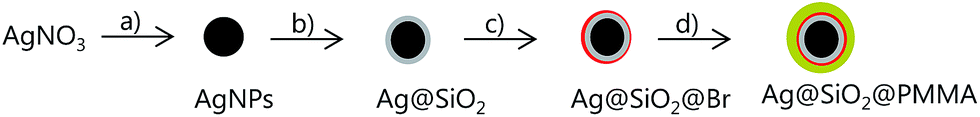DOI:
10.1039/C6RA07397B
(Paper)
RSC Adv., 2016,
6, 44254-44260
Hydrophobization of silver nanoparticles through surface-initiated atom transfer radical polymerization
Received
21st March 2016
, Accepted 28th April 2016
First published on 29th April 2016
Abstract
This work describes the synthesis of poly(methyl methacrylate) (PMMA)-coated Ag@SiO2 core–shell particles by conducting a Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). Silver nanoparticles (AgNPs) were first prepared by the polyol synthesis and subjected to preliminary surface functionalization which includes the encapsulation in an insulating SiO2 shell by an optimized Stöber method and functionalization of the shell with an ATRP initiator. The growth of an organic PMMA shell yields inorganic/organic core–shell particles with an enhanced dispersibility in non-polar solvents.
Introduction
Silver nanoparticles (AgNPs) are widely employed in different scientific fields due to their array of properties. The current application fields of AgNPs include biology,1–3 optics,4–6 catalysis7,8 and electronics.9–12 The properties of AgNPs depend on both their size and shape, and it is essential to gain control over them in order to tune their properties. While the continuous preparation of AgNPs in flow reactors have enabled the control over the size and distribution of the particles,13–16 the polyol synthesis has established itself as a reliable and facile method to prepare different silver nanostructures which includes wires, cubes, spheres, bipyramids and triangles.17 The surface functionalization is essential for both the stability and further use of AgNPs in different application fields. Metal oxides such as TiO2 and SiO2 have been used to encapsulate the AgNPs with an oxide layer in order to produce core–shell particles. While Ag@TiO2 particles exhibit photocatalytic activity,18–20 Ag@SiO2 have been prepared to exploit plasmonic properties.21–23 Furthermore, Ag@SiO2 core–shell particles also feature good dielectric properties with both low conductivities and dielectric losses.24,25 Previous studies on the dielectric properties of core–shell particles revealed control over the dielectric properties with the variation of the silica shell thickness.26–28 Therefore, core–shell particles are promising candidates as high permittivity filler for the preparation of materials with enhanced dielectric properties as well as of permittivity graded materials.29 However, the SiO2 shell is highly hydrophilic and has to be rendered hydrophobic in order to be compatible with polymer matrices such as polydimethylsiloxane (PDMS), polyacrylates or polyolefins. Hybrid organic/inorganic nanocomposites find potential applications in optics, electronics and biomedicine.30,31 While surface hydrophobization can be achieved with alkyl silanes32–35 or by emulsion polymerization,36 this work exploits the possibility of growing an outer polymer layer on the core–shell particles by conducting a Surface-Initiated (SI) Atom Transfer Radical Polymerization (ATRP).37,38 SI-ATRP has previously been used to prepare organic/inorganic hybrid particles. In most of the cases, the growth of polymers such as polystyrenes, polymethacrylates and polyacrylonitriles has been demonstrated on SiO2 particles or Si-surfaces.39–41 Apart from SiO2 nanoparticles, SI-ATRP has also been demonstrated on metal surfaces and particles such as Au, where a thiol-functionalized ATRP-initiator was grafted directly on the particle surface.42–47 The resulting hybrid particles featured good dispersibility and thus compatibility in hydrophobic media as well as high mechanical and thermal stability.30,39,48
Silver is less expensive then Au and is resistant against oxidation due to its high reduction potential. Therefore, the aim of this work was to prepare AgNPs coated with a polymer layer the thickness of which can be fine-tuned by ATRP. The access to such particles will allow the synthesis of permittivity graded materials by layering particles with different shell thickness and by covalently connecting them to one another. Additionally, surface functionalized AgNPs can be used as fillers in composites, whereby the matrix as well as the organic shell of the filler is composed of the same polymer.
Experimental section
Materials and methods
AgNO3, poly(vinyl pyrrolidinone) (PVP), ethylene glycol (EG), tetrahydrofurane (THF), ethanol (EtOH), methanol (MeOH), α-bromoisobutyryl bromide, ammonia (29% solution, NH4OH), triethylamine (TEA), tetraethoxysilane (TEOS), aminopropyl triethoxysilane (APTES), CuCl, N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), ascorbic acid, ethyl-α-bromo isobutyrate (2-(EiB)Br), methyl methacrylate (MMA) were purchased from Aldrich. While EG and MMA were distilled prior use, all other chemicals were used as received. Monomer conversion in the polymerization reaction was monitored and analyzed with a Bruker Avance III 400 NMR spectrometer using a 5 mm BBO Prodigy™ CryoProbe at 400.18, 100.63 and 79.50 MHz, respectively. Gel permeation chromatography (GPC) measurements were done with an Agilent 1100/PSS WinGPC 8.1 system in tetrahydrofurane calibrated with poly(methyl methacrylate) (PMMA) standards (Polymer Standards Service). Prior to the analysis by GPC, the polymers were subjected to cycles of purification which consists of dissolving them in THF followed by the precipitation in MeOH. The nanoparticles were observed with TEM using a Philips CM30 TEM. For the observation of PMMA-coated core–shell particles, the TEM grid was stained with 2 wt% aqueous solution of phosphotungstic acid. UV-vis absorption spectra were recorded with a Cary 50 spectrophotometer, DLS measurements were done with a Malvern Zetasized Nano ZS, the thermogravimetric analysis (TGA) was conducted with a Perkin Elmer TGA7 at a heating rate of 20 °C min−1 under a helium gas flow. The number of particles measured for the statistical determination of the particle size was about 400 for Ag core and about 120–150 for the SiO2 and PMMA-coated particles with the help of the software Image-J and a ruler for Windows, respectively.
Synthesis of AgNPs
In a 100 mL round-bottom flask, PVP (6 g, 54 mmol, Mw = 40 kDa) was dissolved in EG (50 mL) under sonication. After complete dissolution of the polymer, the flask was stirred in an oil bath at 140 °C for 30 min, then an aqueous solution of AgNO3 (1 mL, 1 g mL−1) was injected rapidly and the reaction mixture was stirred further for 1 h at a speed of 900 rpm. The flask was later removed from the oil bath, cooled in a water bath and the reaction mixture was diluted with acetone, centrifuged at 5000 rpm for 0.5 h and decanted. The resulting precipitate was further washed with acetone and water 3 times before redispersion in EtOH. The reaction was repeated 18 times and the particles were redispersed in 400 mL of EtOH. Yield: 9.26 g (82%). Size: 59 ± 22 nm.
Synthesis of Ag@SiO2 core–shell particles
The silica-coating was performed based on existing protocols of the modified Stöber method.26 For the silica-coating, 2 × 190 mL of the AgNPs dispersion in EtOH was transferred each in a 2 L flask, diluted with EtOH (600 mL) and treated with NH4OH (29%, 21.25 mL), ethanolic TEOS solution (2.2 vol%, 45 mL) and stirred at 25 °C for 16 h. The mixture was diluted with acetone, centrifuged and decanted. The particles were redispersed in EtOH and the washing procedure was repeated thrice in order to obtain the core–shell particles (8 g) which were redispersed in EtOH (250 mL). Size: 65 ± 14 nm.
Synthesis of APTES–Br
APTES–Br was prepared according to a previous protocol.49 A mixture containing APTES (10 mL, 43 mmol) and triethylamine (7.8 mL, 57 mmol) in dry THF (200 mL) was treated dropwise with α-bromoisobutyryl bromide (5.55 mL, 51 mmol) at 0 °C. After completing the addition, the mixture was stirred at 0 °C for further 30 min, and then warmed to 25 °C and stirred overnight. The mixture was filtered, concentrated in vacuo to give APTES–Br (13.1 g, 86%) as yellow oil. 1H NMR (CDCl3, 400 MHz): 0.6–0.7 (m, 2H, SiCH2), 1.25 (t, 9H, CH3CH2OSi), 1.63–1.73 (m, 2H, CH2), 2.00 (s, 6H, CCH3), 3.3 (q, 2H, CH2N). 3.85 (q, 6H, CH2CH3), 6.9 (s, 1H, NH).
Synthesis of initiator-coated Ag@SiO2 core–shell particles (Ag@SiO2@Br)
The Ag@SiO2 core–shell particles (230 mL out of the 250 mL stock solution) were transferred to a 500 mL round bottom flask, stirred at 25 °C and treated with triethylamine (5.4 mL, 39.1 mmol) and a mixture of APTES–Br (5.4 g, 14.6 mmol) in EtOH (20 mL). The mixture was stirred for 2 days, then diluted with acetone, centrifuged and decanted. The residue was redispersed in EtOH, diluted with acetone, centrifuged and decanted. The washing cycle was repeated 6 times in order to obtain the initiator-coated core–shell particles (6.4 g, 80%) which were redispersed in EtOH (100 mL).
Surface-initiated (SI) ATRP on initiator coated Ag@SiO2 core–shell particles
The initiator-coated Ag@SiO2 core–shell particles (4 g, ∼0.54 mmol APTES–Br grafted on the surface), MMA (35.7 mL, 336 mmol), 2-(EiB)Br (55 μL, 0.35 mmol) in DMF (10 mL) were transferred to a 250 mL Schlenk flask, and subjected to three freeze–dry–pump–thaw cycles. In a separate flask, a solution of CuCl (80 mg, 0.80 mmol), PMDETA (210 μL, 1 mmol), and ascorbic acid (44 mg, 0.25 mmol) in DMF (15 mL) was degassed with Ar for 30 min and then transferred to the Schlenk flask containing the particles via a cannula. The flask was then placed in a preheated oil bath and stirred at 70 °C. During the reaction, aliquots were taken after defined time intervals for reaction control. The reaction was terminated after 7 h by exposing the reaction to air followed by cooling the reaction mixture down to room temperature. The mixture was dissolved in acetone, centrifuged and decanted. The washing procedure was repeated 7 times until no free polymer chain was detected in the mother liquor after concentration in vacuo. Eventually, PMMA-coated core–shell particles (1.8 g) were obtained after the purification process.
Results and discussion
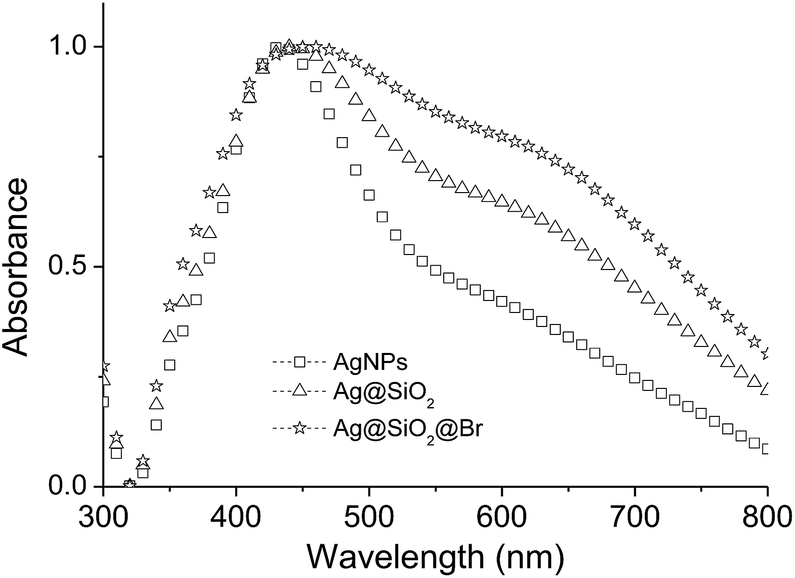
The synthetic procedure to poly(methyl methacrylate) (PMMA)-coated Ag@SiO2 core–shell particles is illustrated in Fig. 1. The AgNPs were prepared by the polyol synthesis, an established method to relatively large amounts of AgNPs per batch reaction.26 The average particle diameter was 59 nm (Fig. 2). The silica-coating was conducted with a modified Stöber method to give core–shell particles in gram quantities with a thin SiO2-shell of ∼3 nm (Fig. 2).19,21,26 The subsequent change in the dielectric medium of the AgNPs (from PVP/EtOH to SiO2) led to a bathochromic shift from 434 to 443 nm in the corresponding UV-vis spectra (Fig. 3). As previously investigated, it is essential to keep the SiO2-shell very thin in order to allow for a maximum increase in the permittivity while at the same time avoiding conductivity.26–28 The thin silica shell seemed to be insufficient in stabilizing the AgNPs from forming some agglomerates which can be attributed to the either strong van der Waals interactions acting between the cores or due to the rather concentrated solution of nanoparticles used which favours aggregation. The formation of some agglomerates between the particles is indicated by the broadening of the peaks in the UV-vis spectra although DLS does not detect it.
 |
| | Fig. 1 General scheme towards the preparation of PMMA-coated Ag@SiO2 core–shell particles: (a) EG, PVP, 140 °C, 1 h; (b) TEOS, NH4OH, EtOH, 25 °C, 16.5 h; (c) APTES–Br, TEA, EtOH, 25 °C, 48 h; (d) MMA, (2-EiB)Br, ascorbic acid, DMF, 70 °C, 7 h. | |
 |
| | Fig. 2 TEM micrographs of AgNPs (a), Ag@SiO2 (b), APTES–Br (c) and PMMA-coated Ag@SiO2 core–shell particles (d). Silver has a higher electron density than silica, therefore it is the dark core, while silica is the grey shell around the dark core. | |
 |
| | Fig. 3 UV-vis spectra of AgNPs, Ag@SiO2, and APTES–Br coated Ag@SiO2 core–shell particles. The spectra were recorded in EtOH. | |
Although the direct growth of polymers on gold nanoparticles (AuNPs) functionalized with thiol-terminated initiators has previously been reported, a main drawback of this approach is the relatively weak Au–S bond (∼40 kcal mol−1) which can undergo bond dissociation at temperatures >60 °C.43 In comparison, the Ag–S bond dissociation energy amounts 52 kcal mol−1, which is a bit higher than the Au–S bond energy but unfortunately will also undergo the same fate at high temperatures.50 Since the polymerization has to be conducted at ambient temperatures in order to circumvent this issue, the choice of polymerizable monomers becomes limited due to the high activation barrier featured by some monomers. To overcome this problem the APTES–Br initiator was chemically grafted to the silica surface of the Ag@SiO2 core–shell particles. The SiO2 shell serves as a protection barrier for the AgNPs towards agglomeration as a result of electrostatic stabilization and allows chemical functionalization of the surface through the presence of the hydroxyl groups.
Similarly, Au@SiO2@PMMA particles have been prepared in the same fashion by Matsui et al.51 Such a three-layered hybrid particle assembly has been used by Liang et al. to produce hairy hybrid microrattles upon etching away the intermediate SiO2 layer.52 Eventually, the Au core is trapped in a hollow cavity surrounded by a polymeric shell consisting either of poly(ethylene glycol) (PEG) or poly(N-isopropylacrylamide). As reported earlier, the use of NH4OH as alkaline catalyst for the initiator fixation induces agglomeration, and therefore TEA was used for this procedure to minimize the risk, and despite the broadened UV-vis signal for the APTES–Br coated core–shell particles (Fig. 3) which is indicative for some agglomeration formation, the DLS measurements and TEM micrographs do not support this findings (Fig. 2 and 4).53 TEA can deprotonate the silanol groups thus enabling the alkoxysilane initiators to attach onto the SiO2 surface.54–56 The immobilization of the initiator on the particle surface was verified by elemental analysis which determined the Br content to be 1.08 wt%, thus giving about 125 μmol of Br per g of nanoparticle. By assuming a spherical shape of the core–shell particles and a silanol group density of 5/nm2,57 the initiator density becomes around 7–8 molecules per nm2 which is higher than reported in the literature and the theoretically possible grafting densities, but could be attributed to the formation of multilayered networks of the APTES–Br on the surface.53,58 However, the calculated polymer chain density was approximately 0.12 chains per nm2, a value which is smaller than the ones reported previously (calculated with the help of the TGA spectra).47,53 The SI-ATRP was performed at 70 °C in order to prevent potential effects of the present copper salts in the solution on the morphology of the AgNPs.59 Free “sacrificial” initiator (2-(EiB)Br) was introduced in order to achieve a degree of control over the polymerization through the accumulation of sufficient amounts of Cu(II) species via radical termination.53,60 The use of CuCl in this work rather than CuBr was attributed to the fact that ATRP reactions conducted with CuBr generally led to higher polymer dispersion index (PDI) compared to the reactions which employed CuCl catalyst.61,62 The polymerization was terminated after 7 h, which led to an overall monomer conversion of around 46%. An increase in the particle size as a result of the growing insulating layer was observed in both the TEM micrographs as well as in the DLS spectra of the corresponding particles at each functionalization stage (Fig. 2 and 4). Unfortunately, it was difficult to differentiate the SiO2- and PMMA-shell from each other through TEM due to the amorphous nature and poor electron density featured by both entities, but an increase in the size of the core–shell particles after the polymerization was observed in the corresponding TEM micrographs (Fig. 2) although the size of the PMMA-coated core–shell particles is much lower than the value obtained from the DLS measurements (Table 1). This can be explained by the fact that the hydrodynamic diameter of the particles is larger in solution due to the swelling of the tethered polymer chains in organic solvents. A similar observation was made by Ohno et al. where the hydrodynamic diameter DH of the particles obtained from DLS measurements surpasses the size determined from the TEM micrographs.53 In contrast, the polymers collapse in the dry state and form compact core–shell structures thus leading to the smaller particle size observed by TEM. Additionally, some chains might undergo interdigitation as a result of attractive forces occurring between them thus leading to a compression of the PMMA shell.51
 |
| | Fig. 4 Hydrodynamic size measured by DLS of AgNPs, Ag@SiO2, APTES–Br, and PMMA-coated Ag@SiO2 core–shell particles recorded in EtOH. | |
Table 1 Determined size parameters of AgNPs, Ag@SiO2 core–shell particles (before and after immobilization of APTES–Br) and PMMA-coated Ag@SiO2 core–shell particles
| Entry |
Size by TEM (nm) |
Size by DLSa (nm) |
λmax (nm) |
| Mean numerical diameter. Measured in EtOH. Measured in THF. |
| AgNPs |
59 ± 22 |
75 ± 29b |
434b |
| Ag@SiO2 |
65 ± 14 |
83 ± 35b |
443b |
| Ag@SiO2@Br |
— |
86 ± 32b |
453b |
| Ag@SiO2@PMMA |
84 ± 16 |
342 ± 99c |
460c |
Concerning the image analysis performed on the TEM micrographs of the Ag@SiO2@PMMA core–shell particles, the average diameter of the particles was increased by ∼20 nm, therefore the resulting polymer shell composed of PMMA chains in the dry state is estimated to be around 10 nm thick (Fig. 5). An overview of the determined parameters for the particles after each functionalization step is shown in Table 1. The corresponding UV-vis spectrum of the PMMA-coated core–shell particles was recorded in THF and is shown in Fig. 6. For comparison, the spectrum of the APTES–Br coated core–shell particles was also recorded in THF and showed a broad red shoulder very likely due some aggregated particles. After the polymerization on the APTES–Br coated core–shell particles, the maximum absorption peak λmax initially experiences a blue shift from 457 to 455 nm which could be explained by the increase in the interparticle distance through the growth of the polymer chains as well as the change in the dielectric medium of the particles (εPMMA < εSiO2).63 The narrower peaks of the PMMA-coated Ag@SiO2 core–shell particles compared to the uncoated counterpart in the UV-vis spectra could be indicative for a better dispersion of the particles in THF as a result of rendering the surface hydrophobic. The absorption peak of the coated particles becomes broader with increasing the polymerization time to 420 min which is likely due to aggregated particles due to cross-linking of the PMMA chains.
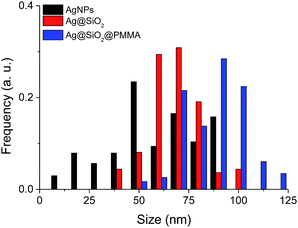 |
| | Fig. 5 Particle size distribution of AgNPs, Ag@SiO2 core–shell particles and PMMA-coated Ag@SiO2 core–shell particles obtained from the image analysis of TEM micrographs. | |
 |
| | Fig. 6 UV-vis spectra of APTES–Br coated Ag@SiO2 core–shell particles and PMMA-coated Ag@SiO2 core–shell obtained from the aliquots taken after a prescribed amount of time. The spectra were recorded in THF. | |
In Fig. 7, water/toluene extraction tests of the particles after each corresponding functionalization step are illustrated. The particles remained hydrophilic even after the immobilization of the APTES–Br initiator which is in agreement with the literature.54 As expected, the grafting of PMMA on the initiator-coated Ag@SiO2 core–shell particles led to a transfer of the particles into the hydrophobic phase due to the hydrophobic polymer chains on the surface. The PMMA-coated core–shell particles were dispersible in a variety of organic solvents which includes toluene, THF and acetone. In order to control and analyse the polymerization, aliquots were taken out of the reaction solution after defined time intervals. The reaction was terminated after a monomer conversion of 46% (Fig. 8). The pattern of the plot depicts an initial steep increase followed by a decrease of the slope after 1 h and is indicative for the potential occurrence of chain termination as described elsewhere.40
 |
| | Fig. 7 Water/toluene extraction tests of AgNPs (a), Ag@SiO2 (b), APTES–Br-coated Ag@SiO2 (c), and PMMA-coated Ag@SiO2 core–shell particles (d). The organic phase is above the aqueous phase. | |
 |
| | Fig. 8 Semi-logarithmic plot of the monomer conversion versus the polymerization reaction time. | |
It is well known that the molecular weight and molecular weight distribution of polymers formed by ATRP on surfaces are basically identical to the values obtained from polymers that were prepared with free “sacrificial” initiators.47,64 Therefore, the characteristics of the isolated polymer formed from the reaction of the free, “sacrificial” initiator should be representative for the grafted polymers. GPC measurements gave a molecular weight Mn = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 for the free polymer after 7 h of polymerization (Fig. 9 and 10). Unfortunately, the PDI deteriorated during the course of the polymerization reaction, namely from 1.6 to 2.6 despite the careful choice of reaction parameters. This might be the direct result of the increase viscosity of the system during the course of the reaction which hinders the stirring thus affecting parameters like uniform heat transfer. On the other hand, this can also be interpreted as the direct result of branching and cross-linking between the polymers in the system which could explain the formation of ensembles of the particles as explained earlier.40 This phenomenon likely occurs extensively after proceeding the reaction for more than 1 h and possibly explains the deterioration in the PDI.
000 g mol−1 for the free polymer after 7 h of polymerization (Fig. 9 and 10). Unfortunately, the PDI deteriorated during the course of the polymerization reaction, namely from 1.6 to 2.6 despite the careful choice of reaction parameters. This might be the direct result of the increase viscosity of the system during the course of the reaction which hinders the stirring thus affecting parameters like uniform heat transfer. On the other hand, this can also be interpreted as the direct result of branching and cross-linking between the polymers in the system which could explain the formation of ensembles of the particles as explained earlier.40 This phenomenon likely occurs extensively after proceeding the reaction for more than 1 h and possibly explains the deterioration in the PDI.
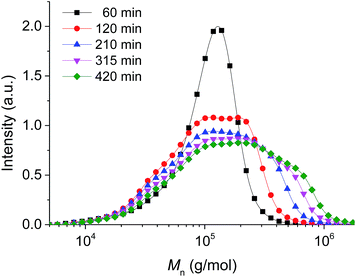 |
| | Fig. 9 Molecular weight (Mn) of free, sacrificial PMMA obtained from GPC measurements of aliquots taken after a prescribed amount of reaction time. | |
 |
| | Fig. 10 The development of the molecular weight Mn and the polymer dispersity index (PDI) with time. | |
The TGA measurements under He atmosphere were performed for Ag@SiO2, APTES–Br, and PMMA-coated Ag@SiO2 core–shell particles. The results shown in Fig. 11 illustrate an increase in the amount of decomposed materials and a decrease in the residual mass with each functionalization step due to the degradation of the organic components. The TGA curves of the PMMA-coated core–shell particles show different stages of degradation, a feature observed for the decomposition of PMMA prepared through radical initiation. The different stages of decomposition are a result of different polymer degradation mechanisms found in PMMA which are described elsewhere.65–68 Overall, the residual mass after the degradation of PMMA-coated Ag@SiO2 core–shell particles is about 79.9 wt%, indicating a polymer content of 14.2 wt% by taking the residual mass of APTES–Br functionalized core–shell particles after thermal degradation into account (Fig. 11). In volumetric terms, the Ag content in the core–shell particles is still around 20–30 vol%, and is within the vicinity of the percolation threshold described for Ag-based nanocomposites.13,69
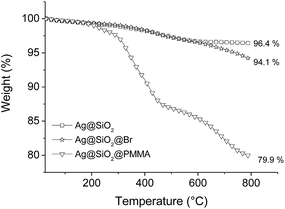 |
| | Fig. 11 TGA curves of Ag@SiO2, APTES–Br, and PMMA-coated Ag@SiO2 core–shell particles. | |
Conclusions
In summary, PMMA-coated Ag@SiO2 core–shell particles which exhibit enhanced solubility in organic, non-polar solvents have been prepared in gram scale by using surface-initiated ATRP. They can serve as an alternative set of core–shell particles to be used for the preparation of nanocomposites in a hydrophobic polymer matrix. The prospective interdigitation of the surface polymers with the polymer chains of the matrix can lead to enhanced packing densities within the composite system beneficial for properties such as air resistance or thermal stability. The enhanced dispersibility of the particles can lead to a homogenous dispersion of the particles into the polymer matrix and avoid the formation of micron-sized agglomerates which are likely to occur when the particles are dispersed into an incompatible matrix. Preliminary results show that this synthesis can be extended to a wide range of monomers.
Acknowledgements
Our special thanks go out to S. Dünki and B. Fischer for conducting the GPC and TGA measurements, respectively. We also gratefully acknowledge Swiss National Science Foundation (SNF132101), (SNFS 150638), and Swiss Federal Laboratories for Materials Science and Technology (Empa, Dübendorf) for financial support.
References
- J. Reszczyńska, A. Jurek, I. Łącka, E. Skwarek and A. Zaleska, Adv. Mater. Sci., 2010, 10, 12–20 Search PubMed.
- A. Goyal, A. Kumar, P. K. Patra, S. Mahendra, S. Tabatabaei, P. J. J. Alvarez, G. John and P. M. Ajayan, Macromol. Rapid Commun., 2009, 30, 1116–1122 CrossRef CAS PubMed.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS PubMed.
- B. J. Wiley, Y. Chen, J. M. McLellan, Y. Xiong, Z.-Y. Li, D. Ginger and Y. Xia, Nano Lett., 2007, 7, 1032–1036 CrossRef CAS PubMed.
- D. D. Evanoff and G. Chumanov, ChemPhysChem, 2005, 6, 1221–1231 CrossRef CAS PubMed.
- D. Adam and R. P. V. D. McFarland, Nano Lett., 2003, 3, 1057–1062 CrossRef.
- R. Xu, D. Wang, J. Zhang and Y. Li, Chem.–Asian J., 2006, 1, 888–893 CrossRef CAS PubMed.
- Z.-J. Jiang, C.-Y. Liu and L.-W. Sun, J. Phys. Chem. B, 2005, 109, 1730–1735 CrossRef CAS PubMed.
- J. Lu, K.-S. Moon and C. P. Wong, J. Mater. Chem., 2008, 18, 4821–4826 RSC.
- L. Qi, B. I. Lee, S. Chen, W. D. Samuels and G. J. Exarhos, Adv. Mater., 2005, 17, 1777–1781 CrossRef CAS.
- L. Lu, K.-S. Moon, J. Xu and C. P. Wong, J. Mater. Chem., 2006, 16, 1543–1548 RSC.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS PubMed.
- J. E. Q. Quinsaat, A. Testino, S. Pin, T. Huthwelker, P. Bowen, H. Hofmann, C. Ludwig and D. M. Opris, J. Phys. Chem. C, 2014, 118, 11093–11103 CAS.
- M. Nishioka, M. Miyakawa, H. Kataoka, H. Koda, K. Sato and T. M. Suzuki, Nanoscale, 2011, 3, 2621–2626 RSC.
- H. Mehenni, L. Sinatra, R. Mahfouz, K. Katsiev and O. M. Bakr, RSC Adv., 2013, 3, 22397–22403 RSC.
- J. Huang, L. Lin, Q. Li, D. Sun, Y. Wang, Y. Lu, N. He, K. Yang, X. Yang and H. Wang, Ind. Eng. Chem. Res., 2008, 47, 6081–6090 CrossRef CAS.
- B. Wiley, Y. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067–1076 CrossRef CAS PubMed.
- Q. Dong, H. Yu, Z. Jiao, G. Lu and Y. Bi, RSC Adv., 2014, 4, 59114–59117 RSC.
- X. Zhang, Y. Zhu, X. Yang, S. Wang, J. Shen, B. Lin and C. Li, Nanoscale, 2013, 5, 3359–3366 RSC.
- J. Qi, X. Dang, P. T. Hammond and A. M. Belcher, ACS Nano, 2011, 5, 7108–7116 CrossRef CAS PubMed.
- H. Baida, P. Billaud, S. Marhaba, D. Christofilos, E. Cottancin, A. Crut, J. Lermé, P. Maioli, M. Pellarin, M. Broyer, N. Del Fatti, F. Vallee, A. Sanchez-Iglesias, I. Pastoriza-Santos and L. M. Liz-Marzan, Nano Lett., 2009, 9, 3463–3469 CrossRef CAS PubMed.
- L. Rainville, M.-C. Dorais and D. Boudreau, RSC Adv., 2013, 3, 13953 RSC.
- W. Wang, L. Zhipeng, G. Baohua, Z. Zhenyu and X. Hongxin, ACS Nano, 2009, 3, 3493–3496 CrossRef CAS PubMed.
- Y. Zhou, L. Wang, H. Zhang, Y. Bai, Y. Niu and H. Wang, Appl. Phys. Lett., 2012, 101, 012903 CrossRef.
- X. Liang, S. Yu, R. Sun, S. Luo, J. Wan, S. Yu, R. Sun, S. Luo, X. Liang, J. Wan and Z. Zhuang, J. Mater. Res., 2012, 27, 991–998 CrossRef CAS.
- J. E. Q. Quinsaat, F. A. Nüesch, H. Hofmann and D. M. Opris, RSC Adv., 2013, 3, 6964–6971 RSC.
- D. M. Opris, J. E. Q. Quinsaat, S. Dünki, Y. S. Ko, M. Alexandru, C. Racles and F. A. Nüesch, Proc. SPIE, 2015, 94300A Search PubMed.
- J. E. Q. Quinsaat, M. Alexandru, F. A. Nüesch, H. Hofmann, A. Borgschulte and D. M. Opris, J. Mater. Chem. A, 2015, 3, 14675–14685 CAS.
- J. Shimomura, Y. Fujii, N. Hayakawa, M. Hanai and H. Okubo, IEEE Conf. Electr. Insul. Dielectr. Phenom., 2010, 978 Search PubMed.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS PubMed.
- H. K. Cho, H.-J. Cho, S. Lone, D.-D. Kim, J. H. Yeum and I. W. Cheong, J. Mater. Chem., 2011, 21, 15486–15493 RSC.
- T. I. Suratwala, M. L. Hanna, E. L. Miller, P. K. Whitman, I. M. Thomas, P. R. Ehrmann, R. S. Maxwell and A. K. Burnham, J. Non-Cryst. Solids, 2003, 316, 349–363 CrossRef CAS.
- V. Gun'ko, M. Vedamuthu, G. Henderson and J. Blitz, J. Colloid Interface Sci., 2000, 228, 157–170 CrossRef PubMed.
- C. H. Lee, S. H. Park, W. Chung, J. Y. Kim and S. H. Kim, Colloids Surf., A, 2011, 384, 318–322 CrossRef CAS.
- R. L. Kaas and J. L. Kardos, Polym. Eng. Sci., 1971, 11, 11–18 CAS.
- Z. F. Huang, X. Y. Qu and Z. Chen, J. Appl. Polym. Sci., 2015, 132, 41919 Search PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- C. M. Hui, J. Pietrasik, M. Schmitt, C. Mahoney, J. Choi, M. R. Bockstaller and K. Matyjaszewski, Chem. Mater., 2014, 26, 745–762 CrossRef CAS.
- I. Mora-Barrantes and J. Valentín, J. Mater. Chem., 2012, 22, 1403–1410 RSC.
- T. Von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497–7505 CrossRef CAS PubMed.
- T. E. Patten and K. Matyjaszewski, Adv. Mater., 1998, 10, 901–915 CrossRef CAS.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Nano Lett., 2002, 2, 3–7 CrossRef.
- H. Dong, M. Zhu, J. A. Yoon, H. Gao, R. Jin and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 12852–12853 CrossRef CAS PubMed.
- S. Nuss and H. Böttcher, Angew. Chem., Int. Ed., 2001, 40, 4016–4018 CrossRef CAS.
- D. Li, Y. J. Jang, J. J.-E. Lee, S. T. Kochuveedu and D. H. Kim, J. Mater. Chem., 2011, 21, 16453–16460 RSC.
- S. Chakraborty, S. W. Bishnoi and H. Pe, J. Phys. Chem. C, 2010, 114, 5947–5955 CAS.
- K. Ohno, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2002, 35, 8989–8993 CrossRef CAS.
- J. Zhang, L. Jiang, K. Pan, Z. Yi and Y. Dan, Polym. Int., 2014, 63, 413–419 CrossRef CAS.
- Y. Sun, X. Ding, Z. Zheng, X. Cheng, X. Hu and Y. Peng, Eur. Polym. J., 2007, 43, 762–772 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, USA, 2007 Search PubMed.
- J. Matsui, S. Parvin, E. Sato and T. Miyashita, Polym. J., 2010, 42, 142–147 CrossRef CAS.
- G. L. Li, L. Q. Xu, K. G. Neoh and E. T. Kang, Macromolecules, 2011, 44, 2365–2370 CrossRef CAS.
- K. Ohno, T. Akashi, Y. Huang and Y. Tsujii, Macromolecules, 2010, 43, 8805–8812 CrossRef CAS.
- M. Husseman and E. Malmström, Macromolecules, 1999, 32, 1424–1431 CrossRef CAS.
- G. Carrot, S. Diamanti, M. Manuszak, B. Charleux and J. P. Vairon, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4294–4301 CrossRef CAS.
- K. Matsuura, K. Ohno, S. Kagaya and H. Kitano, Macromol. Chem. Phys., 2007, 208, 862–873 CrossRef CAS.
- N. J. Fernandes, J. Akbarzadeh, H. Peterlik and E. P. Giannelis, ACS Nano, 2013, 7, 1265–1271 CrossRef CAS PubMed.
- V. V. Naik, M. Crobu, N. V. Venkataraman and N. D. Spencer, J. Phys. Chem. Lett., 2013, 4, 2745–2751 CrossRef CAS.
- Y. Sun, B. Mayers, T. Herricks and Y. Xia, Nano Lett., 2003, 3, 955–960 CrossRef CAS.
- G. Louis Chakkalakal, M. Alexandre, C. Abetz, A. Boschetti-de-Fierro and V. Abetz, Macromol. Chem. Phys., 2012, 213, 513–528 CrossRef.
- S. Edmondson and W. T. S. Huck, J. Mater. Chem., 2004, 14, 730–734 RSC.
- U. Chatterjee, S. K. Jewrajka and B. M. Mandal, Polymer, 2005, 46, 1575–1582 CrossRef CAS.
- K. H. Su, Q. H. Wei, X. Zhang, J. J. Mock, D. R. Smith and S. Schultz, Nano Lett., 2003, 3, 1087–1090 CrossRef CAS.
- K. Ohno, T. Morinaga, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2005, 38, 2137–2142 CrossRef CAS.
- T. Kashiwagi, A. Inaba, J. E. Brown, K. Hatada, T. Kitayama and E. Masuda, Macromolecules, 1986, 19, 2160–2168 CrossRef CAS.
- A. Inaba, T. Kashiwagi and J. E. Brown, Polym. Degrad. Stab., 1988, 21, 1–20 CrossRef CAS.
- T. Kashiwagi and A. Inabi, Polym. Degrad. Stab., 1989, 26, 161–184 CrossRef CAS.
- L. E. Manring, Macromolecules, 1991, 24, 3304–3309 CrossRef CAS.
- S. Rajesh, K. Sonoda, A. Uusimaki, K. H. Yang, H. Y. Lu and H. Jantunen, J. Mater. Sci.: Mater. Electron., 2012, 24, 191–195 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.