
Asymmetric catalytic sulfoxidation by a novel VIV8 cluster catalyst in the presence of serum albumin: a simple and green oxidation system†
Jie Tangac,
Peng-Fei Yaoa,
Xiao-Ling Xua,
Hai-Ye Lia,
Fu-Ping Huang*a,
Qing-Qing Niea,
Mei-Yi Luoc,
Qing Yua and
He-Dong Bian*ab
aState Key Laboratory for the Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmacy, Guangxi Normal University, Guilin, 541004, P. R. China. E-mail: huangfp2010@163.com
bSchool of Chemistry and Chemical Engineering, Guangxi University for Nationalities, Key Laboratory of Chemistry and Engineering of Forest Products, Nanning, 530008, P. R. China. E-mail: gxunchem@163.com
cGuilin Normal College, Guilin 541001, P. R. China
First published on 28th April 2016
Abstract
A novel VIV8 cluster formulated as [V8O12(OH)4(CH3O)4(DAC)4]·7CH3OH (1) (DAC = 1,2-diaminocyclohexane) has been constructed successfully. Enantioselective oxidation of a series of alkyl aryl sulfides catalyzed by 1 is tested in an aqueous medium in the presence of serum albumin. The catalytic procedure is found to be simple and environmentally friendly. The influences of the parameters such as concentration of catalyst and oxidant, pH, and reaction time on the thioanisole as models are investigated. Under optimum conditions, 1 exhibits high conversion (up to 99%), excellent chemoselectivity (≥90% in all cases) and moderate enantioselectivity (up to 75% ee). After binding with serum albumin, the catalytic activity of 1 is promoted. The bovine serum albumin (BSA) and pig serum albumin (PSA) molecules have a more positive effect on the catalytic activity.
Introduction
Polyoxometalates (POMs) stimulate many current research activities in broad fields of science, such as catalysis,1–7 materials,8 medicine,9 magnetism,10–13 and photochemistry.14,15 This can be attributed to their favorable chemical and physical properties, such as redox potentials,16–18 acidities,2 solubility in various media,19 and impressive sensitivity to light and electricity.20 Notably, their thermal,8 hydrolytic,21 and oxidative redox stabilities22 make them competitive candidates to rival organometallic complexes and enzymes for deployment in organic oxidation processes.23–25The use of POMs for carrying out asymmetric catalysis26 remains a major challenge because most POM anions possess high symmetry.18 To date, much effort has been devoted to exploring the preparation of chiral POM-based systems,18 as well as the enantioselectivity achievable with enantiopure POMs for asymmetric transformations.27–30 Nevertheless, there are still many shortcomings, such as low efficiency,27 instability,31 use of toxic organic solvents,28 rapid racemization in solution,31 and complicated synthetic steps to obtain POMs with chiral structures.
Serum albumins (SAs) are the major soluble protein constituents of the circulatory system and have many physiological functions.32 They are generally robust, abundant, readily accessible, inexpensive, and easy to handle.33,34 In addition, SAs have the unique property of specific binding sites for hydrophobic organic molecules,35 and as additives presenting a chiral environment36 may be capable of inducing enantioselectivity. For example, Manfred T. Reetz and Ning Jiao carried out Diels–Alder reactions in water with phthalocyanine–CuII complexes in the presence of SAs, which afforded the products with up to 98% ee.37 In situ generated dioxiranes have been used to oxidize a series of prochiral sulfides to the corresponding sulfoxides with up to 89% ee in the presence of bovine serum albumin (BSA) as a chiral auxiliary.38 However, to the best of our knowledge, few studies have demonstrated the catalysis systems based on POMs with SAs as chiral additives for obtaining enantiomerically pure products.
Chiral sulfoxides and their derivatives are an important class of compounds that are extensively used as chiral synthetic intermediates and auxiliaries for the preparation of biologically active molecules, both in academia and in industry.39 Furthermore, compounds with sulfur chirality play a crucial role in many biochemical processes40 and are widely exploited in the pharmaceutical industry.41 The selective oxidation of organic sulfides to sulfoxides without any over-oxidation to sulfones,42–44 as well as devising environmentally benign catalytic oxidations, are still key challenges in the synthesis of enantiopure sulfoxides.
Herein, we report a new VIV8 cluster formulated as [V8O12(OH)4(CH3O)4(DAC)4]·7CH3OH (1) (DAC = 1,2-diaminocyclohexane) with a novel distorted shuriken-like topology containing two V4 butterfly subunits arranged vertically. This cluster has been used as a homogeneous catalyst for asymmetric sulfoxidation reactions in water in the presence of SAs (Scheme 1). Satisfactory chemoselectivities (>90%) for sulfoxides were obtained in the presence of different SAs. Compared to related chiral POM systems,31,45,46 this system has the advantages of inexpensive chiral ligands, green reaction media, and extremely simple working procedures. Consequently, this VIV8 cluster in conjunction with SAs represents a promising type of POM catalytic system for diverse asymmetric reactions, making it potentially useful in chemical and biological synthesis.
 | ||
| Scheme 1 Oxidation of sulfides catalyzed by 1 in the presence of SA. | ||
Experimental section
Materials
All reagents and solvents for synthesis and analysis were commercially available. Bovine serum albumin (BSA) was provided by Sigma-Aldrich, human serum albumin (HSA), porcine serum albumin (PSA), rabbit serum albumin (RSA) and sheep serum albumin (SSA) were obtained from Zhejiang, China. All the sulfides were purchased from Energy Chemical.Physical measurements
UV-Vis spectra were recorded on a Cary 100 UV-visible spectrophotometer. The FT-IR spectra were recorded on a Perkin-Elmer Spectrum One FT-IR spectrophotometer with a germanium attenuated total reflection (ATR) accessory, a DTGS KBr detector and a KBr beam splitter ratio. Elemental analyses for C, H and N were carried out on a model 2400 II, Perkin-Elmer elemental analyzer. ESI-MS (electrospray ionization mass spectrum) spectra were recorded on a Bruker HCT Electrospray Ionization Mass Spectrometer. HPLC experiments were carried out using UV2302II/P2302II high performance liquid chromatograph. 1H and 13C NMR spectra were recorded on a Bruker NMR.Syntheses of complex 1
X-ray crystallographic determination
All reflection data were collected on an Agilent Supernova diffractometer (Mo, λ = 0.71073 Å) at room temperature. A semiempirical absorption correction by using the SADABS program was applied, and the raw data frame integration was performed with SAINT.47 The crystal structures were solved by the direct method using the program SHELXS-97 (ref. 48) and refined by the full-matrix least-squares method on F2 for all non-hydrogen atoms using SHELXL-97 (ref. 49) with anisotropic thermal parameters. All hydrogen atoms were located in calculated positions and refined isotropically, except the hydrogen atoms of water molecules were fixed in a difference Fourier map and refined isotropically. The details of the crystal data were summarized in Table 1, and selected bond lengths and angles for compounds 1 are listed in Table S1.†| Complex | 1 |
| Empirical formula | C35H96N8O27V8 |
| Formula weight (M) | 1468.72 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 14.1442 (11) |
| b (Å) | 14.9100 (11) |
| c (Å) | 16.0774 (13) |
| α (°) | 117.151 (8) |
| β (°) | 92.303 (6) |
| γ (°) | 91.772 (6) |
| V/(Å 3) | 3009.9 (4) |
| Z | 2 |
| Dc (g cm−3) | 1.621 |
| F(000) | 1524 |
| θ range for data collection (°) | 3.4–26.4 |
| Reflections collected/unique | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 154/12271 [R(int) = 0.032] 154/12271 [R(int) = 0.032] |
| Goodness-of-fit on F2 | 1.046 |
| Final R indices [I > 2σ(I)] | R1 = 0.0604, ωR2 = 0.1433 |
| R indices (all data) | R1 = 0.0753, ωR2 = 0.1554 |
Characterization of 1 in the absence of SA
A 33.3 μM solution of the 1 in 0.05 M phosphate-buffered saline (PBS), pH 7.45, was prepared by mixing 100 μL of a 1 × 10−3 M solution of the 1 and 2900 μL of 0.05 M PBS, pH 7.45. In parallel, a 33.3 μM solution of SA-1 in 0.05 M PBS, pH 7.45, was obtained by mixing 100 μL of a 1 × 10−3 M solution of 1 and 100 μL of a 1 × 10−3 M solution of SA (1.0 equiv.) in 2800 μL of 0.05 M PBS, pH 7.45. The UV/Vis spectra of these solutions were then recorded between 250 and 500 nm.General procedure for oxidation of sulfides
The SA (2.7 μmol, 176 mg) was mixed for 10 min at room temperature with 1 (2.7 μmol, 0.4 mg) through a simple mechanical stirring in 2 mL 50 mM PB (phosphate buffer) solution. 0.27 mmol sulfide was added to the reaction mixture and the stirring was continued for 5 hours. The oxidant (1.5 equiv.) was then added to the solution in one portion and the reaction mixture stirred for another 10 h at room temperature. Then, the solution was quenched with sodium sulfite solution and extracted with dichloromethane (2 mL × 5). Finally, the combined organic solutions were dried over by anhydrous sodium sulfate, filtered, and evaporated under vacuum. The same procedure was followed with complex 1 alone or without 1. A sample of the crude reaction mixture re-dissolved in a minimum amount of isopropanol before being analyzed by HPLC with Chiralcel OB-H (4.6Φ mm × 250 mm) column (Daicel Chemical Industries, Tokyo) at room temperature. Further purification was achieved by chromatography on silica gel then was taken for the NMR analysis. The enantiomeric excess (ee) and chemoselectivity were calculated using the formulas: ee% = [peak area (S − R)/(S + R)] × 100%; chemoselectivity% = [peak area SO/(SO + SO2)] × 100%, SO = sulfoxide, SO2 = sulfone. Conversions were based on sulfide substrate; yields were referred to isolated product after column chromatography and were based on substrate; the absolute configurations of the sulfoxides were assigned by comparison of HPLC elution orders reported in the literature.50,51XRPD results
To confirm whether the crystal structures are truly representative of the bulk materials, X-ray powder diffraction (XRPD) experiments had also been carried out for 1. The XRPD experimental and computer-simulated patterns of the corresponding complexes are shown in Fig. S2.† Although the experimental patterns have a few unindexed diffraction lines and some are slightly broadened in comparison with those simulated from the single crystal models, it can still be considered favorably that the bulk synthesized materials and the as-grown crystals are homogeneous for 1.Results and discussion
Description of the crystal structures
(Table 1). The asymmetric unit contains a [V8O12(OH)4(CH3O)4(DAC)4] unit (Fig. 1e) as well as seven lattice methanol molecules. And the [V8O12(OH)4(CH3O)4(DAC)4] unit consists of eight V(IV) ions, four 1,2-diaminocyclohexane ligands, four μ2-OH− anions, four μ2-CH3O− anions, four μ3-O2− anions, four μ2-O2− anions, and eight terminal O2− anions (Fig. 1e and f). Four external V(IV) ions (V1, V2, V5 and V8) of the V8 octamer are six-coordinate with a distorted octahedral [VN2O4] geometry formed by the coordination of four O atoms (one terminal O2− anion, one μ2-OH− anion, one μ3-O2− anion, and one μ2-CH3O− anion), and two N atoms from a DAC ligand. The distorted quartet cone coordination sphere of the internal V4 is [VO5], in which two μ3-O2− anions, one μ2-OH− anion and one μ2-CH3O− anion lie in the equatorial plane and one terminal O2− anion locate in the axial position. The arrangement of eight vanadium atoms supported by four μ2-OH− anions, four μ2-CH3O− anions, and four μ3-O2− anions displays a distorted shuriken topology which can be described as consisting of five edge-sharing distorted V4 tetrahedron (Fig. 1b) with the adjacent V⋯V distance range of 2.9012 (4)–3.1951 (5) Å. To the best of our knowledge, very rare of metallic skeletons with this topology have been observed in the polynuclear metal complexes.52–54
 | ||
| Fig. 1 (a) The simple structural anatomy of the V8 unit; (b) the V8 skeleton with shuriken topology which can be described as consisting of five edge-sharing distorted V4 tetrahedron; (c) the two V4 butterfly subunits with vertical arrangement; (d) the arrangement of eight vanadium atoms supported by four μ2-OH− anions, four μ2-CH3O− anions, and four μ3-O2− anions, and the distribution of the eight terminal O2− anions; (e) a view of the V8 cluster; (f) the V8 cluster with polyhedron mode; (g) the space-filling plot of the V8 cluster, and it's the size. | ||
From another perspective, 1 could also view as an unusual linked-butterfly structure (Fig. 1a and c). In the V4 butterfly subunit, the four V centers are connected by two μ3-O2− anions and two μ2-CH3O− anions (Fig. 1d). Different with the Mn8 octamers with linked-butterfly structure reported by Christou et al.,55,56 the two V4 butterfly subunits which are connect each other by four μ2-OH− anions (Fig. S1d†) arrange vertically.
ESI-MS solution analysis
Mass spectroscopy is a powerful tool for probing the properties of inorganic clusters in solution as well as investigating the supramolecular self-assemblies of inorganic coordination architectures, as reported by Cronin and co-workers.57–59 In our work, the electrospray mass spectrometric experiment of 1 was taken to explore their solution stability in water.The ESI mass spectrum of 1 is shown in Fig. 2. The main ion peak observed at a weight of 1246 which is isotopically resolved and agree with the theoretical distributions (see Fig. 1, inset) results from obtaining a proton of the [V8O12(OH)4(CH3O)4(DAC)4] unit to form [V8O12(OH)4(CH3O)4(DAC)4 + H]+. These results support the opinion that the VIV8 cluster of 1 remains stable in solution.60–62
 | ||
| Fig. 2 The ESI mass spectrum of 1 in the scale of m/z = 400–3000; insert: the black spectra is the experimentally collected for low resolution, and the red spectra is the predicted isotopic envelopes. | ||
Binding of complex 1 with the serum albumin
UV/Vis spectra (Fig. 3) of 33.3 μM solutions of the 1 were recorded in 50 mM PBS buffer, pH 7.45, in the absence and presence of equimolar SAs (BSA, HSA, RSA, PSA, SSA) in order to examine the extremely simple noncovalent binding.38 Comparing the spectrum of the 1 with those recorded in the presence of SAs (Fig. 3), it can be seen that PSA and BSA caused bathochromic shifts of 10 nm (from 254 to 264 nm) and 6 nm (from 254 to 260 nm), respectively, accompanied by strong increases in intensity. It is thus clear that stronger association complexes of the 1 were formed with PSA and BSA. On the contrary, in the presence of SSA, the spectrum of the 1 was largely unchanged, suggesting weaker binding.37 | ||
| Fig. 3 UV/Vis spectra of 1 (33.3 μM) in the absence and presence of SAs (33.3 μM). | ||
Asymmetric sulfoxidation
Taking the aspect of green chemistry into account, a preliminary investigation on the oxidation of methyl phenyl sulfide (PhSMe) catalyzed by 1 was carried out in the presence of BSA (1:BSA molar ratio 1:1) employing the environmentally benign oxidant H2O2 (ref. 64) (1.5 equiv.) (PhSMe and BSA were chosen for these preliminary studies as a well-known model substrate63 and serum albumin, respectively). Control reactions without 1 or BSA were performed under the same conditions. As shown in Fig. 4, the best result was obtained for 1 in the presence of BSA. Satisfactory yield (93%) and chemoselectivity (98%) were obtained, albeit only with 10% ee. To rule out possible catalysis by BSA alone, the reaction was repeated in the absence of 1. Under otherwise identical conditions, inferior catalytic activity (30% yield and 87% chemoselectivity) was observed. A control experiment without BSA afforded the sulfoxide with 52% chemoselectivity in 51% yield, but the major product was essentially racemic (ee < 1%). Thus, comparison of the results of these reactions clearly demonstrates that the catalysis was mainly due to complex 1, and that the enantiomeric excess was determined by the protein.65–67
 | ||
| Fig. 4 Controlled experiments for oxidation of PhSMe at room temperature for 10 h. | ||
Disappointingly, unsatisfactory enantioselectivity was obtained with H2O2. Therefore, sulfoxidation reactions were evaluated with other oxidants. Among these oxidants, sodium hypochlorite (NaClO) and sodium periodate (NaIO4) gave lower yields and product selectivities, and the ee values obtained were only 15% and 3%, respectively (Fig. 5). However, tert-butyl hydroperoxide (TBHP) effectively oxidized PhSMe affording a yield (92%) comparable to that with H2O2, but with somewhat higher selectivity (97%) and ee (20%). Interestingly, the opposite major enantiomer was produced (R-configured sulfoxide with H2O2; S-configured sulfoxide with TBHP). Thus, the nature of the oxidizing agent determines the enantiomeric excess of the reaction product in some cases.
 | ||
| Fig. 5 Screening of oxidants for oxidation of PhSMe catalyzed by 1 in PB (pH 5.1) at room temperature after 10 h. The mol ratios of oxidant:PhSMe:BSA:1 (2.7 μmol) were 150:100:1:1. | ||
The reaction conditions for the oxidation of PhSMe were further optimized by studying four different parameters, namely the catalyst concentration, the oxidant concentration, reaction time, and pH. Our results showed that the sulfoxide yield and selectivity were strongly affected by the catalyst concentration (Fig. 6). PhSMe was obtained in excellent yield with high selectivity and the maximum enantioselectivity using 0.5 mol% 1 within 10 h. At higher catalyst concentration, the yield and selectivity of the sulfoxide were much lower and the enantioselectivity decreased to 9%.
 | ||
| Fig. 6 Effect of the concentration of 1 on the oxidation of PMS using TBHP in PB (pH 5.1) at room temperature after 10 h. The mol ratios of TBHP:PhSMe (2.7 μmol) were 150:100. | ||
The effects of the TBHP concentration on the catalytic activity and enantioselectivity were studied by employing various TBHP/PhSMe molar ratios in the presence of 0.5 mol% 1 (Table 2). Increase of this ratio significantly increased the conversion and sulfoxide yield, which were maximized at 1.5 equiv. of TBHP. It is noteworthy that at a TBHP:PhSMe molar ratio of 2.5:1 (entry 4), significant decreases in both selectivity (78%) and yield (76%) were observed. Compared with the results at a 2:1 molar ratio, the conversion was almost the same and the ee was slightly higher. These experimental results clearly indicated that the reaction occurred with over-oxidation,68,69 that is, a kinetic resolution process70–72 was operative in the presence of excess oxidant.
| Entry | TBHP (mmol) | Conversion (%) | Yield (%) | Chemoelectivity (%) | ee (%) |
|---|---|---|---|---|---|
| a All reactions were carried out in PB buffer (2 mL, pH 5.1) at room temperature for 10 h. The mol ratios of PhSMe:BSA:1 (1.35 μmol) were 100:0.5:0.5. |
|||||
| 1 | 0.27 | 73 | 69 | 95 | 31 |
| 2 | 0.41 | 98 | 97 | 99 | 35 |
| 3 | 0.54 | 96 | 93 | 97 | 34 |
| 4 | 0.689 | 97 | 76 | 78 | 38 |
Next, complex 1 was used to catalyze the oxidation of PhSMe in the presence of BSA under the above optimized reaction conditions over a period of 22 h. From the reaction time profile of the oxidation of PhSMe (Fig. 7), it can be seen that the conversion increased steadily up to 8 h and attained a maximum value of 99%. Notably, chemoselectivity (98–99%) and enantioselectivity (35–36%) remained nearly constant throughout the course of the reaction, confirming that the obtained enantioselectivity was completely induced during the sulfide oxidation process and was not due to a concomitant kinetic resolution.73
 | ||
| Fig. 7 Effect of the reaction time on the oxidation of PhSMe using TBHP in PB (pH 5.1) at room temperature. The mol ratios of TBHP:PhSMe:BSA:1 (1.35 μmol) were 150:100:0.5:0.5. | ||
Under the above optimized reaction conditions, the oxidation of PhSMe was performed at different pH values (Fig. 8). The oxidation of PhSMe in PBS (pH 4.7) gave a significantly higher enantioselectivity (up to 57% ee) compared to the other reactions (3–33% ee), albeit with a lower yield. Increasing or decreasing the pH from 4.7 significantly reduced the conversion, yield, and, enantioselectivity, possibly because complex 1 became less stable.74
 | ||
| Fig. 8 Effect of pH values on the oxidation of PhSMe using TBHP at room temperature after 8 h. The mol ratios of TBHP:PhSMe:BSA: 1 (1.35 μmol) were 150:100:0.5:0.5. | ||
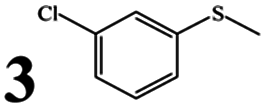
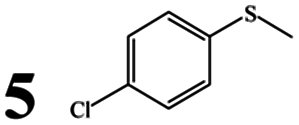
We then tested the catalytic activity of complex 1 in the presence of BSA and other commercially available serum albumins, HSA, PSA, RSA, and SSA, in reactions of a series of alkyl aryl sulfoxides (1–10). The results, which are summarized in Table 3, showed that chemoselectivity was almost unaffected by the albumin source or the substrate (>90% sulfoxide selectivity in each case). However, in terms of conversion and yield, PSA, RSA, and SSA led to poor results, whereas BSA and HSA performed relatively well (typically >90% conversion/yield). Furthermore, the albumin source had a highly significant effect on the ee values, with those for all of the substrates averaging 45.1%, 24.3%, 42%, 7.8%, and 3.8% for 1 in the presence of BSA, HSA, PSA, RSA, and SSA, respectively. The experimental results and spectral studies discussed above clearly indicate that the enantioselectivity was associated with the binding strength between 1 and SA. The high enantioselectivities induced by the PSA-1 and BSA-1 systems indicate that 1 was strongly anchored to the protein. However, in the cases of HSA, RSA, and SSA, this effect was very small, leading to lower ee values (typically <10% ee with SSA), implying weaker binding. Thus, it seems that increasing the interaction of the metal complex with the protein leads to better enantioselectivities.75 Compared with the other proteins, RSA provided the opposite major enantiomer in many cases. For example, opposite results in terms of the configuration were obtained for 2-Cl-, 3-Cl- and 4-Cl-phenyl methyl sulfide (substrates 2, 3 and 5) with RSA; 3-Br-phenyl methyl sulfide (substrate 4) with PSA; vinyl phenyl sulfoxide (substrate 9) with SSA.
| Substrate | Complex 1: conversion (%), chemoselectivity (%), yield (%), ee (%) (configuration) | ||||
|---|---|---|---|---|---|
| BSA | HSA | PSA | RSA | SSA | |
| a Reactions were performed in 2 mL PB pH 4.7 at room temperature for 8 h. The ratios of TBHP: substrate: SA: 1 (2.7 μmol) were 150:100:0.5:0.5. |
|||||
 |
71, 99, 70, 77, R | 90, 98, 88, 35, R | 75, 99, 74, 41, R | 77, 99, 76, 5, R | 82, 99, 81, 2, R |
 |
99, 100, 99, 23, R | 90, 100, 90, 3, R | 95, 95, 90, 36, R | 95, 95, 90, 6, S | 88, 92, 81, 1, R |
 |
98, 97, 95, 19, R | 95, 96, 91, 3, R | 90, 98, 88, 23, R | 90, 98, 88, 2, S | 95, 92, 87, 1, R |
 |
98, 98, 96, 75, R | 94, 97, 91, 56, R | 92, 97, 89, 62, S | 92, 96, 88, 10, R | 91, 98, 89, 9, R |
 |
97, 98, 95, 41, R | 96, 98, 94, 2, R | 95, 99, 94, 60, R | 95, 100, 95, 9, S | 95, 97, 92, 3, R |
 |
95, 100, 95, 57, R | 94, 100, 94, 21, R | 94, 100, 94, 46, R | 94, 94, 88, 4, R | 97, 98, 95, 13, R |
 |
99, 98, 97, 58, R | 99, 98, 97, 34, R | 99, 98, 97, 50, R | 95, 98, 93, 3, R | 99, 100, 99, 3, R |
 |
99, 98, 97, 64, R | 99, 98, 97, 67, R | 95, 98, 93, 68, R | 89, 98, 87, 2, R | 96, 98, 94, 4, R |
 |
91, 100, 91, 26, S | 97, 95, 92, 18, S | 95, 90, 85, 25, S | 95, 91, 86, 29, S | 87, 97, 85, 1, R |
 |
98, 96, 94, 11, S | 94, 98, 92, 4, S | 90, 96, 86, 9, R | 95, 99, 94, 8, R | 71, 99, 76, 1, R |
Irrespective of electron-withdrawing and/or electron-donating groups and their position on the aromatic ring (entries 2–8), good conversions (88–99%) and yields (81–99%) were obtained. Nevertheless, the data suggested that electron-donating groups had a more pronounced effect on the enantioselectivity of the product. The average ee values obtained for electron-deficient 4-Cl- and 4-Br-phenyl methyl sulfides (substrates 5 and 6) were 23% and 28%, respectively. In comparison, electron-rich 4-Me-phenyl methyl sulfide and 4-OMe-phenyl sulfide (substrates 7 and 8) gave average ee values of 30% and 41%, respectively. Sulfoxidations of sterically bulky substrates, such as vinyl phenyl sulfoxide and methyl benzyl sulfoxide (substrates 9 and 10), were successfully achieved with high chemoselectivities. However, lower enantioselectivities were observed in the oxidation of these sterically bulky substrates to the corresponding sulfoxides, possibly due to the increased steric demand.76 Significantly, the oxidation-sensitive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond was not sacrificed under the mild oxidation conditions, producing the requisite sulfoxide in high yield (>85%). Notably, the results indicate that the steric effect had a stronger influence on the configuration. For example, except for vinyl phenyl sulfoxide and methyl benzyl sulfoxide, the products were obtained in the R form in the presence of BSA or HSA. In particular, for vinyl phenyl sulfoxide, the opposite configuration was obtained compared to the reactions of other sulfides in the presence of the same SA (except SSA). The mechanistic details of this reaction are still under investigation.
C double bond was not sacrificed under the mild oxidation conditions, producing the requisite sulfoxide in high yield (>85%). Notably, the results indicate that the steric effect had a stronger influence on the configuration. For example, except for vinyl phenyl sulfoxide and methyl benzyl sulfoxide, the products were obtained in the R form in the presence of BSA or HSA. In particular, for vinyl phenyl sulfoxide, the opposite configuration was obtained compared to the reactions of other sulfides in the presence of the same SA (except SSA). The mechanistic details of this reaction are still under investigation.
Conclusions
In conclusion, a novel VIV8 cluster has been successfully constructed based on 1,2-diaminocyclohexane. Using this VIV8 cluster as a homogeneous catalyst, we have devised an efficient biomimetic procedure for the oxidation of sulfides to sulfoxides in the presence of serum albumin in water at room temperature. This methodology is simple and environmentally friendly. Its simplicity lies in the preparation of the catalyst from easily available starting materials as well as a facile sulfoxidation procedure by physical stirring at room temperature. In addition to the above-mentioned advantages, the use of eco-friendly reaction media and inexpensive chiral additives make it cost-effective, environmentally benign, and thus compatible with the goals of green sustainable chemistry.Acknowledgements
We gratefully acknowledge the National Nature Science Foundation of China (No. 21361003, 21461003 and 21101035), the Guangxi Natural Science Foundation (2014GXNSFBA118056), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, the Talent's Small Highland project of Guangxi Medicinal Industry (1201), New Century Ten, Hundred, Thousand Talents Project in Guangxi, IRT 1225 and the Foundation of Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Ministry of Education of China (CMEMR2011-20, CMEMR2013-A05).References
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–217 CrossRef CAS PubMed.
- N. Mizuno and K. Kamatal, Coord. Chem. Rev., 2011, 255, 2358–2370 CrossRef CAS.
- G. J. Hutchings, J. Mater. Chem., 2004, 14, 3385–3395 RSC.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- S. Takizawa, T. Katayama and H. Sasai, Chem. Commun., 2008, 4113–4122 RSC.
- A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89–101 CrossRef CAS.
- C. Bolm, Coord. Chem. Rev., 2003, 237, 245–256 CrossRef CAS.
- J. T. Rhule, C. L. Hill and D. Judd, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed.
- J. Oble, B. Riflade, A. Noël, M. Malacria, S. Thorimbert, B. Hasenknopf and E. Lacôte, Org. Lett., 2011, 13, 5990–5993 CrossRef CAS PubMed.
- C. L. Wang, Z.-B. Zhang, J. Fu, Y. Xu and D.-R. Zhu, Chem.–Eur. J., 2012, 18, 11909–11912 CrossRef CAS PubMed.
- M. I. Khan, S. Cevik, D. Powell, S. Li and C. J. O'Connor, Inorg. Chem., 1998, 37, 81 CrossRef CAS PubMed.
- A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239–271 CrossRef.
- M. J. Manos, A. J. Tasiopoulos, E. J. Tolis, N. Lalioti, J. D. Woollins, A. M. Z. Slawin, M. P. Sigalas and T. A. Kabanos, Chem.–Eur. J., 2003, 9, 695–703 CrossRef CAS PubMed.
- L. Chen, F.-L. Jiang, Z.-Z. Lin, Y.-F. Zhou, C.-Y. Yue and M.-C. Hong, J. Am. Chem. Soc., 2005, 127, 8588–8589 CrossRef CAS PubMed.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- J. Forster, B. Rösner, M. M. Khusniyarov and C. Streb, Chem. Commun., 2011, 47, 3114 RSC.
- S. Li, W. Sun, K. Wang, H.-Y. Ma, H.-J. Pang, H. Liu and J.-X. Zhang, Inorg. Chem., 2014, 53, 4541–4544 CrossRef CAS PubMed.
- D.-Y. Du, L.-K. Yan, Z.-M. Su, S.-L. Li, Y.-Q. Lan and E. B. Wang, Coord. Chem. Rev., 2013, 257, 702–717 CrossRef CAS.
- C. L. Hill, J. Mol. Catal. A: Chem., 2007, 262, 1–242 CrossRef CAS.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- R. Neumann, Prog. Inorg. Chem., 1998, 47, 317–370 CrossRef CAS.
- N. Mizuno, K. Kamata and K. Yamaguchi, Surf. Nanomol. Catal., 2006, 463–491 CAS.
- R. Neumann, Prog. Inorg. Chem., 1998, 47, 317–370 CrossRef CAS.
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956 CrossRef CAS.
- N. Mizuno, K. Kamata and K. Yamaguchi, Top. Catal., 2010, 53, 876–893 CrossRef CAS.
- S. K. Maiti, S. Dinda, S. Banerjee, A. K. Mukherjee and R. Bhattacharyy, Eur. J. Inorg. Chem., 2008, 2038–2051 CrossRef CAS.
- S.-Z. Luo, S. J.-Y. Li, H. Xu, L. Zhang and J.-P. Cheng, Org. Lett., 2007, 9, 3675–3678 CrossRef CAS PubMed.
- Q. Gao, S.-M. Lu, Y. Liu and C. Li, Tetrahedron Lett., 2011, 52, 3779–3781 CrossRef CAS.
- R. Ishimoto, K. Kamata, K. Suzuki, K. Yamaguchi and N. Mizuno, Dalton Trans., 2015, 44, 10947–10951 RSC.
- Q. Chen, C. Xin, L.-L. Lou, K. Yu, F. Ding and S.-X. Liu, J. Inorg. Organomet. Polym., 2013, 23, 467–471 CrossRef CAS.
- Y.-Z. Wang, H.-L. Li, W. Qi, Y. Yang, Y. Yan, B. Li and L.-X. Wu, J. Mater. Chem., 2012, 22, 9181–9188 RSC.
- S. Colonna, S. Banfi, R. Annunziata and L. Casella, J. Org. Chem, 1986, 51(6), 891–895 CrossRef CAS.
- V. Oliveri and G. Vecchio, Eur. J. Med. Chem., 2011, 46, 961–965 CrossRef CAS PubMed.
- Y.-J. Hu, Y. Ou-Yang, Y. Zhang and Y. Liu, Protein J., 2010, 29, 234–241 CrossRef CAS PubMed.
- S. Colonna, S. Banfi and R. Annunziata, J. Org. Chem., 1986, 51, 891–895 CrossRef CAS.
- A. Pordea, M. Creus, J. Panek, C. Duboc, D. Mathis, M. Novic and T. R. Ward, J. Am. Chem. Soc., 2008, 130, 8085–8088 CrossRef CAS PubMed.
- M. T. Reetz and N. Jiao, Angew. Chem., Int. Ed., 2006, 45, 2416–2419 CrossRef CAS PubMed.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS PubMed.
- A. Ghorbani-Choghamarani, Z. Darvishnejad and M. Norouzi, Appl. Organomet. Chem., 2015, 29, 170–175 CrossRef CAS.
- M. Sutradhar, L. M. D. R. S. Martins, M. F. C. G. Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2015, 301–302, 200–239 CrossRef CAS.
- K. A. Stingl, K. M. Weiß and S. B. Tsogoeva, Tetrahedron, 2012, 68, 8493–8501 CrossRef CAS.
- P. K. Khatri, S. L. Jain and B. Sain, Ind. Eng. Chem. Res., 2011, 50, 701–704 CrossRef CAS.
- E. Muthuswamy and S. L. Brock, J. Am. Chem. Soc., 2010, 132, 15849–15851 CrossRef CAS PubMed.
- C. Mealli, A. Ienco, A. Poduska and R. Hoffmann, Angew. Chem., Int. Ed., 2008, 47, 2864–2867 CrossRef CAS PubMed.
- C. Jahier, M. Coustou, M. Cantuel, N. D. McClenaghan, T. Buffeteau, D. Cavagnat, M. Carraro and S. Nlate, Eur. J. Inorg. Chem., 2011, 5, 727–738 CrossRef.
- Y. Sasaki, Y. Deguchi, M. Morimoto, Y. Imai, M. Matsunaga, T. Sakane, J. Ichihara and S. Yamaguchi, Mater. Tech., 2009, 27, 191–195 CAS.
- SAINT Software Reference Manual, Bruker AXS, Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, Phase Annealing in SHELX-90: Direct Methods for Larger Structures, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- (a) M. I. Donnoli, S. Superchi and C. Rosini, J. Org. Chem., 1998, 63, 9392–9395 CrossRef CAS; (b) J.-T. Sun, C.-J. Zhu, Z.-Y. Dai, M.-H. Yang, Y. Pan and H.-W. Hu, J. Org. Chem., 2004, 69, 8500–8503 CrossRef CAS PubMed; (c) K. Tanaka, K. Kubo, K. Iida, K. Otani, T. Murase, D. Yanamoto and M. Shiro, Asian J. Org. Chem., 2013, 2, 1055–1060 CrossRef CAS; (d) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS PubMed.
- (a) P. Pitchen, E. Duiiach, M. N. Deshmukh and H. B. Kagan, J. Am. Chem. Soc., 1984, 106, 8188–8193 CrossRef CAS; (b) M. A. M. Capozzi, C. Cardellicchio, F. Naso and P. Tortorella, J. Org. Chem., 2000, 65, 2843–2846 CrossRef CAS PubMed; (c) P. Pitchen, E. Duiiach, M. N. Deshmukh and H. B. Kagan, J. Am. Chem. Soc., 1984, 106, 8188–8193 CrossRef CAS; (d) O. G. Mancheno, J. Dallimore, A. Plant and C. Bolm, Org. Lett., 2009, 11, 2429 CrossRef CAS PubMed; (e) F. A. Davis, R. T. Reddy, W. Han and P. J. Carroll, J. Am. Chem. Soc., 1992, 114, 1428–1437 CrossRef CAS; (f) J. H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef PubMed.
- X.-N. Cheng, W. Xue, J.-B. Lin and X.-M. Chen, Chem. Commun., 2010, 46, 246–248 RSC.
- B. F. Abrahams, T. A. Hudson and R. Robson, J. Am. Chem. Soc., 2004, 126, 8624–8625 CrossRef CAS PubMed.
- B. Moubaraki, K. S. Murray, T. A. Hudson and R. Robson, Eur. J. Inorg. Chem., 2008, 4525–4529 CrossRef CAS.
- M. W. Wemple, H.-L. Tsai, W. E. Streib, D. N. Hendrickson and G. Christou, J. Chem. Soc., Chem. Commun., 1994, 1031–1033 RSC.
- M. W. Wemple, H.-L. Tsai, S. Wang, J. P. Claude, W. E. Streib, J. C. Huffman, D. N. Hendrickson and G. Christou, Inorg. Chem., 1996, 35, 6437–6449 CrossRef CAS PubMed.
- G. J. T. Cooper, G. N. Newton, P. Kögerler, D.-L. Long, L. Engelhardt, M. Luban and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 1340–1344 CrossRef CAS PubMed.
- G. N. Newton, G. J. T. Cooper, P. Kögerler, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2008, 130, 790–791 CrossRef CAS PubMed.
- H. N. Miras, E. F. Wilsonw and L. Cronin, Chem. Commun., 2009, 1297–1311 RSC , and references therein.
- S.-H. Zhang, Y. Song, H. Liang and M.-H. Zeng, CrystEngComm, 2009, 11, 865–872 RSC.
- B.-W. Li, Y.-L. Zhou, Q. Chen and M.-H. Zeng, Polyhedron, 2010, 29, 148–153 CrossRef CAS.
- L.-Q. Wei, B.-W. Li, S. Hu and M.-H. Zeng, CrystEngComm, 2011, 13, 510–512 RSC.
- P. K. Bera, D. Ghosh, S. H. R. Abdi, N. H. Khan, R. I. Kureshy and H. C. Bajaj, J. Mol. Catal. A: Chem., 2012, 361–362, 36–44 CrossRef CAS.
- S. M. G. Piresa, M. M. Q. Simõesa, I. C. M. S. Santosa, S. L. H. Rebelob, M. M. Pereirac, M. G. P. M. S. Nevesa and J. A. S. Cavaleiroa, Appl. Catal., A, 2012, 439–440, 51–56 CrossRef.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS PubMed.
- E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2010, 110, 4303–4356 CrossRef PubMed.
- H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS PubMed.
- S. P. Das, J. J. Boruah, N. Sharma and N. S. Islam, J. Mol. Catal. A: Chem., 2012, 356, 36–45 CrossRef CAS.
- A. Bayat, M. Shakourian-Fard and M. M. Hashemi, Catal. Commun., 2014, 52, 16–21 CrossRef CAS.
- H. Sroura, J. Jalkha, P. L. Mauxa, S. Chevancea, M. Kobeissib and G. Simonneauxa, J. Mol. Catal. A: Chem., 2013, 370, 75–79 CrossRef.
- J.-T. Sun, C.-J. Zhu, Z.-Y. Dai, M.-H. Yang, Y. Pan and H.-W. Hu, J. Org. Chem., 2004, 69, 7058–7065 CrossRef PubMed.
- Z.-W. Yang, C.-F. Zhu, Z.-J. Li, Y. Liu, G.-H. Liu and Y. Cui, Chem. Commun., 2014, 50, 8775–8778 RSC.
- M. I. Donnoli, S. Superchi and C. Rosini, J. Org. Chem., 1998, 63, 9392–9395 CrossRef CAS.
- A. Rezaeifard, R. Haddad, M. Jafarpour and M. Hakimi, ACS Sustainable Chem. Eng., 2014, 2, 942–950 CrossRef CAS.
- E. S. Dagousset, A. Urvoas, K. Chelly, W. Ghattas, J.-D. Maréchal, J.-P. Mahy and R. Ricoux, Dalton Trans., 2014, 43, 8344–8354 RSC.
- K. Tanaka, K. Kubo, K. Iida, K. Otani, T. Murase, D. Yanamoto and M. Shiro, Asian J. Org. Chem., 2013, 2, 1055–1060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1439891. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra08153c |
| This journal is © The Royal Society of Chemistry 2016 |