowski relationship, ζ = ημ/ε, with ka ≫ 1 (where η is the viscosity, ε is the dielectric constant of the medium, k and a are the Debye–Hückel parameter and particle radius, respectively).
Critical micelle concentrations of the co-oligomers were determined from light scattering intensity measurements performed on a Malvern 4700 light scattering instrument (λ = 488 nm) equipped with a Malvern Multi-8 7032 correlator at a scattering angle of 90° at 25 °C.25
Transmission electron microscopy (TEM) measurements were conducted on a FEI Tecnai 20, type Sphera TEM instrument (with a LaB6 filament, operating voltage = 200 kV). 200 mesh copper grids with continuous carbon support layer were used.
Cryogenic transmission electron microscopy (cryo-TEM) measurements were conducted on FEI CryoTitan electron microscope operated at 300 kV, equipped with a field emission gun (FEG), a postcolumn Gatan energy filter (GIF) and a post-GIF 2k × 2k Gatan CCD camera. 200 mesh copper grids with lacey carbon layer were used for analysis. The sample vitrification procedure was performed using an automated vitrification robot (FEI Vitrobot Mark III). A 3 μl sample was applied to a Quantifoil grid (R 2/2, Quantifoil Micro Tools GmbH; freshly glow discharged for 40 seconds just prior to use) within the environmental chamber of the Vitrobot and the excess liquid was blotted away. The thin film thus formed was shot into melting ethane. The grid containing vitrified film was immediately transferred to a cryoholder (Gatan 626) and observed under low dose conditions at −170 °C.
Synthesis of Gibbsite
Gibbsite was synthesized according to the method described by Wierenga et al.26 Briefly, to a 2 L polypropylene bottle 1 L of double deionized water (DDI) was added and acidified by 2.6 g hydrochloric acid. To the solution 19.7 g (0.08 mol) aluminium sec-butoxide and 16.3 g (0.08 mol) aluminium isopropoxide were added and dissolved by stirring for 10 days at room temperature. Subsequently the solution was heated in a water bath at 85 °C for three days. The dispersion was then dialyzed against deionized water for 10 days in regenerated cellulose tubes (Visking, MWCO 12,000–14,000), after which the dispersion was centrifuged at 3000 rpm for 15 min to remove large platelets. The supernatant was then redispersed in water and centrifuged at 10000 rpm for 3 h. Very small platelets were removed by discarding the supernatant and the sediment was redispersed in water to obtain a 1 wt% Gibbsite dispersion of the desired particle size. Synthesised Gibbsite platelets have a ζ potential of +40 mV and a Z-average diameter (Dz) of 100 nm with a poly value of 0.14 at pH ≈ 7.
General procedure for the synthesis of anionic co-oligomers via ATRP
All needles, Schlenk flasks and magnetic stirring bars were dried overnight in an oven at 120 °C and purged with argon during cooling before use. The polymerization was conducted in a Schlenk flask equipped with a magnetic stirring bar. n-Butyl acrylate (12.8 g, 0.1 mol) and tert-butyl acrylate (25.6 g, 0.2 mol) were polymerized in 10 vol% DMF using PMDETA (0.26 g, 0.0015 mol) and CuBr (0.21 g, 0.0015 mol) as a catalyst system and 2-bromo-2-methyl-propionic acid phenyl ester (0.31 g, 0.0015 mol) as initiator.24 This initiator was synthesized according to literature protocol.27 The reaction was carried out at 70 °C and terminated after 3 h by cooling the flask to 0 °C and opening it to the air (Scheme 2). The catalyst was removed by passing the reaction mixture through a column of basic alumina. The co-oligomer was then isolated by pouring the solution into a water–methanol mixture and drying the precipitate under vacuum at room temperature. During the polymerization, samples were taken at regular intervals to measure monomer conversion using gas chromatography and molar mass distributions using SEC. Final conversion: 10%. 1H NMR (400 MHz, CDCl3, δ): 7.41, 7.25, 7.13 (aromatic H), 4.02 (O–CH2), 2.22 (–CH2–CH2–), 1.82–1.52 (–CH2–CH2–C(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)), 1.45 (O–C(CH3)3), 0.96 (–CH3).
O)), 1.45 (O–C(CH3)3), 0.96 (–CH3).
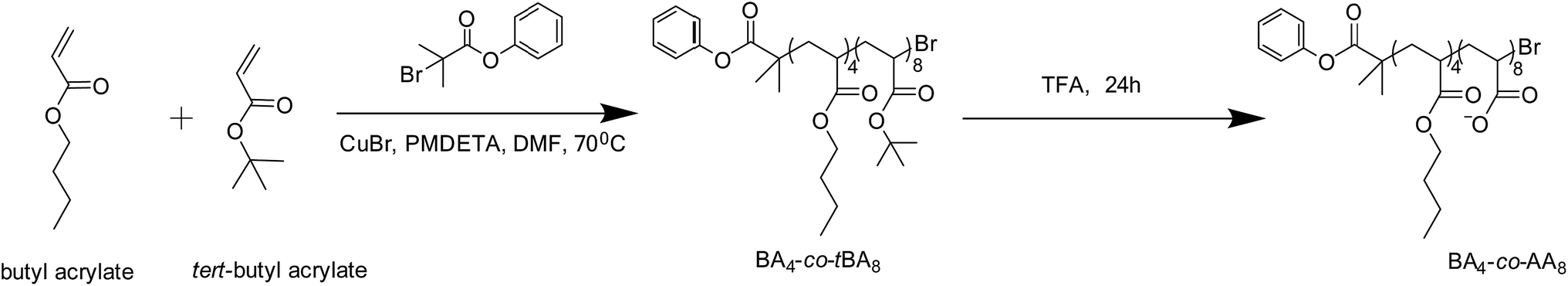 | ||
| Scheme 2 Schematic representation of the synthetic strategy to anionic co-oligomer from butyl acrylate and tert-butyl acylate. | ||
The co-oligomer composition was determined using 1H NMR from the integrated intensity ratios of the area for the aromatic protons (δ = 7.13–7.41) of the end group to the area for methyl group (δ = 0.96) of butyl acrylate or to the area for tert-butyl group (δ = 1.45) of tert-butyl acrylate respectively (see ESI†).
For the hydrolysis of the tert-butyl ester groups, the co-oligomers were dissolved in dichloromethane using a five-fold molar excess of TFA with respect to the tert-butyl groups and stirred at room temperature for 24 h. The hydrolysed product was collected after rotary evaporation of the solvent and TFA, after which the product was washed with dichloromethane and dried at 50 °C under vacuum.28 The degree of hydrolysis was determined from comparison of the integrated signals between 0.8 and 2.8 ppm before and after treatment with TFA.
Adsorption studies
The adsorption procedure was performed as described previously.9,24 In different vials, calculated amounts of anionic co-oligomers were transferred from a 10 mM aqueous stock solution and a total volume of 10 mL was made by adding DDI water. Equal volumes of Gibbsite dispersion (1 wt%) were then added drop wise into these vials under constant stirring. The vials were kept stirring overnight at room temperature. Particle size distributions and ζ-potentials were measured for these samples (see ESI†).Encapsulation experiments
Polymer encapsulated Gibbsite nanoparticles were synthesized by conventional starved feed emulsion polymerization. Briefly, 0.5 mL of a 10 mM aqueous stock solution of anionic co-oligomer and 25 mL of DDI water were transferred into a 50 mL three neck flask after which 2 mL of Gibbsite dispersion (1 wt%) was added drop wise at a rate of 1 mL min−1 using syringe pump NE-1000 under constant stirring at room temperature. The resulting dispersion was then sonicated for 1.5 min using Vibracell tip sonicator at 30% amplitude. 0.09 g of VA-086 initiator (2 wt% based on monomer weight) was added and the reaction mixture was purged with argon for 1 hour after which the flask was heated to 80 °C. Then, 4.5 g of deoxygenated monomer mixture ([MMA]:[BA] = 10:1 w/w) was fed using a syringe pump NE-1000 at a rate of 9 mg min−1. After the completion of monomer addition, the flask was kept stirring at 80 °C for another two hours. During polymerization samples were collected to analyse molar mass and particle size distributions.
Results and discussion
Synthesis and characterisation of anionic co-oligomers
Anionic co-oligomers with different combinations of acrylic acid and butyl acrylate units were synthesized using ATRP (Table 1).| Mna (g mol−1) | Đa | nb | mb | Xnc | CMCd (mM) | iepe (mM) | |
|---|---|---|---|---|---|---|---|
| a Determined by SEC against PS standards.b Determined by 1H NMR.c Number-average degree of polymerization, Xn = n + m.d Determined using scattering intensity from DLS measurements.e Determined by ζ potential measurements. | |||||||
| BA4-co-AA8 | 1.4 × 103 | 1.15 | 4.0 | 8.3 | 12.3 | 0.067 | 0.007 |
| BA8-co-AA16 | 2.3 × 103 | 1.13 | 8.0 | 16.0 | 24.0 | 0.081 | 0.008 |
| BA16-co-AA32 | 4.5 × 103 | 1.28 | 16.2 | 32.3 | 48.5 | 0.100 | 0.013 |
| BA5-co-AA5 | 1.2 × 103 | 1.17 | 5.1 | 5.3 | 10.4 | 0.043 | 0.005 |
| BA20-co-AA20 | 4.2 × 103 | 1.30 | 20.0 | 20.2 | 40.2 | 0.078 | 0.011 |
Estimates for the (effective) critical micelle concentrations of the obtained co-oligomers were determined by measuring the scattering light intensity as a function of co-oligomer concentration.25 Solutions of different co-oligomer concentrations were prepared by dilution of the most concentrated one. The co-oligomer concentration at which the slope of the scattering intensity vs. concentration changes was used to estimate the CMC (see Fig. 1). It should be noted here that due to the molar mass and chemical composition distributions of the prepared co-oligomers we are studying mixed micelles and that the obtained values for the CMC are only rough estimates.
 | ||
| Fig. 1 Scattering intensity from DLS measurement as a function of anionic co-oligomer concentration for BA4-co-AA8. | ||
Values in the range of 0.04 to 0.1 mM (depending on average co-oligomer composition and chain length) were found and are listed in Table 1. In general it can be concluded that the CMC increases with increasing m/n and with increasing n + m at constant m/n, as expected.25,29
Adsorption of these anionic co-oligomers and subsequent encapsulation experiments were carried out at pH = 7, because Gibbsite platelets are completely cationic26 and complete deprotonation of the carboxylic groups in the anionic co-oligomer takes place at pH ≈ 7.30
In order to establish the co-oligomer amount required for the Gibbsite platelets stabilisation, the dependence of the ζ potential and the Z-average diameter on the amount of anionic co-oligomer was studied (see Fig. 2a). Similar to what was observed in previous studies,9,20–24,31 the addition of positively charged Gibbsite platelets to negatively charged co-oligomer results in a decrease in ζ potential (which levels out at higher co-oligomer concentrations) and colloidal instability at the isoelectric point (iep, ζ = 0 mV) as evidenced by a dramatic increase in Dz.
 | ||
| Fig. 2 (a) Effect of anionic co-oligomer concentration on Dz (closed symbols) and ζ (open symbols) of the Gibbsite platelets and (b) isoelectric point as a function of average number of acrylic acid units at pH = 7. Used co-oligomers: (■) BA4-co-AA8, (●) BA8-co-AA16, (★) BA16-co-AA32, (▼) BA5-co-AA5 and (◆) BA20-co-AA20. Note: 1 mg of anionic co-oligomer BA4-co-AA8 corresponds to 5.7 × 10−6 mol of COOH (calculated from Mn = 1.4 × 103 g mol−1 and n(COOH) = 8). | ||
From these ζ-potential measurements the ieps of the different anionic co-oligomers were determined (see ESI†) and the obtained values are listed in Table 1. When comparing these values for the different co-oligomers it was noticed that the co-oligomer concentration at the isoelectric point (ζ = 0 mV) increased with increasing average number of acrylic acid units (Fig. 2b). This observation can be explained by the fact that longer hydrophilic chains will lead to more loop and tail structures on the Gibbsite surface.32,33 Therefore, a higher concentration of long chain co-oligomers is required to reach the isoelectric point.
As in previous studies excellent results in the encapsulation experiments were obtained using a large excess of anionic co-oligomer,9,24 we took the same approach in the current work and used 300 mg of co-oligomer per g of Gibbsite.
Preparation and characterisation of polymer–Gibbsite nanocomposites
Encapsulation experiments were carried out by slowly feeding monomer to the Gibbsite dispersion. Conversion and molar mass distributions were monitored during the polymerization and the results are shown in Fig. 3. | ||
| Fig. 3 (a) Overall conversion vs. time plot (dotted line is monomer feeding profile) and (b) molar mass distributions (scaled to monomer conversion) with overall monomer conversion. Optimized polymerisation conditions: V = 25 mL, T = 80 °C, mGibbsite = 0.02 g, [BA4-co-AA8] = 0.05 mM, VA-086 = 2 wt% based on monomer weight, 4.5 g of monomer mixture ([MMA]:[BA] = 10:1 w/w) was fed at a rate of 9 mg min−1. | ||
From Fig. 3a it is clear that since the overall conversion is the same as monomer feeding profile, we have an instantaneous conversion of 100% and truly starved-feed conditions, which is a prerequisite for efficient encapsulation.34 The molar mass distributions shown in Fig. 3b are typical for a conventional emulsion polymerization. At low M a small peak is observed which corresponds to the anionic co-oligomer. As expected, this peak remains unchanged during the polymerization (even although these co-oligomers were prepared via ATRP, without a catalyst these chains are inactive and will not be chain extended). Overall, the results in Fig. 3 are fully consistent with a conventional starved-feed emulsion polymerization in which the anionic co-oligomers act as efficient stabilizers during the polymerization.
The evolution of the particle morphology during the polymerization was studied using TEM and the results are shown in Fig. 4.
 | ||
| Fig. 4 TEM images of polymer-Gibbsite latex particles for increasing conversion. Conditions as in Fig. 3. | ||
It is clear from these images that the Gibbsite platelets are completely covered by a polymer layer during the course of the polymerization. It should be noted here that platelets with an orientation perpendicular to the electron beam are hard to see because of a reduced diffraction contrast.8 Careful particle counting (>100 particles) showed that all Gibbsite platelets were encapsulated. Only for a very small fraction of particles we could not clearly determine the presence/absence of Gibbsite platelets.
Having established the stabilizing role of the anionic co-oligomer, we studied its effect on Dz and ζ-potential. In Fig. 5 we show the results for Dz and ζ-potential as a function of monomer conversion, expressed here as the overall amount of polymer per g of Gibbsite, for two different co-oligomer concentrations. It is clear from the data in this figure that the particle size is not affected by the co-oligomer amount, but that the ζ-potential is; the lower the amount of co-oligomer, the lower the ζ-potential (in an absolute sense).
 | ||
| Fig. 5 Evolution of (filled symbol) Dz and (empty symbol) ζ-potential at different anionic co-oligomer concentrations: (■) 0.02 mM and (▲) 0.05 mM. Used co-oligomer: BA4-co-AA8. Other conditions as in Fig. 3. | ||
As expected, the particle size increases with increasing polymer content and the fact that there is no effect of the co-oligomer concentration on the size means that there are equal numbers of particles in both systems; this is indeed what is expected if all Gibbsite platelets act as seed for the polymerization and there is no secondary nucleation. This, in turn, explains the observed trend in ζ-potential. The observed decrease in (absolute) ζ-potential is caused by an increase in surface area to be stabilized and, most likely, the fact that the stabilizing moieties are being buried under the forming polymer chains;9,24 obviously in the case of the higher co-oligomer concentration higher (absolute) ζ-potentials are obtained as there are more anionic stabilizers available for the stabilization of the same surface area.
The observed reduction in (absolute) ζ-potential also implies that for a given co-oligomer concentration there exists a maximum amount of polymer per platelet that can be stabilized in a fully batch process before colloidal instability occurs. This is indeed what we observed when we continued feeding the monomer mixture, the latex coagulated. We therefore increased the co-oligomer concentration further from those used in the experiments shown in Fig. 5 (and above the CMC). We did, however, not observe an increase in the amount of polymer per Gibbsite platelet, but the formation of empty latex particles (see ESI†); the amount of empty particles increases with increasing co-oligomer amount as is shown in Fig. 6. This result was not completely unexpected as it is known that in more conventional approaches, efficient encapsulation requires using conditions at free surfactant concentration < CMC to avoid secondary nucleation.34 This is exactly what we observe in our current studies.
 | ||
| Fig. 6 Effect of anionic co-oligomer concentration on fraction of empty particles (%), as determined by particle counting (>100 particles). Used co-oligomer: (■) BA4-co-AA8. Conditions: V = 25 mL, T = 80 °C, mGibbsite = 0.02 g, VA-086 = 2 wt% based on monomer weight, 4.5 g of monomer mixture was fed at a rate of 9 mg min−1. | ||
Overall we can conclude that encapsulation experiments should be carried out using co-oligomer concentrations larger than the minimum co-oligomer concentration required to maintain colloidal stability of the initial Gibbsite platelets (∼0.01 mM under the current conditions) while maintaining the free surfactant concentration below CMC (0.067 mM, so overall ∼0.08 mM under the current conditions). Increasing polymer contents can potentially be obtained by using a co-oligomer feed strategy which ensures that the free co-oligomer concentration remains below CMC. These studies lie beyond the scope of the current paper and are the subject of future work.
Effect of chain length and hydrophilic–lipophilic balance of the anionic co-oligomers
As shown above, the chain length of anionic co-oligomer affects adsorption of the co-oligomer onto the Gibbsite platelets (Fig. 2b) and this in turn could potentially affect the encapsulation process. We therefore studied the effects of chain length and ratio of BA to AA units in the co-oligomer on the resulting composite latex morphology. Encapsulation experiments were carried out using the same conditions as before and the obtained latexes had very similar properties as those before (see Table 2 for Dz and ζ-potential).In all cases, full encapsulation of the Gibbsite platelets occurred as is clearly shown by the TEM images in Fig. 7.
 | ||
| Fig. 7 Cryo-TEM images of final polymer–Gibbsite latex particles obtained by using different anionic co-oligomers. Used co-oligomers: (a) BA4-co-AA8, (b) BA8-co-AA16, (c) BA16-co-AA32, (d) BA5-co-AA5 and (e) BA20-co-AA20. Conditions as in Fig. 3. Please note that the “small particles” in (a–e) are caused by some ice contamination. | ||
It is clear from the results in Fig. 7 and Table 2 that successful encapsulation of Gibbsite is possible for all investigated co-oligomers (with differing AA/BA ratios and overall chain lengths) and that composite latexes are obtained with very similar properties. This also suggests that the possible effects of the chemical composition and molar mass distributions of the prepared co-oligomers are of minor (if any) importance and may even imply that the co-oligomers need not be synthesized by ATRP, but that conventional free-radical polymerization may be sufficient. We are currently investigating this possibility.
Conclusions
In this work, we have presented for the first time a simple procedure for the encapsulation of Gibbsite platelets, based on conventional emulsion polymerisation, without the need for modifying the Gibbsite platelets. In this strategy anionic co-oligomers were used to stabilise the initial positively charged Gibbsite platelets and the resulting nanocomposite latex particles. The main advantage of the developed approach is that it eliminated the necessity of relatively complicated and time-consuming surface modifications, which is commonly required. Using co-oligomer concentrations below CMC, but higher than those required to stabilize the Gibbsite platelets, leads to efficient encapsulation where every latex particle contains a single Gibbsite platelet, independent of the investigated anionic co-oligomer composition or chain length.Acknowledgements
The authors thank the Euro-Asian Cooperation for Excellence and Advancement, Stichting Emulsion Polymerisation and A*STAR Research Attachment Programme for financial support.References
- K. I. Garcia-Chavez, C. A. Harnandez-Escobar, S. G. Flores-Gallardo, F. Soriano-Corral, E. Saucedo-Salazar and E. A. Zaragoza-Contreras, Micron, 2013, 49, 21–27 CrossRef CAS PubMed.
- J. Faucheu, C. Gauthier, L. Chazeau, J.-Y. Cavaille, V. Mellon, F. Pardal and E. Bourgeat-Lami, Polymer, 2010, 51, 4462–4471 CrossRef CAS.
- E. Zengeni, P. Hartmann and H. Pasch, ACS Appl. Mater. Interfaces, 2012, 4, 6957–6968 CAS.
- M.-C. Wang, J.-J. Lin, H.-J. Tseng and S. Hsu, ACS Appl. Mater. Interfaces, 2012, 4, 338–350 CAS.
- X. Huang and W. J. Brittain, Macromolecules, 2011, 34, 3255–3260 CrossRef.
- A. M. van Herk, Macromol. React. Eng., 2015, 10, 22–28 CrossRef.
- Y. Reyes, P. J. Peruzzo, M. Fernandez, M. Paulis and J. R. Leiza, Langmuir, 2013, 29, 9849–9856 CrossRef CAS PubMed.
- L. B. de Paiva, A. R. Morales and F. R. V. Diaz, Appl. Clay Sci., 2008, 42, 8–24 CrossRef.
- S. I. Ali, J. P. A. Heuts, B. S. Hawkett and A. M. van Herk, Langmuir, 2009, 25, 10523–10533 CrossRef CAS PubMed.
- Y. Xi, R. L. Frost and H. He, J. Colloid Interface Sci., 2007, 305, 150–158 CrossRef CAS PubMed.
- A. Bonnefond, M. Micusik, M. Paulis, J. R. Leiza, R. F. A. Teixeira and S. A. F. Bon, Colloid Polym. Sci., 2013, 291, 167–180 CAS.
- N. Negrete-Herrera, J.-L. Putaux, L. David, F. De Haas and E. Bourgeat-Lami, Macromol. Rapid Commun., 2007, 28, 1567–1573 CrossRef CAS.
- S. Cauvin, P. J. Colver and S. A. F. Bon, Macromolecules, 2005, 38, 7887–7889 CrossRef CAS.
- E. Zengeni, P. C. Hartmann and H. Pasch, Macromol. Chem. Phys., 2013, 214, 62–75 CrossRef CAS.
- D. J. Voorn, W. Ming and A. M. van Herk, Macromol. Symp., 2006, 245–246, 584–590 CrossRef.
- D. J. Voorn, W. Ming and A. M. van Herk, Macromolecules, 2006, 39, 4654–4656 CrossRef CAS.
- B. zu Putlitz, K. Landfester, H. Fischer and M. Antonietti, Adv. Mater., 2001, 13, 500–503 CrossRef CAS.
- N. Negrete-Herrera, J.-M. Letoffe, J.-L. Putaux, L. David and E. Bourgeat-Lami, Langmuir, 2004, 20, 1564–1571 CrossRef.
- M. A. Mballa Mballa, J. P. A. Heuts and A. M. van Herk, Colloid Polym. Sci., 2012, 291, 501–513 Search PubMed.
- D. Nguyen, H. S. Zondanos, J. M. Farrugia, A. K. Serelis, C. H. Such and B. S. Hawkett, Langmuir, 2008, 24, 2140–2150 CrossRef CAS PubMed.
- D. Nguyen, C. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 346–352 CrossRef CAS.
- V. T. Huynh, D. Nguyen, C. H. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1413–1421 CrossRef CAS.
- D. Nguyen, C. H. Such and B. S. Hawkett, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 250–257 CrossRef CAS.
- O. P. Loiko, A. B. Spoelstra, A. M. van Herk, J. Meuldijk and J. P. A. Heuts, Polym. Chem., 2016, 7, 3383–3391 RSC.
- A. Muñoz-Bonilla, A. M. van Herk and J. P. A. Heuts, Macromolecules, 2010, 43, 2721–2731 CrossRef.
- A. M. Wierenga, T. A. J. Lenstra and A. P. Philipse, Colloids Surf., 1998, 134, 359–371 CrossRef CAS.
- D. M. Haddleton and C. Waterson, Macromolecules, 1999, 32, 8732–8739 CrossRef CAS.
- O. Colombani, M. Ruppel, F. Schubert, H. Zettl, D. V. Pergushov and A. H. E. Müller, Macromolecules, 2007, 40, 4338–4350 CrossRef CAS.
- M. Zamurovic, S. Christodoulou, A. Vazaios, E. Iatrou, M. Pitsialis and N. Hadjichristidis, Macromolecules, 2007, 40, 5835–5849 CrossRef CAS.
- I. Nishida, Y. Okaue and T. Yokoyama, Langmuir, 2010, 26, 11663–11669 CrossRef CAS PubMed.
- S. Sennato, F. Bordi and C. Cametti, Europhys. Lett., 2004, 68, 296–302 CrossRef CAS.
- D. L. Allara, Polym. Sci. Technol., 1980, 12B, 751–756 CAS.
- R. F. Colletti, H. S. Gold and C. Dybowski, Appl. Spectrosc., 1987, 41, 1185–1189 CrossRef CAS.
- W.-D. Hergeth, P. Starre, K. Schmutzler and S. Wartewig, Polymer, 1988, 29, 1323–1328 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterisation details. See DOI: 10.1039/c6ra16364e |
| This journal is © The Royal Society of Chemistry 2016 |