
Fabrication of rod-like nanocapsules based on polylactide and 3,4-dihydroxyphenylalanine for a drug delivery system
Dongjian Shia,
Lei Zhanga,
Jiali Shena,
Xiaojie Lia,
Mingqing Chen*a and
Mitsuru Akashib
aThe Key Laboratory of Food Colloids and Biotechnology Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, P. R. China. E-mail: mqchen@jiangnan.edu.cn; Tel: +86-510-85917019
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita 565-0871, Japan
First published on 19th November 2015
Abstract
3,4-Dihydroxyphenylalanine (DOPA) has the property of self-polymerization to form a PDOPA polymer with crosslinking structure, and coats onto surfaces of diverse substrates at alkaline pH values. In this study, rod-like nanocapsules were facilely fabricated based on a bio-based polymer by taking advantage of the DOPA properties. A block-like poly(lactide)-b-amidated poly(3,4-dihydroxyphenylalanine) (PLA-b-APDOPA) copolymer was firstly synthesized through an amidation reaction with pre-prepared functional PLA and APDOPA. The DOPA compound and obtained PLA-b-APDOPA copolymer were subsequently coated onto the silica nanorods to get PLA-b-APDOPA/PDOPA@SiO2 nanorods. Afterwards, PLA-b-APDOPA/PDOPA nanocapsules were formed by removal of the silica template. The structure of the copolymer was confirmed by a 1H NMR spectrum. The formed nanorods and nanocapsules were observed by SEM and TEM. The structure and amount of the coated layers were determined by XPS and TGA. The results showed a rough surface of the nanorods after being coated with the polymers and the formation of a thin PDOPA layer and a thick PLA-b-APDOPA layer on the silica surface. Moreover, the formed nanocapsules had good biocompatibility. A model drug was successfully entrapped into the capsules, and could be slowly released from the nanocapsules in vitro depending on the pH buffer. The obtained rod-like nanocapsules could be used as carriers in biomedical fields.
Introduction
Spherical nanoparticles (NPs) with multi-functionalities have been developed as nano-carriers for sustained release, molecular targeting, and environmental responsiveness in biomedical fields1–4 due to their high drug loading and cellular uptake efficacies. Bio-inspired from viruses with characteristic rod-like shapes for efficiently entering cells, rod-like nanoparticles have been developed and are also confirmed to have more excellent cellular uptake efficacy than spherical nanoparticles.5,6 Therefore, various rod-like nanoparticles have been prepared using liposomes, peptides, polylactide (PLA), chitosan, and other polymers.7–10 Among them, PLAs have attracted considerable attention, due to the fact that they are synthesized from renewable resources and are nontoxic after biodegradation, for various applications in industry, agriculture, and biomedical fields.11,12 Up to now, many researchers have reported the preparation of nanoparticles, nanofibers and nanofilms based on PLA.13,14 Particularly, hollow PLA nanomaterials are important in medical applications due to their unique features, such as high surface-to-volume ratio, high structural stability and high drug loading capacity.15,16 Coating PLA onto the surfaces of inorganic nano-templates is a useful method to obtain hollow nanoparticles.17 Introducing PLA onto nanomaterial surfaces can be generally achieved by two methods. The first one is surface-initiated ring-opening polymerization (si-ROP) of lactide after the preliminary surface modification of the nanomaterials.18,19 Mrówczyński et al.20 reported magnetic-PLA core–shell nanoparticles through a chemical linkage by si-ROP of lactide. Priftis’ group21 employed the “grafting from” technique to polymerize lactide onto the surface of carbon nanotubes. Another method is covering the nanomaterial surfaces with a PLA coating by spin coating.22 However, the first process was relatively complicated and resulted in a low induction amount of PLA. The latter process only weakly sticks the PLA and templates together. Therefore, a primary adhesive coating is advised to give a complete cover on the one hand and ensure connectivity to the PLA on the other hand.3,4-Dihydroxyphenylalanine (DOPA) and dopamine, biomolecules that contain the catechol and amine functional groups, are found as modified amino acids in diverse animal species.23–25 Messersmith and other researchers demonstrated PDOPA coating on the surfaces of diverse substrates,26,27 such as metals, metal oxides, ceramics, synthetic polymers, and a wide range of other hydrophilic and hydrophobic materials by self-polymerization of DOPA at alkaline pH values. The PDOPA coating obtained from this methodology is very thin; approximately several nanometers. This thin PDOPA layer will provide important insights into their safety and efficacy as protein and drug carriers, due to the fact that DOPA is an amino acid resource.28 Accordingly, DOPA is indeed a molecule that could anchor to the materials to be adhesively covered on the template surface and connect the PLA polymer. Moreover, the formed crosslinking PDOPA layer could keep the particles stable, even after etching the nano-template. Therefore, by introducing DOPA chains into the PLA polymer, the obtained PLA–PDOPA might be coated onto the surface of silica particles in one step.
It was well documented that silica nanoparticles are not immunogenic or toxic by both in vivo and in vitro tests.29 Rod-like silica nanoparticles are of special interest because of their hydrophilic nature, easy colloidal suspension formation, easy removability by HF, and surface functionalization. Modified silica nanorods have demonstrated potential applications in biomedicine, bioseparation and biocatalysis. Moreover, silica nanorods have been found to have excellent cellular uptake efficacy and a rapid clearance from urine.6,30 Thus, silica nanorods are suitable templates for the preparation of high performance hollow nanorods. However, there are no existing reports of the preparation of PLA–PDOPA copolymers and further fabrication of rod-like nanocapsules using silica nanorods as templates.
Therefore, herein, we synthesized block-like poly(lactide)-b-amidated poly(3,4-dihydroxyphenylalanine) (PLA-b-APDOPA) through an amidation reaction between pre-prepared PLA and APDOPA. Then, DOPA and PLA-b-APDOPA layers were orderly coated onto the surface of silica nanorods. Subsequently, the PLA-b-APDOPA/PDOPA nanocapsules were fabricated by removal of the silica template (Scheme 1). A model drug, ibuprofen, was successfully entrapped into the capsules. The drug-loaded capsules showed slow drug release behavior in vitro.
 | ||
| Scheme 1 Synthetic illustration of PLA-b-APDOPA/PDOPA nanocapsules. | ||
Experimental section
Materials
Lactide was purchased from Tokyo Chemical Industrial Co., Ltd., recrystallized from ethyl acetate, and then dried in vacuo at room temperature for 24 h. 3,4-Dihydroxyphenylalanine (DOPA, 99%), Boc-t-butoxycarbonyl (99%), tert-butyldimethylsilyl chloride (TBDMS, 97%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 99%), tetrabutyl ammoniumfluoride (TBAF, 99%), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 99%), hydroxybenzotriazole (HOBT, 99%), and ibuprofen were obtained from Aladdin Reagent Co., Ltd. (Shanghai, China) and used without purification. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Life Technologies Co., Ltd. Methanol, succinic anhydride, acetonitrile, ethanol, chloroform, dichloromethane (DCM), polyvinylpyrrolidone (PVP, 98%), potassium bisulfate, sodium citrate dehydrate, amino propanol, ammonium hydroxide, dicumyl peroxide, hydrofluoric acid (HF) and triethylamine (TEA) were bought from Sinopharm Chemical Industrial Co., Ltd. and used as received. Dulbecco’s modified eagle medium (DMEM), fetal bovine serum (FBS) and penicillin–streptomycin (P/S) were purchased from Gibco. NIH/3T3 cells were purchased from Biomedical Co., Ltd., China.Synthesis of APDOPA(TBDMS)2
APDOPA(TBDMS)2 was synthesized according to the previous reports.31 Firstly, 3.0 g DOPA was added into a stirred solution of TBDMS (6.28 g) in anhydrous acetonitrile. The colorless suspension was cooled in an ice bath for 10 min and an addition of DBU (6.5 mL) was subsequently dropped into the solution. After further reaction in the ice bath for 4 h, it was stirred at room temperature for an additional 20 h. A colorless solid DOPA(TBDMS)2 was obtained by evaporating the solvent in vacuo, washing with chloroform, and further purification with methanol/acetonitrile using three dissolving/precipitation cycles. The yield was about 40%. 1H NMR (CD3OD): 0.2 (m, 12H), 1.01 (s, 9H), 2.95 (m, 2H), 3.85 (m, 1H), 6.65–6.80 (m, 3H). ESI m/z [M + Na+]: 448.3.Then, 2.12 g of DOPA(TBDMS)2 was added into 50 mL DCM containing 1.91 g of EDC, 2.73 g of HOBT and 10 mL of TEA, and the mixture was kept at room temperature for 24 h. APDOPA(TBDMS)2 was obtained by extraction with DCM three times and drying in vacuo. 1H NMR (CDCl3): 0.18 (m, 12H), 1.03 (s, 9H), 2.90 (m, 2H), 3.90 (m, 1H), 6.65–6.80 (m, 3H). Mn(GPC): 2000, and Mw/Mn: 1.24.
Synthesis of H2N-PLA-COOH
Amino propanol (20 mmol) and Boc t-butoxycarbonyl (22 mmol) were dissolved in chloroform, and then the mixture was placed at room temperature for 24 h. After the reaction finished, Boc-amino propanol was obtained by washing with potassium bisulfate and deionized water several times, and then drying in vacuo at room temperature. Then, L-lactide (3 g) was dissolved in 2 mL dimethylbenzene containing 9 mg of stannous octoate. The Boc-amino propanol (185 mg) was added into the mixed solution, and the reaction was heated to 160 °C and kept at this temperature for 12 h.32 Once the polymerization finished, the reaction mixture was cooled to room temperature and resulted in a solid product. Boc-HN-PLA was finally purified by dissolving in chloroform and precipitating in an excess of diethyl ether three times, and then being dried under vacuum. Boc-HN-PLA (1 g) and succinic anhydride (0.25 g) were added into a flask and reacted for 6 h at 160 °C. The product H2N-PLA-COOH was purified by dissolving in chloroform, precipitating in diethyl ether, and then drying at room temperature under a vacuum. Mn(GPC): 4000, and Mw/Mn: 1.18.Synthesis of a PLA-b-APDOPA block-like copolymer
H2N-PLA-COOH (1.5 g) and APDOPA(TBDMS)2 (0.23 g) were dissolved in methanol. These two solutions were mixed and reacted at 25 °C for 24 h, in the presence of EDC and HOBT as catalysts. A block-like (PLA-b-APDOPA)x (abbreviated as PLA-b-APDOPA) polymer was obtained using TBAF to remove the TBDMS groups, and then was further purified by dissolving in acetone to remove the unreacted PLA and dialysis in ethanol to remove APDOPA.Synthesis of rod-like silica
Polyvinylpyrrolidone (30 g) was dissolved in n-amyl alcohol (300 mL) by sonication for 2 h. Then, ethanol (30 mL), ultrapure water (8.4 mL), and 0.18 mol L−1 sodium citrate dehydrate (2 mL) were added into the PVP solution. After the mixture formation, TEOS (3 mL) and ammonium hydroxide (6.75 mL) were added into the mixture and reacted for 12 h at room temperature. The precipitate nanorods were collected by centrifuging and washing with ethanol and water three times.Polymer coating on silica nanorods
Firstly, 40 mg of the silica nanorods were dispersed in 40 mL Tris buffer (pH = 8.5). DOPA (80 mg) was added and continuously stirred at room temperature for 24 h forming the PDOPA polymer layer. The PDOPA coated silica nanorods (PDOPA@SiO2) were centrifuged, followed by washing with water. PLA-b-APDOPA/PDOPA@SiO2 nanorods were fabricated by coating PLA-b-APDOPA on the surface of the PDOPA@SiO2 nanorods, which was the same process as the PDOPA@SiO2 nanorods.Preparation of PLA-b-APDOPA/PDOPA nanocapsules
PLA-b-APDOPA/PDOPA capsules were obtained after removing the template particles using 2 M HF/8 M NH4F solution at pH 5. The PLA-b-APDOPA/PDOPA@SiO2 nanorods (20 mg) were added into 10 mL of Tris buffer (pH = 5) aqueous solution, and the mixture was stirred at room temperature for 12 h. The nanoparticles were collected by centrifugation and then dispersed in water (10 mL) three times, and then dried at room temperature under a vacuum to obtain the PLA-b-APDOPA/PDOPA nanocapsules.In vitro cell cytotoxicity of the nanocapsules
In this experiment, a positive group, sample group, and negative group were designated. NIH/3T3 cells were firstly placed into a plastic dish with 3.5 cm diameter (5 × 104 cells per well) in complete DMEM (with 10% FBS) and incubated at 37 °C in a 5% CO2 incubator for 24 h. Then, 10 μL of DMSO and the PLA-b-APDOPA/PDOPA nanocapsule suspension was added into 90 μL culture medium, respectively, and these two mixtures were designated as the positive group and sample group. Another 100 μL pure medium was used as the negative group. The cells were further cultured over 4 h and stained with fluorescein diacetate. The cell density that adhered on each scaffold was measured from randomly selected views, which were observed at 100-fold magnification by a fluorescence microscope (Nikon 80i, Japan).The cytotoxicity of the capsules was also investigated by MTT assay. The NIH/3T3 cells were cultured onto a 96-well plate (5 × 104 cells per well) in complete DMEM (with 10% FBS) at 37 °C in a humidified atmosphere of 5% CO2. After incubation for 24 h, the aforementioned media were respectively added into the mixtures of 90 μL medium and 10 μL PLA-b-APDOPA/PDOPA nanocapsule suspension with various concentrations of 0.5, 1 and 2 mg mL−1. For comparison, a positive DMSO group (10% of DMSO concentration) and a negative group (control sample) were designated. Plates were further incubated at 37 °C for 4 h after the addition of 20 μL MTT stock solution. Subsequently, 150 μL DMSO was added into the wells, and then the absorbance was recorded at 560 nm using an Elisa microplate reader (Infinite M200 Pro). All measurements were taken in triplicate. A glass coverslip was used as the control. The percent cell viability was calculated from the ratio of the mean absorbance of the sample to the mean absorbance of the control.
Drug encapsulation into the nanocapsules
A certain amount of the nanocapsules was added into a 2 mL ibuprofen/ethanol solution (2 mg mL−1) for 24 h. Then, the ibuprofen-encapsulated nanocapsules were centrifuged, dried by lyophilization and kept for further study. The supernatant of the ibuprofen solution after separation of the nanocapsules was collected to determine the encapsulation efficiency by UV spectroscopy at a wavelength of 264 nm.| Encapsulation efficiency = (Cibuprofen − Cfree ibuprofen)/Ctotal ibuprofen × 100% |
| Encapsulation content = (Ctotal ibuprofen − Cfree ibuprofen)/Wnanocapsules × 100% |
In vitro drug release
Ibuprofen-encapsulated nanocapsules were dispersed in 5 mL PBS (pH 7.4) at a constant temperature of 37 °C. At regular intervals, 100 μL solution was taken out for UV absorption measurements at a wavelength of 264 nm, and an equal volume of blank PBS was added.Characterization
Fourier transform infrared (FTIR) spectra were recorded with a Nicolet iS50 infrared spectrometer (Thermo Fisher Scientific) in the range of 400–4000 cm−1 with a resolution of 4 cm−1. 1H NMR spectra were recorded on a AVANCE III HD 400 MHz spectrometer (Switzerland) in CDCl3. The purified and dried samples were subjected to thermogravimetric analysis (TGA) under air using a Mettler Toledo TGA instrument at a heating rate of 20 °C min−1 from 50 to 600 °C under oxygen. Transmission electron microscopy (TEM) images were taken with a JEM-2100 field emission microscope. For the TEM observations, samples were dispersed in deionized water and then dried on a holey carbon film Cu grid. Scanning electron microscopy (SEM) images were taken with a Hitachi S-4800 electron microscope. For the SEM observations, samples were dispersed in deionized water and then dried on a silicon slice. Fluorescence microscopy images were taken with a fluorescence microscope (Nikon 80i). X-ray photoelectron spectroscopy (XPS) was carried out using an energy analysis instrument (Thermo ESCALAB 250) with an Al Kα X-ray source and ultrahigh vacuum (∼10 −10 mbar).Results and discussion
Synthesis of PLA-b-APDOPA copolymer
In order to prepare the LA and DOPA block copolymer, amino propanol was used to initiate ring-opening polymerization of the lactide. The obtained H2N-PLA was further carboxylated by succinic anhydride to prepare H2N-PLA-COOH. To avoid the oxidation of the catechol groups of DOPA, the hydroxyl groups were protected by TMDMS before the amidation of DOPA. After obtaining the APDOPA(TMDMS)2, the PLA-b-APDOPA copolymer was synthesized using an amidation reaction as illustrated in Scheme 2, and deprotecting the hydroxyl groups by removing the TBDMS groups with TBAF.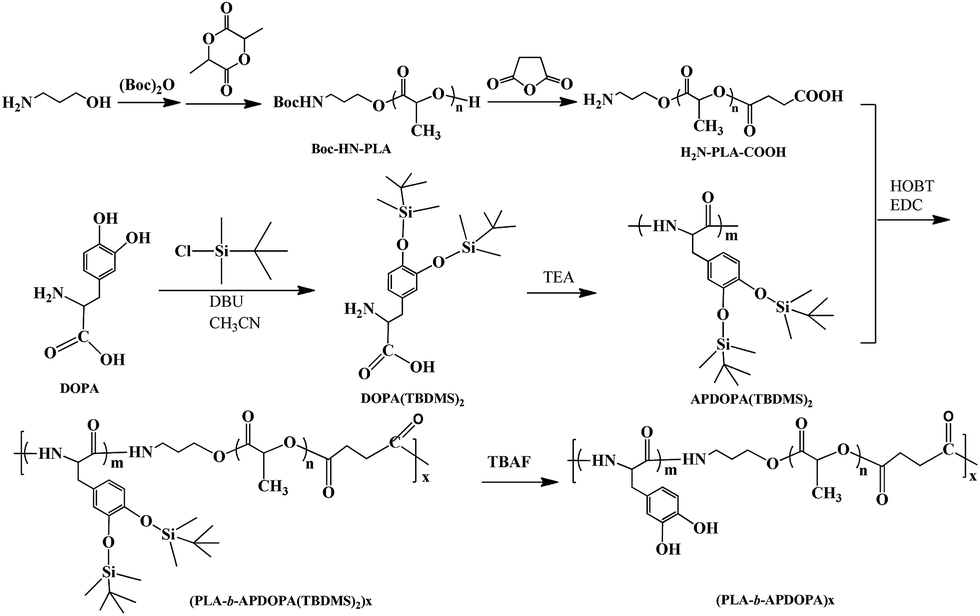 | ||
| Scheme 2 Schematic representation for the synthesis process of (PLA-b-APDOPA)x (abbreviated as PLA-b-APDOPA). | ||
The structure of the PLA-b-APDOPA copolymer was confirmed by a 1H NMR spectrum, as shown in Fig. 1. The chemical shifts at 5.1 and 1.5 ppm were ascribed to the protons of the methine (–C(O)CH–) and methyl groups in the LA units. Peaks appearing at 3.8, 4.2, and 6.8 ppm were assigned to the –CH2Ar, –HNC(H)CO–, and –Ar groups in the DOPA units, respectively. These results indicated the successful preparation of the PLA-b-APDOPA copolymer. A FTIR spectrum also confirmed the polymer structure (data not shown). By comparing the peak area of the methine groups to the benzene groups, the molar ratio of the LA units to DOPA units was found to be 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The molecular weight of the copolymer was 2.1 × 104 and Mw/Mn was 1.44 from GPC measurement.
:1. The molecular weight of the copolymer was 2.1 × 104 and Mw/Mn was 1.44 from GPC measurement.
 | ||
| Fig. 1 1H NMR spectrum of the PLA-b-APDOPA copolymer. | ||
Preparation of PLA-b-APDOPA/PDOPA@SiO2 nanorods
The synthesis of the silica rods related to model growth theory. Fig. 2a shows an SEM image of the silica nanorods with diameters of 200 nm, an average length of 1.5 μm and smooth surfaces. | ||
| Fig. 2 SEM images of the SiO2 nanorods (a), PDOPA@SiO2 nanorods (b), PLA-b-APDOPA/PDOPA@SiO2 nanorods (c), and a TEM image of the PLA-b-APDOPA/PDOPA nanocapsules (d). | ||
In pH 8.5 buffer solution, the catechol groups of DOPA self-polymerized with a crosslinked structure and adhesively coated onto various substrates tightly via covalent bonds, as illustrated in Scheme 1. Since the composition of DOPA chains in the PLA-b-APDOPA was relatively low, DOPA was firstly adhesively coated onto the silica nanorods to enhance the crosslinking degree of the PDOPA layer, prior to the PLA-b-APDOPA coating. During the coating process of DOPA at pH 8.5, the nanorod suspension began to darken, showing a formation of the PDOPA coating. The SEM image of the PDOPA@SiO2 nanorods shows no significant differences in the length and diameter of the nanorods, except that the surface became rough (Fig. 2b).
The PLA-b-APDOPA copolymer was further coated onto the surface of the PDOPA@SiO2 nanorods in the pH 8.5 buffer solution. The diameters of the obtained PLA-b-APDOPA/PDOPA@SiO2 nanorods were slightly bigger (about 210 nm) than those of the PDOPA@SiO2 nanorods, as shown in Fig. 2c. Moreover, the surface of the PLA-b-APDOPA/PDOPA@SiO2 nanorods was also more rough. Previous studies have successfully proved that nano-scale structures with rough surfaces had excellent cellular uptake efficacy. Thus, the PLA-b-APDOPA/PDOPA@SiO2 nanorods might enhance cellular uptake.
To detect the coating amounts of the PDOPA and PLA-b-APDOPA polymers onto the SiO2 nanorods, TGA analysis was performed from 50 to 600 °C under oxygen. As shown in Fig. 3, the decomposition temperature of the SiO2 nanorods in the first stage was at around 100 °C, which contributed to the physically adsorbed water in the nanorods. A second decomposition occurred at 380–500 °C and the weight loss was about 5.9%. After coating with the PDOPA layer, the decomposition behavior was similar to the SiO2 nanorods, and the weight loss of the PDOPA@SiO2 nanorods was 7.48%. Accordingly, the loading amount of PDOPA was 1.57%, suggesting the formed PDOPA layer was very thin. In the case of the PLA-b-APDOPA/PDOPA@SiO2 nanorods, there were two decomposition stages at 200–320 °C and 320–460 °C, which could be assigned to the PLA-b-APDOPA polymers and PDOPA polymers respectively. Surprisingly, the loading amount of the PLA-b-APDOPA polymers was higher at approximately 55.3%.
 | ||
| Fig. 3 TGA curves of the SiO2, PDOPA@SiO2 and PLA-b-APDOPA/PDOPA@SiO2 nanorods. | ||
Coating of the SiO2 nanorods with PDOPA and PLA-b-APDOPA was further confirmed by XPS spectra of the resulting SiO2, PDOPA@SiO2 and PLA-b-APDOPA/PDOPA@SiO2 nanorods. In the XPS spectrum of the SiO2 nanorods (Fig. 4a), the SiO2 nanorod surfaces showed Si2p (∼102 eV), C1s (285 eV), and O1s (532 eV) peaks. The O1s, Si2p peaks correspond to the SiO2 nanorods and the C1s peak might belong to the remaining stabilizer. After the coating of PDOPA, the intensity of the C1s peak increased (Fig. 4b). In addition, a new N1s peak at 399 eV appeared, which belongs to a characteristic component of PDOPA. However, the Si2p peak still existed, which indicated that the PDOPA layer was very thin (which is the same as the results from the TGA curves in Fig. 3) and that the coating of PDOPA on the silica surface was incomplete (also confirmed by the SEM image Fig. 2b). However, after the coating of PLA-b-APDOPA onto the surfaces of the PDOPA@SiO2 nanorods, there were only two peaks, i.e., the C1s and O1s peaks, remaining in the XPS spectrum of the PLA-b-APDOPA/PDOPA@SiO2 nanorods (Fig. 4c). The peak intensities of C1s and O1s increased significantly, while the Si2p and N1s peaks disappeared. These results indicated that the surface component of the resulting nanorods was PLA polymer chains and that the PLA layer was relatively thick. SEM and TGA measurements also indicated a thick coating layer and a large coating amount (55.3% in Fig. 3) of the PLA-b-APDOPA copolymer on the silica nanorods.
 | ||
| Fig. 4 XPS spectra of the SiO2 nanorods (a), PDOPA@SiO2 nanorods (b) and PLA-b-APDOPA/PDOPA@SiO2 nanorods (c). | ||
Formation of the PLA-b-APDOPA/PDOPA nanocapsules
By dispersing the PLA-b-APDOPA/PDOPA@SiO2 nanorods in HF/NH4F (pH = 5), the silica template was etched from the core, and the hollow PLA-b-APDOPA/PDOPA nanocapsules were produced. Fig. 2d shows the morphology of the PLA-b-APDOPA/PDOPA nanocapsules. The core of the nanorods appears white in the image, suggesting capsule formation. Moreover, after removal of the silica nanorods, the length of the nanocapsules became shorter, from an average of 1.5 μm to 500 nm, whereas the diameter of the nanocapsules was broadened from 200 nm to 350 nm, compared with the PLA-b-APDOPA/PDOPA@SiO2 nanorods. These size changes were possibly due to the change of osmotic pressure between the inner and outer regions of the capsules, by a lack of supporting template and to keep the lowest surface energy of the nanocapsules. Moreover, it was the crosslinking bonds in the self-polymerized PDOPA layer that kept the rod-like shape of the nanocapsules and avoided dissociation during the process of silica removing.Cytotoxicity studies of the PLA-b-APDOPA/PDOPA nanocapsules
The cytotoxicity of the PLA-b-APDOPA/PDOPA nanocapsules was estimated using NIH/3T3 cells as the model cells by an MTT assay method, and the results are shown in Fig. 5a. By changing the concentration of the PLA-b-APDOPA/PDOPA nanocapsules from 0.5 to 2 mg mL−1, the cell viability for the nanocapsules was shown to be about 88% independent of the nanocapsule concentration. Thus, these PLA-b-APDOPA/PDOPA nanocapsules exhibited negligible cytotoxicity to the cells. | ||
| Fig. 5 (a) Cell viability of NIH–3T3 cells treated with the PLA-b-APDOPA/PDOPA nanocapsules. (b) Fluorescence microscopy image of NIH–3T3 cells treated with the PLA-b-APDOPA/PDOPA nanocapsules. | ||
Moreover, the morphology of the cell growth was also observed by fluorescence microscopy. Fig. 5b shows the NIH/3T3 cell adhesion on the PLA-b-APDOPA/PDOPA nanocapsules. It is clear that NIH/3T3 cells were shown to be well adhered to and proliferating on PLA-b-APDOPA/PDOPA, indicating the good cell biocompatibility of the PLA-b-APDOPA/PDOPA nanocapsules.
Drug encapsulating and in vitro release
Ibuprofen, as a model hydrophobic drug, was easily encapsulated into the PLA-b-APDOPA/PDOPA nanocapsules due to non-covalent interactions such as hydrophobic interactions, π–π interactions and hydrogen bond interactions. The drug encapsulation content and encapsulation efficiency were measured to be about 12% and 48%, respectively.The in vitro drug release of ibuprofen from the PLA-b-APDOPA/PDOPA nanocapsules was performed at 37 °C in different buffer solutions (pH 7.4 and pH 5.8). As shown in Fig. 6, no dramatic burst release of ibuprofen appeared in pH 7.4 and pH 5.8 buffer solutions. The release rate of ibuprofen in the pH 7.4 buffer was much higher than that in the pH 5.8 buffer, and about 60% and 10% of ibuprofen were respectively released from the nanocapsules within 500 min. The slow release rate might correspond to the crosslinking of the nanocapsules. The difference in drug release behavior between pH 7.4 and pH 5.8 was probably due to the following two reasons. Firstly, ibuprofen is more soluble in neutral and basic solutions than in acidic solution, resulting in the fact that it could come out of the capsules and be dissolved in the buffer solution at pH 7.4. Secondly, interactions such as hydrogen bond interactions between ibuprofen and the nanocapsules at pH 7.4 might be weaker, compared to the interactions at pH 5.8, due to the partial oxidation of DOPA. Moreover, both ibuprofen and DOPA molecular chains contain carboxyl groups, and the electrostatic repulsive force between ibuprofen and nanocapsules is increased in pH 7.4 buffer solution.
 | ||
| Fig. 6 Drug release from encapsulation in PLA-b-APDOPA/PDOPA nanotubes at pH 7.4 and pH 5.8. | ||
Thus, ibuprofen could have controlled release from the PLA-b-APDOPA/PDOPA nanocapsules depending on the pH. Since there are multifunctional groups such as carboxyl, amine and catechol groups in DOPA, DOPA might have strong interaction with various drugs (or cells). Thus, different drugs might show different release behaviours, which would depend on the drug solubility in pH buffers and the interactions between the drugs and the nanocapsules. Thus, we are now intensively further studying the encapsulation and release mechanisms of various drugs in the PLA-b-APDOPA/PDOPA nanocapsules.
Conclusions
A block-like PLA and APDOPA copolymer was synthesized through an amidation reaction between pre-prepared functional PLA and APDOPA. DOPA and the obtained PLA-b-APDOPA copolymer could be adhesively coated onto the surfaces of silica nanorods. SEM images showed the formed nanorods with rough surfaces. XPS and TGA results indicated the formation of a thin PDOPA layer and a thick PLA-b-APDOPA layer on the silica surfaces. PLA-b-APDOPA/PDOPA nanocapsules were formed by removal of the silica template. The nanocapsules had hollow structures and good biocompatibility. Moreover, a model drug was successfully entrapped into the capsules, and could be slowly released from the nanocapsules in vitro depending on the pH buffer. The obtained rod-like nanocapsules have potential applications as carriers in biomedical fields through oral intake or by injection, according to the drug properties.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51173072, 21504032), the Fundamental Research Funds for the Central Universities (JUSRP51408B), MOE & SAFEA for the 111 Project (B13025), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.Notes and references
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed.
- A. Karatrantos, N. Clarke, R. J. Composto and K. I. Winey, Soft Matter, 2015, 11, 382–388 RSC.
- D. J. Shi, M. Matsusaki and M. Akashi, J. Controlled Release, 2011, 149, 182–189 CrossRef CAS PubMed.
- D. J. Shi, M. Matsusaki and M. Akashi, Macromol. Biosci., 2009, 9, 248–255 CrossRef CAS PubMed.
- C. Xu, Y. T. Niu, A. Popat, S. Jambhrunkar, S. Karmakar and C. Z. Yu, J. Mater. Chem. B, 2014, 2, 253–256 RSC.
- X. L. Huang, L. L. Li, T. T. Liu, N. J. Hao, H. Liu, D. Chen and F. Q. Tang, ACS Nano, 2011, 5, 5390–5399 CrossRef CAS PubMed.
- M. M. Fan, W. Z. Zhang, C. Cheng, Y. Liu, B. J. Li, X. Sun and S. Zhang, Part. Part. Syst. Charact., 2014, 31, 994–1000 CrossRef CAS.
- J. M. Williford, Y. Ren, K. Huang, D. Pan and H. Q. Mao, J. Mater. Chem. B, 2014, 2, 8106–8109 RSC.
- H. Luo, G. Xiong, Q. Li, C. Ma, Y. Zhu, R. Guo and Y. Wan, Fibers Polym., 2014, 15, 2591–2596 CrossRef CAS.
- J. Peyre, T. Pääkkönen, M. Reza and E. Kontturi, Green Chem., 2015, 17, 808–811 RSC.
- H. T. Cui, J. Shao, Y. Wang, P. B. Zhang, X. S. Chen and Y. Wei, Biomacromolecules, 2013, 14, 1904–1912 CrossRef CAS PubMed.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- C. Liu, K. W. Chan, J. Shen, H. M. Wong, K. W. K. Yeung and S. C. Tjong, RSC Adv., 2015, 5, 72288–72299 RSC.
- J. B. Fan, Y. Y. Song, H. Li, J. P. Jia, X. Guo and L. Jiang, J. Mater. Chem. B, 2014, 2, 3911–3914 RSC.
- K. Cheng and S. Sun, Nano Today, 2010, 5, 183–196 CrossRef CAS.
- P. L. Lu, Y. C. Chen, T. W. Ou, H. H. Chen, H. C. Tsai, C. J. Wen, C. L. Lo, S. P. Wey, K. J. Lin, T. C. Yen and G. H. Hsiue, Biomaterials, 2011, 32, 2213–2221 CrossRef CAS PubMed.
- F. Wu, X. Lan, D. Ji, Z. Liu, W. Yang and M. Yang, J. Appl. Polym. Sci., 2013, 129, 3019–3027 CrossRef CAS.
- F. H. Chen, Q. Gao, G. Hong and J. Z. Ni, J. Magn. Magn. Mater., 2008, 320, 1921–1927 CrossRef CAS.
- H. F. Wang, C. Wu, X. Liu, J. Sun, G. M. Xia, W. Huang and R. Song, Colloid Polym. Sci., 2014, 292, 2949–2957 CAS.
- R. Mrówczyński, A. Nan, R. Turcu, J. Leistner and J. Liebscher, Macromol. Chem. Phys., 2015, 216, 211–217 CrossRef.
- D. Priftis, N. Petzetakis, G. Sakellariou, M. Pitsikalis, D. Baskaran, J. W. Mays and N. Hadjichristidis, Macromolecules, 2009, 42, 3340–3346 CrossRef CAS.
- Y. F. Zhu, F. Piscitelli, G. G. Buonocore, M. Lavorgna, E. Amendola and L. Ambrosio, ACS Appl. Mater. Interfaces, 2012, 4, 150–157 CAS.
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653–701 CrossRef PubMed.
- S. Moulay, Polym. Rev., 2014, 54, 436–513 CrossRef CAS.
- D. J. Shi, R. J. Liu, W. Dong, X. J. Li, H. J. Zhang, M. Q. Chen and M. Akashi, RSC Adv., 2015, 5, 82252–82258 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. Yu, W. Wei, M. S. Menyo, A. Masic, J. H. Waite and J. N. Israelachvili, Biomacromolecules, 2013, 14, 1072–1077 CrossRef CAS PubMed.
- A. Postma, Y. Yan, Y. Wang, A. N. Zelikin, E. Tjipto and F. Caruso, Chem. Mater., 2009, 21, 3042–3044 CrossRef CAS.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S.-Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- M. J. Sever and J. J. Wilker, Tetrahedron, 2001, 57, 6139–6146 CrossRef CAS.
- C. Deng, X. S. Chen, H. J. Yu, J. Sun, T. C. Lu and X. B. Jing, Polymer, 2007, 48, 139–149 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |