
A biocompatible poly(N-vinylimidazole)-dot with both strong luminescence and good catalytic activity†
Bin Wang,
Hua-Ji Liu* and
Yu Chen*
Department of Chemistry, School of Sciences, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300354, P. R. China. E-mail: liuhuaji@tju.edu.cn; chenyu@tju.edu.cn
First published on 22nd December 2015
Abstract
Poly(N-vinylimidazole) (PVIm) that contains a large amount of bio-active imidazole units was used as the sole carbon source to synthesize PVIm-dot through a one-pot hydrothermal method without any further modification and surface passivation. The measurements of X-ray photoelectron spectroscopy, dynamic light scattering, transmission electron microscopy and X-ray diffraction proved that only a slight carbonization occurred during the hydrothermal treatment of PVIm. The characterizations of 1H NMR, FTIR and thermogravimetric analysis verified that the obtained PVIm-dot well inherited the chemical structure of its precursor PVIm. Unlike PVIm, the obtained PVIm-dot showed an obvious excitation-dependent photoluminescence (PL) behavior, and its PL features were quite stable at different pH values and ionic strength. The PVIm-dot possessed low cytotoxicity and could enter cancer cells, making it a suitable candidate for bio-imaging. Moreover, the PVIm-dot still kept the catalytic activity of its imidazole units. With the catalytic hydrolysis of p-nitrophenyl acetate as the model reaction, it was found that the PVIm-dot showed good catalytic activity in this reaction and its catalytic efficiency was better than PVIm. What's more, the variation of PL intensity during the reaction could be used as a luminescent sensor to monitor the progress of the hydrolysis reaction.
Introduction
During the past two decades, great attention has been paid to fluorescent nanocrystals due to their high quantum yield, tunable florescence colour, good photostability and resistance to metabolic degradation in bio-applications.1–4 Heavy metal quantum dots, such as CdTe, PbTe and CdSe show excellent fluorescent ability, but their long term toxicity and environmental hazard restrict their application.5–7 Extensive efforts have thus been made on the development of non- or low-toxic fluorescent materials as alternatives to semiconductor-based quantum dots. Carbon quantum dots (CQDs), which are always less than 10 nm in size, are particularly encouraging owning to their outstanding good biocompatibility, low toxicity and robust chemical inertness.8–10Many approaches have been developed to synthesize CQDs. Top-down approaches, including laser ablation,11,12 electrochemical oxidation,13 and chemical oxidation14–16 are currently regarded as state of the art methods to synthesize CQDs. However, these approaches always involve rigorous experimental condition and tedious preparation processes. By contrast, bottom-up approaches, including microwave method,10,17 combustion18 and hydrothermal techniques19,20 seem to be much more simple and convenient. The chemical precursors can be used to produce CQDs through the bottom-up approach are various, including glucose,21 chitosan,22 glycol,23 poly(vinyl alcohol),24 polyethylene glycol,25 critical acid,26 amino acid,27 or some natural bio-resources, such as orange juice,28 soy milk29 and so on. Among all these precursor compounds, hydroxyl, primary amino or carboxyl groups are widely existed. During the synthetic processes, they will go carbonization or dehydration. Accompanied with the formation of CQDs, the chemical structure of the parent compounds were sacrificed and seriously damaged. This led that the properties of the precursors vanished and the application fields of these CQDs were only limited in photo-related fields. Further widening the applications of these CQDs, special additives for the surface modification/passivation should be used.9,10,30
Imidazole is one of the most important heterocyclic aromatic compounds and it plays a crucial role in primary biomacromolecules, such as amino acid, nucleic acids and proteins, and also in many pharmaceutical and agrochemical compounds. N-Vinylimidazole (VIm) was frequently used to prepared synthetic macromolecules with imidazole group. Its homo- or copolymers had many potential applications, such as fuel cells,31 membranes for metal ion removal,32,33 gene delivery,34,35 ionic liquid,36,37 and catalysts.38,39 Herein, we reported a simple hydrothermal approach to prepare polymer dots derived from poly(N-vinylimidazole) (PVIm). The synthesis was conducted at mild condition, and did not need any additives for the surface modification/passivation. The resultant product (PVIm-dot) could be well dispersed in aqueous solution and displayed an excitation tunable luminescence. PVIm-dot possessed low cytotoxicity and could enter the cancer cell, making it a suitable candidate for bio-imaging. What's more exciting, the resultant PVIm-dot inherited the chemical structure of its PVIm precursor. Therefore, PVIm-dot still kept the function of imidazole, such as catalysis. To our best knowledge, highly luminescent polymer carbon dot preserving the chemical properties of the precursors has never been reported. The PVIm-dot prepared here was the first polymer dot that was not only luminescent, but also kept the catalytic activity of its precursor.
Experimental
Materials
N-Vinylimidazole (VIm, 99%) was purchased from Aldrich and purified by vacuum distillation before use. Azoisobutyronitrile (AIBN) was purchased from Fluka and recrystallized from methanol solution. p-Nitrophenyl acetate (NPA, 98%) was purchased from TCI and used as received. High glucose Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), phosphate buffer saline (DPBS), trypsin, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT, 98%), and any other reagents and solvents were purchased from Beijing Solarbio Science & Technology Co., Ltd.Characterization
1H NMR spectra were recorded at 25 °C on a Varian INOVA 400 MHz spectrometer. Fourier transform infrared (FTIR) spectra were recorded on a Japan Shimadzu FTIR-8400S, and the scanning range was 4000–400 cm−1. Ultraviolet-visible (UV-vis) absorption spectra were collected using a Purkinje General (China) T6 UV/Vis spectrophotometer. Fluorescence spectra were recorded using a Varian Cary Eclipse photoluminescence spectrometer with a scan rate of 600 nm min−1. Fluorescence decay spectra were performed with a Fluorolog 3 (HOR1BA JOB1N YVON). X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI 5000 Versa Probe photoelectron spectrometer with Alfa X-ray radiation as the X-ray source for excitation. Dynamic light scattering (DLS) measurement were performed using a Malvern Nano ZS instrument at 25 °C with 633 nm He–Ne laser light and light collection at 90°. Transmission electron microscopy (TEM) studies were performed with a JEOL JEM-2100F instrument at a voltage of 200 kV. The X-ray diffraction (XRD) spectrum was recorded on a BDX3300 X-ray diffractometer (Beijing University Instrument Factory) using Cu Kα radiation (wavelength 0.1542 nm) and a scan step of 0.02° at 25 °C. Thermogravimetric analysis (TGA) was carried out on Netzsch STA 409 PC/PG in the temperature range of 30 to 700 °C at a heating rate of 10 °C min−1 and nitrogen flow rates of 30 mL min−1. The cells were imaged under an FV 1000S-IX81 confocal laser scanning microscope (Olympus).Polymerization
A typical procedure for polymerization was as follows: VIm (2 g, 21.3 mM) and AIBN (24 mg) were dissolved in 10 mL of methanol, and the mixture was degassed through three cycles of vacuum–nitrogen purge and then immersed in an oil bath thermostated at 70 °C for 12 hours. The resultant solution was dialyzed in methanol for 48 hours, and the dialysis solvent was changed every 12 hours. After the dialysis, the solvent was removed under vacuum and the polymer was dried under vacuum at room temperature overnight.Synthesis of PVIm-dot
Four hundred milligram of polymer was dissolved in 10 mL of deionized water. The solution was transferred to a 25 mL-high-pressure vessel and heated to 200 °C for 8.5 hours. After the vessel was cooled to room temperature, the brown solution was filtered with a 0.22 μm filter, and then the solution was freeze-dried for 36 hours.Catalytic reaction
The hydrolysis of NPA was conducted in a quartz cuvette using a UV-vis spectrophotometer equipped with a temperature controller. 2 mL of PVIm-dot with a concentration of 0.5 mg mL−1 in aqueous solution was kept in the cuvette at 30 °C for 30 min, and then 5 μL of NPA solution (0.4 M, in acetonitrile) was promptly injected into the cuvette with an injector. The reaction mixture was immediately agitated with a glass pipette for 5 s, and the optical density at 402 nm of the solution was detected by the UV-vis spectrometer. The fluorescence intensity of the PVIm-dot during the reaction was record by a Varian Cary Eclipse photoluminescence spectrometer at an excitation wavelength of 345 nm.Measurement of fluorescence quantum yield
The quantum yield of PVIm-dot was determined by a comparative method. Quinine sulfate in 0.1 M H2SO4 (literature quantum yield: 54%) was selected as a standard sample to calculate the QY of test sample which was dissolved in deionized water at different concentrations. All the absorbance values of the solutions at excitation wavelength were measured with UV-vis spectrophotometer. Photoluminescence (PL) emission spectra of all the samples were recorded by photoluminescence spectrometer at an excitation wavelength of 345 nm. The integrated fluorescence intensity is the area under the PL curve in the wavelength range from 370 to 600 nm. Then a graph was plotted using the integrated fluorescence intensity against the absorbance and trend line was added for each curve with intercept at zero. Absolute values were calculated according to the following equation:| ΦX = ΦST(GradX/GradST)(ηX2/ηST2) |
Cell culture
HeLa cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco) with high glucose, containing 10% fetal bovine serum (FBS) at 37 °C in 5% CO2 humidified atmosphere.Cell viability assay
The cytotoxicity of PVIm-dot was assessed through MTT assay. HeLa cells were seeded in a 96-well plate at a density of 1.5 × 104 cells per well and incubated for 24 hours. Then the culture medium in each well was replaced by 150 μL of fresh medium which contained different amount of PVIm-dot. After incubation for 24 hours, 20 μL of MTT (5 mg mL−1 in DPBS) was added to each well and incubated for another 4 hours. Finally all medium was removed and 150 μL of DMSO was added, followed by shaking for 15 min. The absorbance for each well was measured at 492 nm using an Anthos 2010 microplate reader, and pure DMSO was used as a blank and non-treated cell was used as a control. The relative cell viability (mean% ± SD, n = 5) was expressed as Abssample/Abscontrol × 100%.Cellular imaging
Hela cells were seeded over a glass coverslip in a 6-well plate at a density of 2 × 105 cells per well and incubated for 12 hours. Then the culture medium in each well was replaced with 2 mL of fresh medium which contain 100 μg of PVIm-dot. After being incubated for another 12 hours, the medium was removed and the cells were washed with DPBS three times, and then fixed with 4% paraformaldehyde solution in PBS for 10 minutes. The samples were observed under an FV1000S-IX81 confocal laser scanning microscope with the excitation wavelength of 405 nm.Results and discussion
Synthesis and characterization of PVIm-dot
Imidazole, a nitrogen rich heterocyclic aromatic compound, which is thought hard to be carbonized and dehydrated, is quite stable at high temperature.40 Herein, PVIm was used as a sole carbon source to synthesize the polymer dot through a one-pot hydrothermal method without the involvement of acid, alkali, salt, organic solvent and any further modification/passivation. The aqueous solution of PVIm is colorless and transparent. After the hydrothermal treatment, the solution changes from colorless to brown, where the as-prepared PVIm-dot is freely dispersed in the aqueous solution with a transparent appearance without further ultrasonic dispersion (see Scheme 1).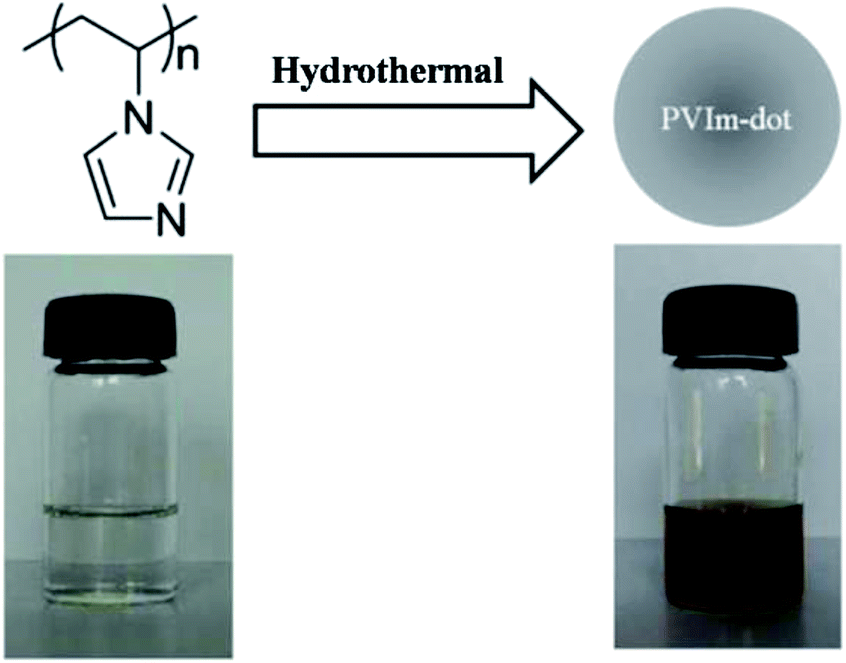 | ||
| Scheme 1 Scheme for the hydrothermal conversion and the photographs of PVIm aqueous solution before and after the hydrothermal reaction. | ||
The aqueous solution of PVIm is non-emissive in the visible region under UV light, whereas the hydrothermal treatment leads to the appearance of strong fluorescence under UV light. The occurrence of strong fluorescence implies that PVIm must experience some chemical reactions, such as oxidation or carbonization, under the hydrothermal condition, resulting in the formation of emissive PVIm-dot. In order to find the best reaction condition to generate more PVIm-dots and/or PVIm-dot with more efficient emitting property, different hydrothermal temperatures and time were investigated. Fig. 1a shows that increasing the hydrothermal temperature from 140 to 200 °C leads to a significant increase of the PL intensity. This means that a higher hydrothermal temperature endows the product stronger PL intensity. Since 200 °C reaches the maximum work temperature of our oven, we set the best hydrothermal temperature as 200 °C. When increasing the hydrothermal time from 3 to 8.5 hours, the PL intensity of the product shows an obvious increase, but further prolonging the reaction time to 11 hours, the PL intensity does not increase much (Fig. 1b). So the optimal hydrothermal condition was set as 200 °C and 8.5 hours.
 | ||
| Fig. 1 PL spectra of PVIm-dot prepared (a) at different hydrothermal temperature after 4.5 h and (b) after different hydrothermal time at 200 °C (concentration of PVIm is 1 mg mL−1; excitation wavelength is 330 nm). | ||
DLS was used to monitor the change of the particle size of PVIm during the hydrothermal process (Fig. 2a). It is found that no obvious aggregation occurs, and the diameter of PVIm-dot is always in the range of 5–8 nm. Fig. 2b shows the transmission electron microscopy (TEM) image of the as-prepared PVIm-dot. It can be seen that the image of PVIm-dot is not so clear. This is because the carbonization of PVIm during the hydrothermal process is not so sufficient and only a slight carbonization occurs. Moreover, the high resolution TEM (HRTEM) image of the PVIm-dot does not reveal any clear lattice fringes, indicating the amorphous nature of the PVIm-dot. At the same time, the XRD pattern of the PVIm-dot shows a broad peak at 2θ = 23° (Fig. 2c), revealing an amorphous carbon phase,41 which agrees well with the HRTEM analysis.
 | ||
| Fig. 2 (a) DLS curves of PVIm-dot prepared after different hydrothermal time, (b) TEM image of PVIm-dot, and (c) XRD pattern of PVIm-dot. | ||
The structure and components of PVIm and PVIm-dot were also identified by XPS. The PVIm starting material (Fig. 3a–c) shows a single C 1s peak with a binding energy of 284.8 eV, which is deconvoluted into two peaks associated with C–C/C–H carbon (284.7 eV) and C–N from the imidazole functionality (286 eV) (Fig. 3a). The N 1s spectrum (Fig. 3b) demonstrates the doping of both pyridinic (398.4 eV) and pyrrolic (400.3 eV) N atoms.2 There is also a signal of weak intensity corresponding to O 1s at a binding energy of 531.5 eV (Fig. 3c), and this is thought to be due to the slow oxidation of the polymer surface due to the aging. In comparison to the XPS data of the starting material, the hydrothermal product PVIm-dot shows many similarities (Fig. 3d–f). The intensity of N 1s signal has a high N 1s/C 1s ratio (0.27), indicating that this sample has photoelectron signal intensity close to that of the PVIm starting material. The main difference between the two samples can be seen when examining the O 1s signals. In contrast to the starting material, the hydrothermal product PVIm-dot shows a strong signal with a binding energy of 531.6 eV which is due to the carbonyl oxygen. This is thought to due to the oxidation process which occurs during the hydrothermal of PVIm. From the XPS analysis, it is clear that in the synthesized material, much of the starting material functionality is maintained. New oxygen functionalities indicate that some carbonyl groups decorate the PVIm-dot surface.
 | ||
| Fig. 3 XPS data for the PVIm starting materials: (a) C 1s, (b) N 1s, (c) O 1s and PVIm-dot, (d) C 1s, (e) N 1s, (f) O 1s. | ||
The starting PVIm and its hydrothermal product PVIm-dot were also investigated using 1H NMR, FTIR and TGA. Fig. 4a shows the 1H NMR spectra of PVIm and PVIm-dot in deuterated water. For PVIm, the signals corresponding to the backbone protons appear between 1.8 and 3.7 ppm, while the signals around 6.4 and 7.2 ppm belong to the three protons in the imidazole ring.40 For the hydrothermal product PVIm-dot, the 1H NMR spectrum do not show any change to its precursor polymer and the signals of the protons in the imidazole ring and the protons in the backbone all exist, indicating that the hydrothermal process does not damage the chemical structure of PVIm. The FTIR spectrum of PVIm and PVIm-dot are showed in Fig. 4b. For PVIm, the FTIR spectrum exhibits the following characteristic bands: 3110 cm−1 (C–H ring stretching mode), 2945 cm−1 (CH and CH2 the main chain stretching modes), 1650 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ring stretching modes) and 1499 cm−1 (C–C, CN ring stretching modes). For PVIm-dot, its FTIR spectrum is almost the same as PVIm. According to Fig. 4c, the TGA curves of PVIm-dot is similar to PVIm, the thermal decomposition of the two samples take place in one major step in the temperature range of 360–480 °C, and the maximum decomposition temperature is 440 °C. These test results fully indicate that the hydrothermal product PVIm-dot has a similar chemical structure to its precursor, and the structure information of PVIm has been inherited by PVIm-dot sufficiently.
C ring stretching modes) and 1499 cm−1 (C–C, CN ring stretching modes). For PVIm-dot, its FTIR spectrum is almost the same as PVIm. According to Fig. 4c, the TGA curves of PVIm-dot is similar to PVIm, the thermal decomposition of the two samples take place in one major step in the temperature range of 360–480 °C, and the maximum decomposition temperature is 440 °C. These test results fully indicate that the hydrothermal product PVIm-dot has a similar chemical structure to its precursor, and the structure information of PVIm has been inherited by PVIm-dot sufficiently.
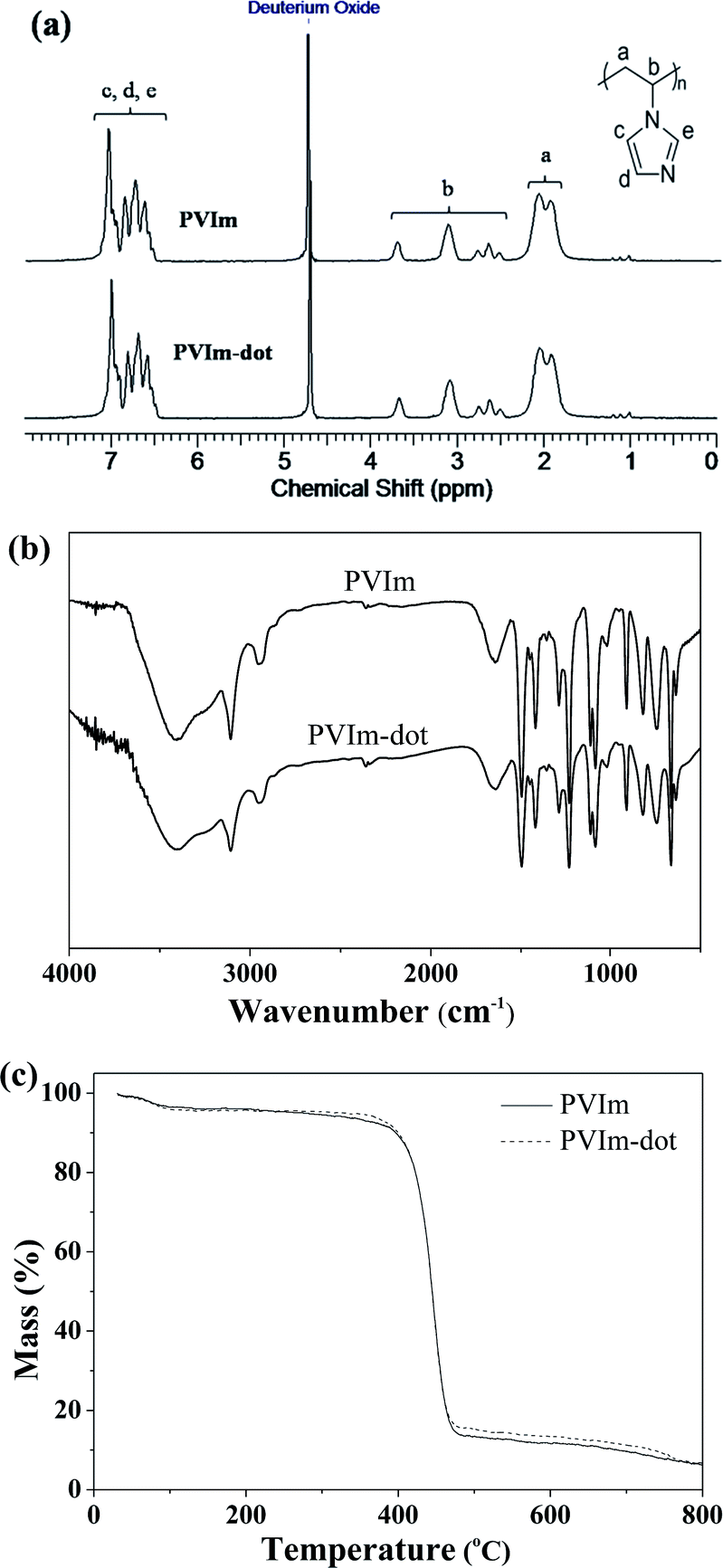 | ||
| Fig. 4 (a) 1H NMR spectra, (b) FTIR spectra and (c) TGA curves of the precursor polymer PVIm and its hydrothermal product PVIm-dot. | ||
Photoluminescence property of PVIm-dot
The UV-vis absorption spectra of PVIm-dot and its PL excitation and emission spectra are shown in Fig. 5a. A strong UV-vis absorption peak at 335 nm is found, which is similar to other common carbon dots. In the fluorescence spectra, the optimal excitation and emission wavelength of PVIm-dot center at 335 nm and 429 nm, showing a blue color under a hand-held UV lamp. The inset in Fig. 5a shows the UV optical images of the polymer dot under illumination of visible and UV light. PVIm-dot exhibits a bright blue photoluminescence, which is strong enough to be easily recognized by naked eyes. Its excitation-dependent PL behavior was investigated. When the excitation wavelengths change from 330 nm to 465 nm, the emission peak position, as being showed in Fig. 5b, is red-shifted from 425 nm to 545 nm, and the PL intensity decreases remarkably, indicating a strong dependence on the excitation wavelengths. This phenomenon is quite similar to other fluorescent carbon dots. Although, up to date, the origins of PL in polymer dots are not very clear, we reason that this excitation dependence phenomenon may come from the emissive traps, electronic conjugate structures, and free zigzag sites of PVIm-dot.30 Using quinine sulfate as a reference, the PL quantum yield of the PVIm-dot prepared under the optimal condition is measured to be 8.0% (ESI, Fig. S1†), which is much higher than some other polymer dot based on poly(vinyl alcohol) or polysaccharides.22,24 We also examined the polymer dot fluorescence lifetime at 339 nm excitation (Fig. 5c). The resultant average life time is 8.8 ns and as far as we know, it is much longer than other carbon dots ever reported.42 | ||
| Fig. 5 (a) PL and UV-vis spectra of PVIm-dot at a concentration of 1 mg ml−1, (b) PL spectra of PVIm-dot with the increase of excitation wavelength from 330 to 465 nm with a 15 nm interval, and (c) the time-resolved fluorescence decay of PVIm-dot. | ||
PVIm-dot processes excellent solubility in water and the fluorescent property of PVIm-dot at different ionic strengths are monitored. When increasing the concentration of KCl from 0 to 2.0 M, there is only a slight decrease in its PL intensity (ESI, Fig. S2†), which is beneficial for the use of PVIm-dot in salt solutions. Another interesting phenomenon is the pH-dependent PL behavior of PVIm-dot (ESI, Fig. S3†). PVIm-dot shows the strongest PL intensity at pH = 7. When increasing the pH from 7 to 13, there are no changes in its characteristic peak and its PL intensity just decreases a little. But when adjusting the pH of the aqueous solution of PVIm-dot from 7 to 2, the PL intensity decreases significantly and the emission peak shows a blue-shifting from 440 nm to 421 nm. This is possibly caused by the quaternization of the imidazole group in the polymer chain in acidic condition.
The application of PVIm-dot
Through the above discussion, it is clear that during the hydrothermal reaction, slight carbonization and oxidation occur in the PVIm precursor. The resultant PVIm-dot acquires a strong and stable PL ability, and at the same time, the chemical structure of the precursor polymer is finely preserved in PVIm-dot. In order to verify whether the properties of PVIm had been fully remained by PVIm-dot, herein, a typical reaction that the hydrolysis of p-nitrophenyl acetate (NPA) catalyzed by PVIm-dot was conducted (Scheme 2). | ||
| Scheme 2 The hydrolysis reaction of p-nitrophenyl acetate catalyzed by PVIm-dot. | ||
PVIm is well known to act as a selective catalyst for esterolysis via the formation of catalyst–substrate complexes.43 In this section, PVIm and its hydrothermal product with different hydrothermal time for catalyzing the hydrolysis of NPA at 30 °C were conducted. UV-vis spectrum was used to monitor the progress of catalytic reaction through tracing the rate of absorbance increasing at 402 nm due to the release of p-nitrophenol in its ionized form (ESI, Fig. S4†).38,39 The intensity of absorbance at 402 nm with time was given in Fig. 6a. One can see that at the beginning of the reaction, the absorbance increases linearly with time, therefore the slope of the curve herein can be used to represent the reaction rate. Interestingly, with the increase of hydrothermal time of PVIm-dot, its reaction rate for catalyzing the hydrolysis of NPA increases. That means that the PVIm-dot with a longer hydrothermal time has a higher catalytic efficiency. This observation might arise from the adsorption of substrates at the carbonization center of the PVIm-dot which was formed during the hydrothermal process via hydrophobic association.44 Therefore, it can be deduced that the hydrothermal product PVIm-dot fully inherits the catalysis ability of its precursor PVIm, and even shows better catalytic efficiency than PVIm.
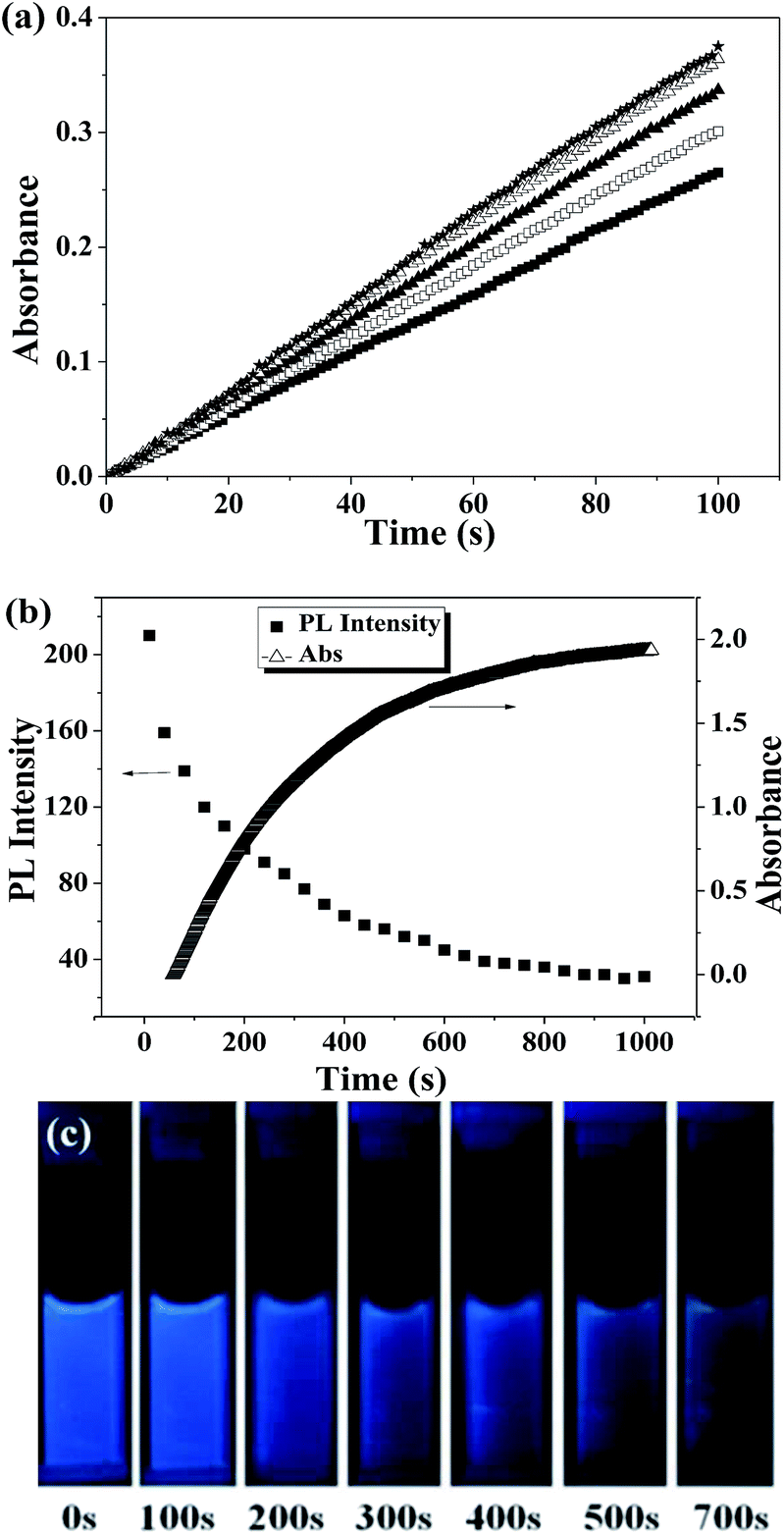 | ||
| Fig. 6 (a) Time dependence of the solution absorbance recorded during the hydrolysis of NPA catalyzed with PVIm-dot prepared for the different time (■ 0 h, □ 2 h, ▲ 4 h, △ 6 h, ★ 8.5 h) of hydrothermal treatment, (b) time dependence of the solution absorbance and PL intensity during the hydrolysis reaction (PVIm-dot prepared under 8.5 h of hydrothermal treatment was used as the catalyst), and (c) photography of the reaction system under a UV lamp. | ||
PVIm-dot can be also used as a PL sensor to monitor the hydrolysis of NPA. First, the variation of absorbance at 402 nm with time is shown in Fig. 6b. At the beginning, the absorbance increases rapidly. As the reaction goes on, the reactant NPA is consumed, and the increasing tendency of absorbance slows down. At last, the hydrolysis reaction is over, and the absorbance does not increase any more. Correspondingly, the PL intensity of the catalyst PVIm-dot is also given in Fig. 6b. At the beginning of the hydrolysis reaction, the PL intensity decreases rapidly, and then slows down, and at last remains constant. This is attributed to the quenching effect of hydrolysis product p-nitrophenol. It is worth pointing out that the PL intensity at different time has good linear relationship with the absorbance, and follows the following formula:
| Ipl = −80A + 212 |
As the relationship between absorbance and the mount of p-nitrophenol follows the Lambert–Beer law, the previous formula can be transformed into the following style:
| c = (212 − Ipl)/80εl |
Fluorescent CDs have drawn much attention especially for their application as optical imaging agents because of their excellent biocompatibility, low toxicity, and chemical stability. PVIm-dot in water carries certain amount of positive charges since it has many basic imidazole groups. Therefore it is possible for PVIm-dots to be adsorbed by the negatively-charged membrane surface of cell due to the electrostatic interaction. In the present research, PVIm-dot (QY of 8.0%) was used as an optical imaging agent to label HeLa cells (the QY of PVIm-dot is not very high but is adequate for bioimaging applications24). Low toxicity is a key point in bioimaging. The MTT assay method was used to access the cytotoxicity of PVIm-dot. HeLa cells were cultured in the medium with a certain concentration of PVIm-dot for 24 hours. The results are shown in Fig. 7a. The viability of HeLa cells shows slight decrease with the increase of the PVIm concentration. But even when the concentration of PVIm-dot is 500 μg mL−1, the cell viability is almost 90%, which verifies the low toxicity of PVIm-dot. HeLa cells were cultured in the medium containing 100 μg mL−1 of PVIm-dot for 12 hours, and the bright field image of the fluorescence microscope (Fig. 7b) proves that these cells are healthy. The HeLa cells that were treated with pure DMEM medium do not show any fluorescence under 365 nm excitation, and correspondingly, the HeLa cells which were treated with medium contain PVIm-dot show bright blue color under UV excitation (Fig. 7c). The superposition of bright field image and fluorescent field image of HeLa cells treated with PVIm-dot shows that the emissive PVIm-dots enter inside the cells, but outside the cell nucleus. So the PVIm-dot can serve as a promising candidate for in vivo imaging and biosensors because of its low toxicity and good biocompatibility.
 | ||
| Fig. 7 (a) Cellular toxicity of PVIm-dot on HeLa cell viability, (b) bright field image, (c) fluorescent field image, and (d) superposition of bright field image and fluorescent field image of HeLa cells treated with PVIm-dot for 12 h. | ||
Conclusions
In this work, PVIm-dot was prepared through a simple hydrothermal method using PVIm as a sole carbon source without additives (acid, alkali, or salt). Under high temperature and pressure in the reaction vessel, PVIm experienced a slight carbonization and oxidation to form the final PVIm-dot. The as-prepared PVIm-dot inherited the chemical structure and physical properties of its precursor PVIm, and showed a strong PL behavior in aqueous solution. It also had a high yield and good water solubility and suitable for use in different pH values and KCl aqueous solutions. PVIm-dot could be used to catalyze the hydrolysis of NPA just like PVIm precursor, but showed better catalytic activity. Moreover, it could be also used as a luminescent sensor to monitor the hydrolysis process of NPA. PVIm-dot possessed low cytotoxicity, and could enter the cancer cells, making it a suitable candidate for bio-imaging.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (20804027, 21274106).Notes and references
- X. Liu, R. Deng, Y. Zhang, Y. Wang, H. Chang, L. Huang and X. Liu, Chem. Soc. Rev., 2015, 44, 1479–1508 RSC.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889–896 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–545 CrossRef CAS PubMed.
- E. M. Ali, Y. Zheng, H.-h. Yu and J. Y. Ying, Anal. Chem., 2007, 79, 9452–9458 CrossRef CAS PubMed.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS PubMed.
- C. Dong, H. Qian, N. Fang and J. Ren, J. Phys. Chem. B, 2006, 110, 11069–11075 CrossRef CAS PubMed.
- X. Jia, J. Lia and E. Wang, Nanoscale, 2012, 4, 5572–5575 RSC.
- X. Liu, H.-J. Liu, F. Cheng and Y. Chen, Nanoscale, 2014, 6, 7453–7460 RSC.
- J.-Y. Yin, H.-J. Liu, S. Jiang, Y. Chen and Y. Yao, ACS Macro Lett., 2013, 2, 1033–1037 CrossRef CAS.
- S.-L. Hu, K.-Y. Niu, J. Sun, J. Yang, N.-Q. Zhao and X.-W. Du, J. Mater. Chem., 2009, 19, 484–488 RSC.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- Q.-L. Zhao, Z.-L. Zhang, B.-H. Huang, J. Peng, M. Zhang and D.-W. Pang, Chem. Commun., 2008, 41, 5116–5118 RSC.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- M. J. Krysmann, A. Kelarakis and E. P. Giannelis, Green Chem., 2012, 14, 3141–3145 RSC.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 34, 5118–5120 RSC.
- S. C. Ray, A. Saha, N. R. Jana and R. Sarkar, J. Phys. Chem. C, 2009, 113, 18546–18551 CAS.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- H. Ding, J.-S. Wei and H.-M. Xiang, Nanoscale, 2014, 6, 13817–13823 RSC.
- Z.-C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X.-H. Zhang, H. Tan, A. Yuang, C. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- Y. Yang, J. Cui, M. Zheng, C. Hu, S. Tan, Y. Xiao, Q. Yang and Y. Liu, Chem. Commun., 2012, 48, 380–382 RSC.
- Y. Lei, J. Weihua, Q. Lipeng, J. Xuewei, Z. Daiying, W. Dongkai and Y. Li, Nanoscale, 2015, 7, 6104–6113 RSC.
- S. Zhu, J. Zhang, L. Wang, Y. Song, G. Zhang, H. Wang and B. Yang, Chem. Commun., 2012, 48, 10889–10891 RSC.
- R.-J. Fan, Q. Sun, L. Zhang, Y. Zhang and A.-H. Lu, Carbon, 2014, 71, 87–93 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- H. Huang, C. Li, S. Zhu, H. Wang, C. Chen, Z. Wang, T. Bai, Z. Shi and S. Feng, Langmuir, 2014, 30, 13542–13548 CrossRef CAS PubMed.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatr, Chem. Commun., 2012, 48, 8835–8837 RSC.
- C. Zhu, J. Zhai and S. Dong, Chem. Commun., 2012, 48, 9367–9369 RSC.
- C. Liu, P. Zhang, X. Zhai, F. Tian, W. Li, J. Yang, Y. Liu, H. Wang, W. Wang and W. Liu, Biomaterials, 2012, 33, 3604–3613 CrossRef CAS PubMed.
- Y. S. Guan, H. T. Pu, M. Jin, Z. H. Chang and D. C. Wan, Fuel Cells, 2010, 10, 973–982 CrossRef CAS.
- M. Takafuji, S. Ide, H. Ihara and Z. Xu, Chem. Mater., 2004, 16, 1977–1983 CrossRef CAS.
- Z. Ajji and A. M. Ali, J. Hazard. Mater., 2010, 173, 71–74 CrossRef CAS PubMed.
- S. Santanakrishnan and R. A. Hutchinson, Macromol. Chem. Phys., 2013, 214, 1140–1146 CrossRef CAS.
- S. Asayama, S. Nishinohara and H. Kawakami, Bioconjugate Chem., 2011, 22, 1864–1868 CrossRef CAS PubMed.
- J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS.
- S. Soll, M. Antonietti and J. Yuan, ACS Macro Lett., 2012, 1, 84–87 CrossRef CAS.
- Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu and S. Liu, Macromolecules, 2007, 40, 3538–3546 CrossRef CAS.
- B. Wang, H.-J. Liu, T.-T. Jiang, Q.-H. Li and Y. Chen, Polymer, 2014, 55, 6036–6043 CrossRef CAS.
- C. Fodor, J. Bozi, M. Blazsó and B. Iván, Macromolecules, 2012, 45, 8953–8960 CrossRef CAS.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS PubMed.
- H. Liu, Z. He, L.-P. Jiang and J.-J. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 4913–4920 CAS.
- W. D. Horrocks and G. N. L. Mar, J. Am. Chem. Soc., 1963, 85, 3512–3513 CrossRef CAS.
- I. M. Okhapkin, E. E. Makhaeva and A. R. Khokhlov, Adv. Polym. Sci., 2006, 195, 177–210 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Quantum yield measurement; effect of salt and pH on the photoluminescence; time dependent UV-vis spectra of the hydrolysis of NPA catalyzed with PVIm-dot. See DOI: 10.1039/c5ra20640e |
| This journal is © The Royal Society of Chemistry 2016 |