
In situ formed graphene/ZnO nanostructured composites for low temperature hydrogen sulfide removal from natural gas
Sunil P. Lonkar,
Vishnu Pillai,
Ahmed Abdala and
Vikas Mittal*
Department of Chemical Engineering, The Petroleum Institute, P.O. box 2533, Abu Dhabi, UAE. E-mail: vmittal@pi.ac.ae; Tel: +971-26075491
First published on 22nd August 2016
Abstract
Nanostructured composites of graphene and highly dispersed sub-20 nm sized ZnO nanoparticles (TRGZ) were prepared via a novel method combining freeze-drying and thermal annealing. A direct synthesis implies thermal reduction of graphite oxide and in situ ZnO formation which acted as mutual spacers preventing restacking of the graphene layers and eventual aggregation of the nanoparticles at moderate temperatures. A series of compositions with different weight ratios of ZnO nanoparticles were prepared and used as a reactive sorbent in low temperature hydrogen sulfide (H2S) removal from natural gas. The composite sorbent having a ZnO mass ratio of 45.1 wt% showed a significantly greater H2S adsorption capacity (3.46 mmol g−1) than that of pure ZnO (1.06 mmol g−1), indicating that hybridization of ZnO with grpahene significantly improved the H2S removal ability. Such enhancement was mainly attributed to the higher surface area, greater pore volume and unique morphology at the nanoscale in the graphene–ZnO hybrid.
Introduction
The combination of multi-dimensional nanomaterials often leads to the formation of multi-functional and hierarchical composites with exceptional properties. Recently, numerous studies focusing on composites of graphene with various inorganic nanostructures have attracted a great deal of interest due to a combination of novel properties and potential applications.1–4 Among these, nanohybrids of graphene with nanosized zinc oxide (ZnO) are widely studied due to their exceptional combination of properties. Graphene, a two dimensional material having a single layer of an sp2 network of carbon atoms possesses several intriguing and peculiar properties such as high charge mobility (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1), surface area (2630 m2 g−1), thermal conductivity (2000 to 5000 W m K−1) and optical properties.5 On the other hand, owing to its exceptional chemical and physical properties, zinc oxide, a functional n-type semiconductor with a wide band gap of 3.37 eV, has received significant interest in various areas of research.6 Hence, combination of these two as nanohybrids of graphene and ZnO leads to the generation of materials for variety of applications such as sensors,7,8 photocatalysis,9–11 solar cells,12,13 water treatment,14 energy storage,15,16 adsorbent materials17,18 and many more.
000 cm2 V−1 s−1), surface area (2630 m2 g−1), thermal conductivity (2000 to 5000 W m K−1) and optical properties.5 On the other hand, owing to its exceptional chemical and physical properties, zinc oxide, a functional n-type semiconductor with a wide band gap of 3.37 eV, has received significant interest in various areas of research.6 Hence, combination of these two as nanohybrids of graphene and ZnO leads to the generation of materials for variety of applications such as sensors,7,8 photocatalysis,9–11 solar cells,12,13 water treatment,14 energy storage,15,16 adsorbent materials17,18 and many more.
Hydrogen sulphide (H2S) is one of the common and undesirable sulphur component often found in natural gas, syngas, biogas and other industrial gases.19,20 Due of its highly toxicity, offensive odour and acidic nature, H2S can pose serious threats to health and environment. Moreover, trace amounts of H2S can cause catalyst poisoning and pipeline corrosion.21 Its oxidation in the atmosphere to SO2 results in an acid rain formation. Therefore, development of efficient adsorbent materials with excellent desulfurization performance is highly essential. Owing to its favourable thermodynamics in sulphidation reaction, zinc oxide (ZnO) is considered as a very effective sorbent for removal of H2S from various gas streams and has also attracted significant attention due to its thermal stability and non-pyrophoric property.22–24 ZnO reacts with H2S to form insoluble zinc sulphide.25 The mechanism involves dissociation of H2S into H+ and HS−, followed by adsorption of HS− on ionic solid such as ZnO which later converts into ZnS by proton transfer from H2S to the chemisorbed OH groups on the Zn–O surface.26 As this reaction is exothermic, a lower temperature favours the forward reaction, resulting in high efficiency of desulfurization.27 The kinetics ZnO sulphidation reaction with H2S was predicted that the ZnO–H2S reaction proceeds with surface reaction, ion migration and completed by sulphidation penetration.28 In the further advancement of the field, the hybrid nanostructures involving ZnO are of special interest in the development of solid sorbents for H2S removal as they are capable of offering high sorption capacity.29–31 The hybrid materials based on graphene and nanoscale ZnO are promising in desulfurization because of excellent surface properties of graphene and catalytic properties of ZnO nanoparticles, which can be integrated together to enhance the overall performance of the sorbent.18,32,33 These studies concluded that the degree of oxygen functional groups and surface area of graphene in conjunction with particle size and distribution of ZnO are determining factors for low temperature H2S adsorption. However, most of these studies report graphene/ZnO composite synthesis that involves chemical reduction of GO using toxic reducing agents and conditions not suitable on industrial scales. Moderate to severe synthetic conditions including use of different solvents, microwaves, etc. under variable pH conditions, temperatures and pressures were employed. Moreover, ZnO particle size in the composites is occasionally found to be ≥100 nm with non-uniform particle dispersion. An in situ growth of ZnO nanoparticles with simultaneous graphene oxide reduction to obtain graphene/ZnO nanohybrids can offer scalable and facile route for the straight forward synthesis of nanostructured graphene/ZnO composites suitable for low temperature H2S adsorption.
In this work, preparation of graphene/ZnO nanostructured composites is reported via in situ process under thermal conditions using zinc hydroxyacetate as a Zn precursor. This simple and environment friendly method can be easily scaled up to obtain high quality graphene/ZnO composite materials suitable for wide range of applications. Finally, the performance of these hybrid materials as low temperature H2S sorbent was evaluated. A series of graphene/ZnO composites were prepared and their effectiveness towards H2S adsorption was assessed and activities were compared with pristine ZnO sorbent.
Experimental
Materials
Graphite powder (Sigma Aldrich, 10 mesh), sulfuric acid (Sigma Aldrich, ACS reagent, 95.0–98.0%), hydrochloric acid (Sigma-Aldrich, ACS reagent, 37%), potassium permanganate (Fischer Scientific, ≥99%), hydrogen peroxide (Sigma-Aldrich, 30 wt% in H2O), sodium hydroxide (Sigma-Aldrich, ACS reagent, ≥97.0%), zinc acetate dehydrate (Sigma-Aldrich, ACS reagent, ≥98%) and phosphoric acid (Sigma-Aldrich, ACS reagent, ≥85 wt% in H2O), were used. Zinc hydroxyacetate was prepared by procedure presented elsewhere.34Synthesis of graphite oxide (GO)
Graphite oxide was prepared from natural graphite by using improved synthesis proposed by Tour et al.35 In brief, mixture of concentrated sulfuric acid (270 mL) and phosphoric acid (33 mL) was added to a 5 L Erlenmeyer flask placed in an ice-bath. About 5 g of natural flake graphite (10 mesh) was dispersed in the cold sulfuric acid with an overhead stirrer. Subsequently, 2.7 g of KMnO4 was added slowly over 15–20 min, and the resulting one-pot mixture was stirred for 72 h at room temperature to allow the oxidation of graphite. The colour of the mixture changed from dark purple-green to dark brown. Later, about 35% hydrogen peroxide (H2O2) solution was added to terminate the oxidation process, and the colour of the mixture changed to bright yellow, indicating a high oxidation level of graphite. As-synthesized graphite oxide was suspended in water containing 1 M dilute hydrochloric acid to give a yellow-brown dispersion, which was subjected to repeated washing with de-ionized water until a pH of 4–5 was achieved. To ensure complete removal of the residual salts and acids, dialysis process was used.Preparation of graphene/ZnO nanohybrids (TRGZ)
Aqueous dispersions of GO (200 mg) and stoichiometric quantity of zinc hydroxyacetate was prepared under ultra-sonication for 30 min and rapidly mixed at room temperature in a round-bottomed flask followed by stirring. The resulting homogenous mixture was freeze-dried at −90 °C to obtain GO–Zn salt composite. Further, the composites were thermally annealed in a tube furnace at 400 °C for 2 h under argon atmosphere with a heating rate of 5 °C min−1 to finally obtain thermally reduced graphene oxide (TRG)/ZnO composite (Scheme 1). A colour change from light brown to black was also noticed. Stoichiometric quantity of zinc hydroxyacetate viz. 18.7 mg, 56.4 mg and 94 mg was used in order to obtain TRG/ZnO composites with 10, 30, and 50 wt% ZnO loading and abbreviated as TRGZ-1, TRGZ-2 and TRGZ-3, respectively. The pristine ZnO nanoparticles were prepared by thermal annealing of zinc hydroxyacetate following the similar procedure that was used for TRGZ hybrids.34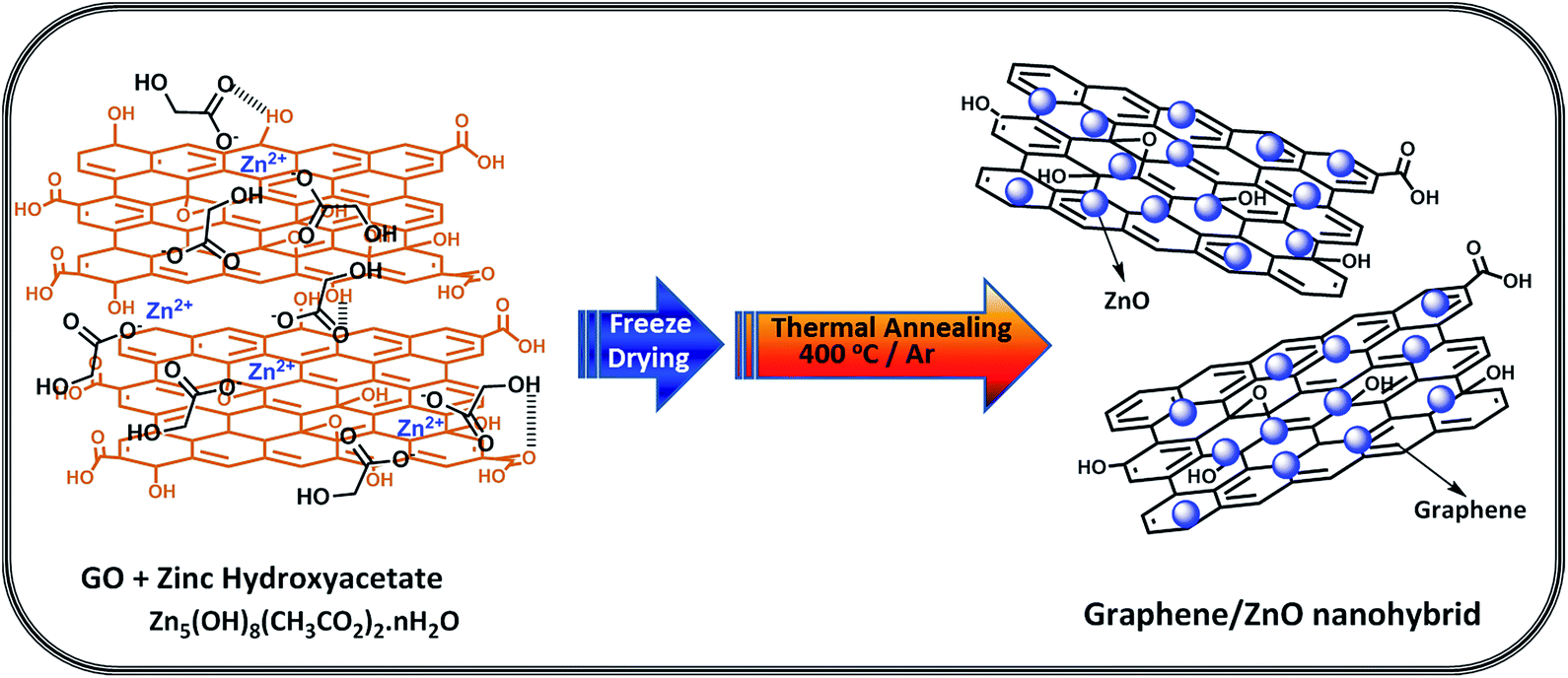 | ||
| Scheme 1 Schematic representation of interaction between GO and zinc hydroxyacetate and TRG/ZnO nanohybrid synthesis. | ||
Characterization
TRG/ZnO nanohybrids were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), scanning electron microscopy (SEM), Raman spectroscopy, Fourier transform infra-red spectroscopy (FT-IR) thermogravimetric analysis (TGA) and N2 physisorption. XRD was performed using CuKα radiation (X'Pert Pro X-ray diffractometer from Philips) at angle range (2θ 5–60°). The XPS measurements were performed on an SSX-100 system (Surface Science Laboratories, Inc.) equipped with a monochromated Al Kα X-ray source, a hemispherical sector analyzer (HSA) and a resistive anode detector. TEM analyses were performed using FEI Tecnai G20 with 0.11 nm point resolution and operated at 200 kV using Gatan digital camera. SEM (1540 XB Zeiss) coupled with energy dispersive X-ray analysis (EDX) was used to define the particle size and the structure of the nanohybrids. LabRAM HR (Horiba Scientific) was used to obtain Raman spectra. Typically, a 50× objective was used with 633 nm excitation line. TGA analysis was carried out on Discovery TGA (TA instruments) using the temperature range from 50 to 800 °C at a ramp rate of 10 °C in an air atmosphere (30 mL min−1). N2 physisorption was carried out at liquid N2 temperature with a Micromeritics ASPS 2010 analyser to examine the porosity and surface area of the hybrids. The samples were pre-treated at 100 °C in a high vacuum for 24 h before N2 adsorption.H2S sorption studies
H2S sorption experiments were carried out at room temperature (30 °C) and 290 psig pressure. The sorption tube was made of glass with an outer of diameter of 8 mm and the height of 20 mm, into which ∼0.5 g of the adsorbent was packed. For adsorption, a model gas mixture containing (99.4% of CH4, 0.41% of CO2 and 0.15% of H2S) was passed through the adsorption cell with a flow rate of 60 mL min−1. The gas mixture was delivered through the books mass flow controller at fixed flow rate (Fig. 1). The analysis of the breakthrough gas was performed using a quadrupole mass spectrometer. Helium was used as marker gas. The breakthrough and saturation capacity (denoted as Cap (BT), mmol g−1, STP) for H2S was calculated according to the following equation:36where BT is breakthrough time, the time (min) when the H2S concentration reached 1% (i.e. 15 ppmv); FR is the flow rate (mL min−1); Vmol is the molar volume (24.4 mL mol−1 at STP); W is the weight of the sorbent (in g) and

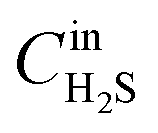 is the initial concentration of the H2S in test gas mixture, respectively. The point at which H2S concentration reached 0.1% of initial concentration was considered as breakthrough point.
is the initial concentration of the H2S in test gas mixture, respectively. The point at which H2S concentration reached 0.1% of initial concentration was considered as breakthrough point.
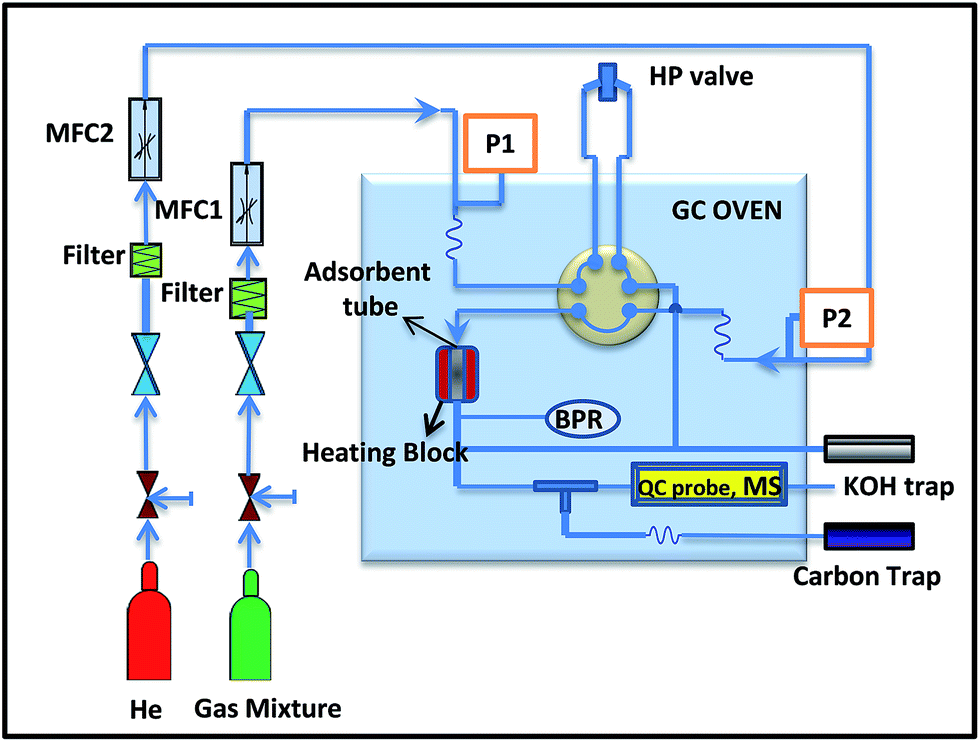 | ||
| Fig. 1 Schematic representation of the fixed-bed flow system for adsorption measurements. | ||
Results and discussion
Morphology
As illustrated in Scheme 1, various functional groups (e.g., carbonyl, hydroxyl, and epoxy groups) on GO surface can attach zinc hydroxyacetate through the electrostatic attraction to become the nuclear site of a “seed” on GO support. On thermal treatment, GO can undergo deoxygenation and exfoliation to obtain thermally reduced graphene oxide (TRG) with controlled residual oxygenated groups and large specific surface area.37 Simultaneously, zinc salt gradually decomposes into the ZnO seeds which can interact with active sites that are generated on the surface of the TRG, which after prolonged thermal annealing converts into uniformly dispersed nanostructured composites (TRGZ). It was expected that the gases released during thermal decomposition of zinc salt can also contribute to the exfoliation of GO to TRG and in return the large graphitic sheets can control the ZnO size at nanoscale.The morphology of the resulting nanohybrids was elucidated using TEM and SEM analysis. The typical TEM images of ZnO nanoparticles and TRGZ composites are shown in Fig. 2. It can be seen that the thermal annealing of zinc hydroxyacetate under controlled conditions resulted into hexagon shaped ZnO nanoparticles with average size of ∼100 nm. A significant change in morphology from hexagonal to spherical was observed after hybridizing with graphene. Also, a substantial decrease in the size of ZnO particles was noted in presence of graphene, thus, confirming its control on the size and dispersion of the ZnO seeds. From the TEM images, it could be observed that a large number of uniformly dispersed ZnO nanoparticles with average size below 20 nm were formed on the surface of graphene. For TRGZ-1, the average size of ZnO particles was below 10 nm with more isolated particulates which can be attributed to the lower Zn salt concentration.
 | ||
| Fig. 2 TEM images of (a) pristine ZnO nanoparticles, (b) TRGZ-1, (c) TRGZ-2, and (d) TRGZ-3 graphene/ZnO nanohybrids. | ||
For higher ZnO loaded composites namely, TRGZ-2 and TRGZ-3, a slight increase in ZnO size as well as number of particles was observed. At higher Zn salt concentrations, though the size controlling effect of graphene layers was found to reduce, however, the distribution and average size were still uniform. Overall, the TEM images clearly revealed that the nanostructured TRGZ composites with a uniform ZnO dispersion were successfully prepared. Further, the average crystalline size of the pristine ZnO nanoparticles and in situ formed graphene supported ZnO nanoparticles was estimated from Scherrer's formula.38 It was found that the in situ generated ZnO nanoparticles in TRGZ composites forms smaller crystallites in compared to the pristine ZnO (23.4 nm). Amongst, TRGZ composites, TRGZ-1 possesses smallest ZnO crystallites size (10.9 nm) and increases with the ZnO content for TRGZ-2 (14.4 nm) and TRGZ-3 (15.8 nm), respectively. These results are in good agreement with above mentioned TEM results.
As shown in Fig. 3, SEM analyses also confirmed the nanostructured morphology of the TRGZ composites. Uniform nano-assemblies of ZnO were visible in case of highly loaded TRGZ composites. The Zn elemental mapping (Fig. 3 inset) clearly indicated the uniform dispersion of the zinc species i.e. ZnO, along with increase in density for higher ZnO loadings. Overall, the interactions between functional groups on the graphene surface and the ZnO seeds significantly contributed to the formation of the hierarchically structured TRG/ZnO hybrids.
 | ||
| Fig. 3 SEM images of from left to right, TRGZ-1, TRGZ-2 and TRGZ-3 composites (inset Zn element mapping). | ||
Structural and chemical characterization
Fig. 4A shows the XRD patterns of the ZnO and TRGZ hybrids indicating characteristic diffraction peaks primarily indexed to wurtzite structure of ZnO for all the samples.39 Similarly, the diffraction pattern of TRGZ-1 (Fig. 4B) exhibited a new broad peak at 2θ = 24.33° corresponding to the (002) peak of thermally reduced graphite oxide.40 Moreover, a diffraction peak broadening and a shift towards lower 2θ values compared with that of as prepared pristine TRG signified the intercalation of ZnO nanoparticles into TRG layers. No diffraction peaks related to TRG were detected in TRGZ-2 and TRGZ-3, implying that at higher ZnO loadings, the reduced graphene oxide sheets were completely exfoliated due to the large evolution of gases during thermal decomposition of zinc hydroxyacetate which ultimately resulted into larger ZnO nano-assemblies on TRG surface. In addition, no characteristic peaks assigned to graphite oxide (the characteristic peak at around 2θ = 9.7°) were observed in TRGZ composites, indicating the successful thermal reduction of the layered GO in the hybrids. Fig. 4C also shows the XRD profiles of GO and TRG samples. The characteristic peak at 9.7° is assigned to the (001) plane of GO, while after thermal exfoliation of GO to TRG, the diffraction peak at 9.7° disappeared and a new broad peak at 25.9° was observed, which was assigned to the graphene (002) planes.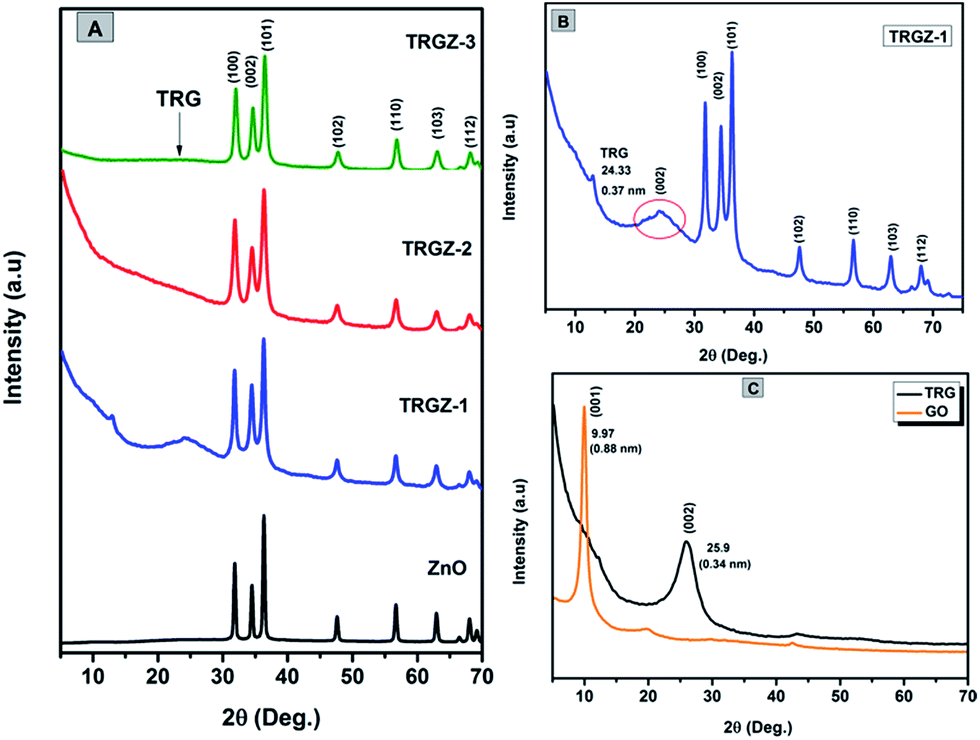 | ||
| Fig. 4 XRD diffraction patterns of (A) ZnO, TRGZ-1, TRGZ-2 and TRGZ-3 (B) TRGZ-1 and (C) GO and TRG samples. | ||
XPS was used to monitor the chemical states after thermal reduction in GO and its interaction with in situ formed ZnO nano-assemblies. Fig. 5A shows the C 1s and O 1s chemical states of GO and TRGZ hybrids.
 | ||
| Fig. 5 XPS spectra (A) C 1s region of GO and TRZ-1, (B) survey spectra of TRGZ-1, and (C) high resolution spectra of Zn 2p region, for TRGZ-1 composite. | ||
It can be seen that XPS spectra of GO can be fitted into three peaks, corresponding to carbon atoms in different functional groups: sp2 carbon (C–C, 284.9 eV), carbon in C–O bonds (287.2 eV), and carbonyl carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 288 eV), respectively.41 For TRGZ hybrids, the peak intensity of the C–C bond was significantly increased while oxygen-bonded carbons (C–O and CO) were considerably reduced, indicating a sufficient reduction of graphite oxide into graphene during the thermo-annealing process.42 Further, the survey spectra of TRG/ZnO nanohybrid (Fig. 5B) shows the characteristic peaks of Zn 2p1/2, Zn 2p3/2, O 1s, and C 1s only and no peaks of other elements were observed which ensured the single phase, contamination free in situ growth of ZnO nano-assemblies on TRG sheets. The high-resolution scan of Zn 2p, shown in Fig. 5C, identified the exact peak location of Zn 2p3/2 at 1022.6 eV. Hence, XPS findings confirmed the successful formation ZnO/TRG nanohybrids under one-step thermo-annealed process.
O, 288 eV), respectively.41 For TRGZ hybrids, the peak intensity of the C–C bond was significantly increased while oxygen-bonded carbons (C–O and CO) were considerably reduced, indicating a sufficient reduction of graphite oxide into graphene during the thermo-annealing process.42 Further, the survey spectra of TRG/ZnO nanohybrid (Fig. 5B) shows the characteristic peaks of Zn 2p1/2, Zn 2p3/2, O 1s, and C 1s only and no peaks of other elements were observed which ensured the single phase, contamination free in situ growth of ZnO nano-assemblies on TRG sheets. The high-resolution scan of Zn 2p, shown in Fig. 5C, identified the exact peak location of Zn 2p3/2 at 1022.6 eV. Hence, XPS findings confirmed the successful formation ZnO/TRG nanohybrids under one-step thermo-annealed process.
Raman spectroscopy is a powerful and non-destructive technique to extract useful information about graphene and its related materials.43 Generally, graphitic materials show the characteristic D and G bands corresponding to k-point phonons of A1g symmetry and E2g phonon of sp2 carbon which are assigned to local defects and disorder especially at the edges of graphene and graphite platelets.44 Fig. 6 shows the Raman spectra of the GO, TRG and the corresponding TRGZ hybrids with different ZnO loading. The pristine GO exhibited G and D bands at Raman shifts of 1584 and 1333 cm−1, respectively, with an intensity ratio, ID/IG = 1.42. These bands shifted to 1349 and 1589 cm−1 after the thermal treatment of GO. Also, trivial increase in ID/IG was observed which indicated a decrease in the size of the in-plane sp2 domains, which was mainly attributed to the removal of the oxygen functional group in GO during thermal reduction process.42 For TRG/ZnO composites, further increase in ID/IG ratio was noted which signified the simultaneous GO reduction and ZnO nanoparticle formation implying successful synthesis of TRG/ZnO hybrids. Fig. 7 also shows the FT-IR spectra of the as-prepared GO, TRG, ZnO and TRGZ samples. GO exhibited characteristic IR peaks at 1620 cm−1 corresponding to the remaining sp2 character, the absorption peaks at 1722 cm−1 and 1392 cm−1 were ascribed to CO stretching of COOH groups, tertiary C–OH groups vibrations and epoxy symmetrical ring deformation vibrations, respectively.45 Furthermore, the broad peak at 3330 cm−1 was attributed to the hydroxyl stretching vibrations of the C–OH groups, and the band at 1050 cm−1 was assigned to C–O stretching vibrations mixed with C–OH bending.
 | ||
| Fig. 6 Raman spectra of GO, TRG and TRG/ZnO composites. | ||
 | ||
| Fig. 7 FT-IR spectra of GO, TRG, ZnO and TRG/ZnO composites. | ||
In TRG, the characteristic features of GO almost disappeared, indicating the reduction of GO to graphene under thermal treatment. For ZnO, the absorption band at 440 cm−1 resulted due to stretching modes of Zn–O.34 In the FT-IR spectrum of TRGZ composites, only characteristic bands corresponding to the TRG and ZnO were reflected indicating the formation of TRG/ZnO nanohybrids. Further, absence of characteristic absorbance of CH3COO− assigned to raw material zinc hydroxyl acetate confirmed the successful preparation of TRGZ composites. The increment in the intensity of Zn–O peak with the content of ZnO was also noted for these composites.
TGA was used to determine ZnO loading and thermal stability of the resulting TRG/ZnO nanostructured composites under oxidative environment. As shown in Fig. 8, all the composites exhibited weight loss weight around 400 °C which was attributed to the removal of oxygen-containing groups and the decomposition of carbon framework of graphene from the composites. The thermal stability of graphene in case of TRGZ-2 was improved which is attributed to the higher loading and better dispersion of nanosized ZnO. Based on the residues and considering weight loss in pristine ZnO, the weight percentage of ZnO in composites was calculated to be TRGZ-1 (8.89%), TRGZ-2 (25.34%) and TRGZ-3 (45.13%), respectively.
 | ||
| Fig. 8 TGA thermograms of GO, TRG, ZnO and TRG/ZnO composites. | ||
N2 adsorption–desorption isotherms were used to investigate microstructure and exposed surface area in the resulting TRGZ composite materials. As shown in Fig. 9, ZnO and TRGZ composites yielded a type-IV isotherm with a hysteresis loop in the relative pressure range of 0.8–1.0, indicating the presence of inhomogeneous mesopores. The pore size distribution curve calculated from the adsorption branch by the BJH method displayed relatively small pores with a typical size of 1–5 nm. Also, it was observed that the TRGZ-3 composite had a BET surface area of 119.3 m2 g−1, which is almost 10 times higher than that of the pure ZnO nanoparticles (12.4 m2 g−1). The higher surface area for TRGZ composite could be attributed to the presence of high surface area TRG support, leading to more surface active sites and the increased adsorption of reactants during the composite formation. The surface area and pore properties for other composites are tabulated in Table 1.
 | ||
| Fig. 9 BET N2 isotherms for ZnO, TRG and TRG/ZnO hybrids. | ||
| Nanohybrids | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | BJH pore diameter (Å) |
|---|---|---|---|
| ZnO | 12.4 | 0.083 | 31.25 |
| TRG | 223.3 | 0.93 | 39.50 |
| TRGZ-1 | 256.7 | 1.23 | 39.25 |
| TRGZ-2 | 207.9 | 1.35 | 37.80 |
| TRGZ-3 | 119.3 | 0.80 | 38.05 |
H2S adsorption breakthrough tests
As prepared TRG/ZnO nanostructured sorbents were evaluated for removal of H2S at room temperature (30 °C) in the presence of CH4 and compared with that of the pristine ZnO nanoparticles and TRG under similar conditions. A dynamic H2S breakthrough test was used (Fig. 1) and the resulting breakthrough curves for tested adsorbents are shown in Fig. 10. The breakthrough capacities for the samples are tabulated in Table 2. Firstly, to gain insights about the contribution of CH4 sorption on the material, CH4 breakthrough curve of TRGZ-3, was measured under the same conditions mentioned above. The results indicated that saturation of the sorbent bed was completed in only 10 min.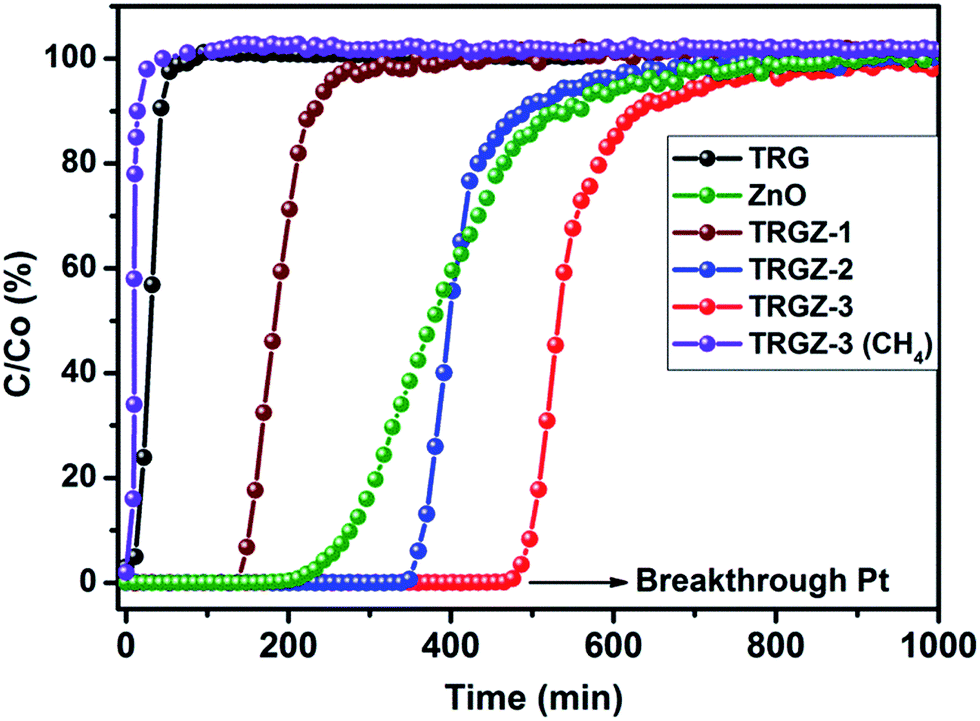 | ||
| Fig. 10 H2S breakthrough curves for TRG/ZnO composites treated at ambient conditions with different ZnO ratio and CH4 breakthrough curves for TRGZ-3 composite. | ||
| Sample | H2S Cap (BT) (mmol per g-sorb) | H2S Cap (BT) (mg H2S per g-sorb) |
|---|---|---|
| TRG | 0.09 | 3 |
| TRGZ-1 | 1.06 | 35 |
| TRGZ-2 | 2.68 | 88 |
| TRGZ-3 | 3.46 | 114 |
| ZnO | 1.63 | 53 |
This indicated that CH4 adsorption on the adsorbent was negligible. A remarkable difference in H2S breakthrough curves was observed for the TRGZ hybrid composites as compared to pristine ZnO nanoparticles (Fig. 10). TRGZ-3 and TRGZ-2 sorbents exhibited highest H2S breakthrough capacity of 114 mg per g-sorbent and 88 mg per g-sorbent, respectively, as compared to 53 mg per g-sorbent for pristine ZnO. It was noteworthy that TRGZ composites contained a lesser amount of the active component, ZnO. Amongst TRGZ composites, TRGZ-1 had the lowest H2S sorption performance, as its breakthrough capacity was measured to be 35 mg per g-sorbent. As expected, pure TRG showed negligible breakthrough capacity at 3 mg per g-sorbent due to absence of any active site for H2S adsorption. The high H2S sorption capacities for TRGZ-2 and TRGZ-3 sorbents can be attributed to the sub-20 nm ZnO particles which enhanced the reactivity with H2S at low temperature in conjunction with the high surface area of TRG which provided more active sites. Also, the residual oxygen-containing functional groups on the basal planes of TRG (as confirmed by XPS and FT-IR results) play a critical role in promoting oxygen activation by accelerating the electron transfer, thereby promoting the activity of the terminal groups in surface reaction.46 In addition, these functional groups help the distribution of active ZnO particles on the surface. So the possible mechanism involves the initial physisorption of H2S molecules by oxygenated functional groups of graphene (Fig. 11I) which later can reach to the finely dispersed active ZnO nanoparticles and get chemisorbed through reactive adsorption (Fig. 11II).47,48
 | ||
| Fig. 11 Schematic representation for possible H2S adsorption on TRGZ sorbent. | ||
In present work, the higher H2S sorption by TRGZ-3 (mg per g sorbent) over pristine ZnO is attributed to the considerably large surface area (190.3 m2 g−1), which was almost 15 times that of ZnO (Table 1). Moreover, the pore volume of TRGZ-3 (0.80 cm3 g−1) was higher than that of ZnO (0.083 cm3 g−1). Whereas, TRGZ-2 possess higher surface properties over TRGZ-3 but showed lower H2S capacity (88 mg per g sorbent).
This difference in adsorption behaviour further indicate that not only high surface area, but also the content of nanosized ZnO active component is determining factor in low temperature H2S adsorption by using graphene/ZnO nanostructured adsorbents. The H2S breakthrough curve for ZnO was significantly broad and might have high saturation capacity as compared to the TRGZ composites due to the differences in pore structure. As the sorption of sulphur compound by metal oxides is a process of volume expansion, the sorbent becomes more compact and even pore closure occurs in some severe cases during the reaction.49 This prevents gaseous reactive molecules from reaching the interior fresh active sites, leading to a very low uptake and utilization of sorbent, especially at ambient temperature. In contrast, TRGZ sorbents due to larger surface areas and smaller size ZnO nanoparticles along with high pore volumes did not suffer from this and exhibited improved H2S adsorption. Overall, the higher amount of the ZnO active component in conjunction with high surface area and good dispersion due to the graphene layers are the critical factors for enhanced low temperature H2S adsorption.
Further, the EDX spectra of H2S treated TRGZ composites were recorded to monitor the changes in elemental composition, as shown in Fig. 12 for TRGZ-3 hybrid. The EDX spectrum exhibited a strong peak for sulphur element indicating the reactive adsorption of H2S on the sorbent. The quantitative results of the S/Zn ratio were calculated from the area of the corresponding spectral K lines and the amount of S in the composites was observed to be around 10 wt% which was in agreement with the breakthrough calculations. The sulphur element mapping image (inset in Fig. 12) also conformed the reactivity of the uniformly dispersed ZnO. However, to further elaborate this study, it would be interesting to investigate the reactivity of these adsorbents at higher temperatures which is under investigation.
 | ||
| Fig. 12 EDX spectrum of TRGZ-3 and sulfur elemental mapping (inset) after H2S adsorption. | ||
Conclusions
In summary, a simple one-step, scalable method was demonstrated to prepare TRG-ZnO composites with formation of sub-20 nm ZnO particles uniformly dispersed on the graphene surface. The exfoliated structure in graphene and nanosized morphology of ZnO could be controlled through mutual interactions during the in situ synthesis process. Due to nano-porous hierarchical structure with large specific surface area and incorporation of the active ZnO nanoparticles, the resultant hybrid materials also showed substantial H2S adsorption at room temperature. TRGZ composite with 45.1 wt% ZnO exhibited almost double the H2S adsorption capacity as compared to pure ZnO nanoparticles prepared under similar conditions. Hence, considering nanosized ZnO can enhance the reactivity with H2S at low temperature and high surface areas can provide more active sites, this study leads to generation of sorbent materials for effective H2S removal.Acknowledgements
Authors acknowledge Mr Samuel Stephen, Ms Anjana Tharalekshmy and Ms Sabna Khadar for their assistance in TEM and BET, Raman, SEM analysis, respectively.References
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483–3507 CAS.
- J. Zhu, M. Chen, Q. He, L. Shao, S. Wei and Z. Guo, RSC Adv., 2013, 3, 22790–22824 RSC.
- S. Wang, J. Li, X. Zhou, C. Zheng, J. Ning, Y. Zhong and Y. Hu, J. Mater. Chem. A, 2014, 2, 19815–19821 CAS.
- G. Cheng, X. Yu, M. D. Zhou and S. Y. Zheng, J. Mater. Chem. B, 2014, 2, 4711–4719 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- A. Kolodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef CAS.
- K. Y. Hwa and B. Subramani, Biosens Bioelectron, 2014, 62, 127–133 CrossRef CAS PubMed.
- A. T. E. Vilian, M. Rajkumar, S. M. Chen, C. C. Hu and S. Piraman, RSC Adv., 2014, 4, 48522–48534 RSC.
- S. Ameen, M. S. Akhtar, H. K. Seo and H. S. Shin, Mater Lett, 2013, 100, 261–265 CrossRef CAS.
- B. Weng, S. Q. Liu, Z. R. Tang and Y. J. Xu, RSC Adv., 2014, 4, 12685–12700 RSC.
- Y. Liu, Y. Hu, M. Zhou, H. Qian and X. Hu, Appl. Catal., B, 2012, 125, 425–431 CrossRef CAS.
- J. Chen, C. Li, G. Eda, Y. Zhang, W. Lei, M. Chhowalla, W. I. Milne and W. Q. Deng, Chem Commun, 2011, 47, 6084–6086 RSC.
- Z. Y. Zhan, L. X. Zheng, Y. Z. Pan, G. Z. Sun and L. Li, J Mater Chem, 2012, 22, 2589–2595 RSC.
- D. Y. Fu, G. Y. Han, Y. Z. Chang and J. H. Dong, Mater Chem Phys, 2012, 132, 673–681 CrossRef CAS.
- C. T. Hsieh, J. S. Lin, Y. F. Chen, C. Y. Lin and W. Y. Li, Mater Chem Phys, 2014, 143, 853–859 CrossRef CAS.
- A. Ramadoss and S. J. Kim, Mater Chem Phys, 2013, 140, 405–411 CrossRef CAS.
- J. F. Wang, T. Tsuzuki, B. Tang, X. L. Hou, L. Sun and X. G. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 3084–3090 CAS.
- H. S. Song, M. G. Park, E. Croiset, Z. W. Chen, S. C. Nam, H. J. Ryu and K. B. Yi, Appl Surf Sci, 2013, 280, 360–365 CrossRef CAS.
- D. Panza and V. Belgiorno, Process Saf. Environ. Prot., 2010, 88, 420–424 CrossRef CAS.
- L. C. Yang, X. M. Ge, C. X. Wan, F. Yu and Y. B. Li, Renewable Sustainable Energy Rev., 2014, 40, 1133–1152 CrossRef CAS.
- W. R. P. J. Kidnay and D. G. McCartney, Fundamentals of Natural Gas Processing, CRC Press, Boca Raton, FL, 2nd edn, 2011 Search PubMed.
- H. F. Garces, H. M. Galindo, L. J. Garces, J. Hunt, A. Morey and S. L. Suib, Microporous Mesoporous Mater., 2010, 127, 190–197 CrossRef CAS.
- H. F. Garces, A. E. Espinal and S. L. Suib, J Phys Chem C, 2012, 116, 8465–8474 CAS.
- I. Rosso, C. Galletti, M. Bizzi, G. Saracco and V. Specchia, Ind Eng Chem Res, 2003, 42, 1688–1697 CrossRef CAS.
- A. Samokhvalov and B. J. Tatarchuk, Phys Chem Chem Phys, 2011, 13, 3197–3209 RSC.
- O. Mabayoje, M. Seredych and T. J. Bandosz, J. Colloid Interface Sci., 2013, 405, 218–225 CrossRef CAS PubMed.
- C. H. Shao, A. X. Jiang, F. Li, B. Yan and B. B. Zhou, Chin. J. Inorg. Chem., 2005, 21, 1149–1154 CAS.
- J. Sun, S. Modi, K. Liu and R. Lesieur, Energy Fuels, 2007, 21, 1863–1871 CrossRef CAS.
- M. Mureddu, I. Ferino, E. Rombi, M. G. Cutrufello, P. Deiana, A. Ardu, A. Musinu, G. Piccaluga and C. Cannas, Fuel, 2012, 102, 691–700 CrossRef CAS.
- R. Portela, F. Rubio-Marcos, P. Leret, J. F. Fernandez, M. A. Banares and P. Avila, J. Mater. Chem. A, 2015, 3, 1306–1316 CAS.
- M. Dancila, G. Voicu and E. Vasile, Rev. Chim., 2014, 65, 1521–1524 CAS.
- H. S. Song, M. G. Park, W. Ahn, S. N. Lim, K. B. Yi, E. Croiset, Z. Chen and S. C. Nam, Chem. Eng. J., 2014, 253, 264–273 CrossRef CAS.
- H. S. Song, M. G. Park, S. J. Kwon, K. B. Yi, E. Croiset, Z. Chen and S. C. Nam, Appl Surf Sci, 2013, 276, 646–652 CrossRef CAS.
- A. Moezzi, A. McDonagh, A. Dowd and M. Cortie, Inorg Chem, 2013, 52, 95–102 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- X. X. Wang, X. L. Ma, X. C. Xu, L. Sun and C. S. Song, Top Catal, 2008, 49, 108–117 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- R. N. Moussawi and D. Patra, RSC Adv., 2016, 6, 17256–17268 RSC.
- H. M. Ju, S. H. Choi and S. H. Huh, J. Korean Phys. Soc., 2010, 57, 1649–1652 CrossRef CAS.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- D. Zhan, Z. H. Ni, W. Chen, L. Sun, Z. Q. Luo, L. F. Lai, T. Yu, A. T. S. Wee and Z. X. Shen, Carbon, 2011, 49, 1362–1366 CrossRef CAS.
- M. Cheng, R. Yang, L. C. Zhang, Z. W. Shi, W. Yang, D. M. Wang, G. B. Xie, D. X. Shi and G. Y. Zhang, Carbon, 2012, 50, 2581–2587 CrossRef CAS.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett, 2007, 7, 238–242 CrossRef CAS PubMed.
- J. Song, X. Wang and C.-T. Chang, J. Nanomater., 2014, 2014, 276143 Search PubMed.
- M. Seredych, O. Mabayoje and T. J. Bandosz, Langmuir, 2012, 28, 1337–1346 CrossRef CAS PubMed.
- O. Mabayoje, M. Seredych and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2012, 4, 3316–3324 CAS.
- C. Petit, B. Mendoza and T. J. Bandosz, ChemPhysChem, 2010, 11, 3678–3684 CrossRef CAS PubMed.
- L. Neveux, D. Chiche, D. Bazer-Bachi, L. Favergeon and M. Pijolat, Chem. Eng. J., 2012, 181, 508–515 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |