
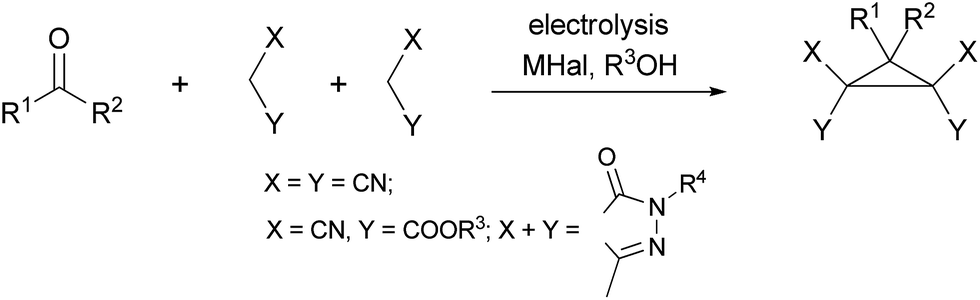
The first electrocatalytic stereoselective multicomponent synthesis of cyclopropanecarboxylic acid derivatives†
Anatoly N. Vereshchagin*,
Michail N. Elinson and
Mikhail P. Egorov
N. D. Zelinsky Institute of Organic Chemistry, Leninsky pr. 47, Moscow, 119991, Russian Federation. E-mail: vereshchagin@ioc.ac.ru
First published on 9th November 2015
Abstract
The one-pot electrocatalytic domino transformation of aldehydes and two different C–H acids – alkyl cyanoacetate and dialkyl malonate in the presence of sodium bromide–sodium acetate as a double mediatory system in alcohol in an undivided cell (simple beaker) results in stereoselective formation of trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylates in 52–87% yields. This protocol uses group-assisted purification (GAP) chemistry in which the pure products were simply isolated by filtration from the reaction mixture. Developed electrocatalytic process allows to obtain multigramme-scale amount of trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylates.
Introduction
Cyclopropane is a basic structural element in a wide range of natural compounds and occupies a significant place in synthetic organic chemistry.1 The cyclopropyl group is also a vital structural unit in many synthetic and naturally occurring compounds, exhibiting a wide spectrum of biological properties ranging from enzyme inhibition to herbicidal, antibiotic, antitumour, antiviral and antidopamine activities.2,3 Cyclopropanecarboxylic acid derivatives are successfully used in agriculture and medicine.4,5 It has recently been found that substituted 1,1,2,2-tetracyanocyclopropanes inhibit RGS-proteins (regulators of G-protein signaling)5a and protein autosplicing and used for the treatment of tuberculosis.5b
Though the methods of cyclopropanes synthesis have been documented a long time ago, all of them consist of two main groups: (1) intramolecular cyclization or (2) interaction of two different molecules (addition of carbenes to olefins or Michael initiated ring closure (MIRC) are the most known examples of this type).6 The well-known method of MIRC synthesis of substituted cyclopropanes involves addition of halosubstituted C–H acid anions, generated by the treatment of the corresponding C–H acid with base, to the conjugated activated olefins followed by cyclization with elimination of halogen anion.7
Electrosynthesis is a competitive method of modern organic chemistry.8a The importance of electrochemical synthesis can hardly be overestimated because of its great and, in some cases, unique possibilities for performing various transformations of organic compounds.8b
The next essential step in the cyclopropane ring construction was electrochemical modification, which was performed with C–H acids instead of halogenated ones and using halogen, generated in situ on a mediatory system in an undivided cell (Fig. 1).9
 | ||
| Fig. 1 Undivided three-necked cell. | ||
This modification allowed to create a novel approaches to the synthesis of substituted cyclopropanes: (1) electrolysis of activated olefins (alkylidenemalonates,10a alkylidencyanoacetates10b or alkylidenemalononitriles10c,d) and C–H acids (malonic esters,10b,d cyanoacetic esters,10e malononitrile10c) including heterocyclic C–H acids (barbituric acids11a and pyrazoline-5-ones11b), Scheme 1; (2) electrolysis of carbonyls and two equivalents of C–H acids,12 Scheme 2; (3) electrolysis of carbonyls and two different C–H acids (electrocatalytic multicomponent synthesis of cyclopropanes), Scheme 3.13
 | ||
| Scheme 1 Electrolysis of activated olefins and C–H acids. | ||
 | ||
| Scheme 2 Electrolysis of carbonyls and C–H acids. | ||
 | ||
| Scheme 3 Electrocatalytic multicomponent synthesis of cyclopropanes. | ||
Results and discussion
In the present study, we report the first electrocatalytic stereoselective multicomponent synthesis of cyclopropanes via one-pot electrolysis of aromatic aldehydes, alkyl cyanoacetates and dialkyl malonates in the presence of mediator in an undivided cell. In order to find optimal conditions, the electrolysis of benzaldehyde 1a, methyl cyanoacetate 2a and dimethyl malonate 3a in methanol was selected as a model reaction (Scheme 4, Table 1). | ||
| Scheme 4 Electrocatalytic stereoselective multicomponent synthesis of trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylates 4a–m. | ||
| Entry | Mediator | Premixing time at rt (min) | Electricity passed (F mol−1) | Time of the electrolysis (min) | Temperature of the electrolysis (°C) | Yield of 4ab (%) | Current efficiency (%) |
|---|---|---|---|---|---|---|---|
| a 50 ml beaker. Benzaldehyde 1a (15 mmol, 1.59 g), methyl cyanoacetate (15 mmol, 1.49 g), dimethyl malonate (15 mmol, 1.98 g), NaHal (5 mmol, 0.41 g), NaOAc (5 mmol), methanol (35 ml), iron cathode (5 cm2), graphite anode (5 cm2), current density 100 mA cm−2.b Isolated cyclopropane.c The yields given in parenthes were determined from 1H NMR spectroscopic and GLC data. | |||||||
| 1 | NaBr | 0 | 3.0 | 144 | 0 | 43 (65)c | 29 |
| 2 | NaBr | 0 | 3.0 | 144 | 10 | (44)c | |
| 3 | NaBr | 0 | 3.0 | 144 | −10 | (26)c | |
| 4 | NaBr | 0 | 3.5 | 168 | 0 | 41 (60)c | 23 |
| 5 | NaBr–NaOAc | 0 | 3.0 | 144 | 0 | 72 | 48 |
| 6 | NaBr–NaOAc | 15 | 3.0 | 144 | 0 | 84 | 56 |
| 7 | NaBr–NaOAc | 30 | 3.0 | 144 | 0 | 87 | 58 |
| 8 | NaI–NaOAc | 30 | 3.0 | 144 | 0 | 72 | 48 |
Our regular electrocatalytic transformations were usually carried out in an undivided three-neck cell (Fig. 1). However, there are problems with stirring in case of formation of insoluble products or large-scale electrolyses. To resolve this problem in the present study we used simple beaker as an undivided cell (Fig. 2).
 | ||
| Fig. 2 (a) Start point of domino process, (b) reaction mixture after premixing (the white precipitate is methyl benzylidenecyanoacetate 5). | ||
As it follows from our results, the proposed electrocatalytic process was carried out at one stage by co-electrolysis of benzaldehyde, methyl cyanoacetate and dimethyl malonate in the presence of NaBr as mediator passing 3.0 F mol−1 electricity. In all cases, high conversion of benzaldehyde 1a and methyl cyanoacetate 2a was observed. Optimal temperature of the process is 0 °C. The yield decreases with the raise of temperature up to 10 °C (Table 1, entry 2), most likely due to various side oligomerization processes, which are typical for cyanoacetic derivatives in the presence of bases.14 Temperature lowering to −10°С (entry 3) also leads to yield drop. In this case methyl benzylidenecyanoacetate (the product of benzaldehyde and methyl cyanoacetate condensation) was detected in the reaction mixture by NMR spectroscopy (25%).
The use of NaBr–NaOAc mediatory system resulted in increasing the yield of cyclopropane 4a from 43 up to 72% (entry 5). In double mediatory system, NaOAc is a catalyst of Knoevenagel condensation of aldehyde and methyl cyanoacetate. In the absence of electricity, benzaldehyde and methyl cyanoacetate under the action of NaOAc undergo condensation into the corresponding methyl benzylidenecyanoacetate 5 during 30 min.
We have established that a 30 min stirring of benzaldehyde 1a, methyl cyanoacetate 2a and malonate 3a in methanol in the presence of NaBr–NaOAc at ambient temperature followed by cooling down to 0 °C resulted in the formation of precipitate of methyl benzylidenecyanoacetate 5 (Fig. 2). Subsequent passing of electricity at 0 °C affords the cyclopropane 4a in 87% yield (Table 1, entry 7), which was isolated without chromatography and recrystallization by simple filtration from reaction mixture. NaBr–NaOAc is more efficient mediatory system than NaI–NaOAc for the studied electrocatalytic process (Table 1, entry 8).
Under the optimal conditions the electrocatalytic stereoselective multicomponent transformation of an aldehydes 1a–k, alkyl cyanoacetates 2a,b and a malonates 3a,b afforded the corresponding trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylates 4a–m in 52–86% substance yields and 32–58% current efficiency (Scheme 4, Table 2).
| Entry | Aldehyde | Ar | R | Electricity passed (F mol−1) | Time of the electrolysis (min) | Product | Yield of 4b (%) | Current efficiency (%) |
|---|---|---|---|---|---|---|---|---|
| a 50 ml beaker. Aldehyde 1 (15 mmol), alkyl cyanoacetate 2 (15 mmol), malonic ester 3 (15 mmol), NaBr (5 mmol, 0.51 g), NaOAc (5 mmol, 0.41 g), alcohol (35 ml). Premixing for 30 min at rt then electricity was passed at 0 °C (the conversion of starting material was checked by GLC), iron cathode (5 cm2), graphite anode (5 cm2), current density 100 mA cm−2.b Isolated cyclopropane.c Scale of electrolysis: 1a (50 mmol), 2a (50 mmol), 3a (50 mmol), NaBr (5 mmol, 0.51 g), NaOAc (5 mmol, 0.41 g), methanol (50 ml). | ||||||||
| 1 | 1a | C6H5 | Me | 3.0 | 144 | 4a | 86 | 57 |
| 2c | 1a | C6H5 | Me | 3.5 | 530 | 4a | 87 | 50 |
| 3 | 1a | C6H5 | Et | 3.0 | 144 | 4b | 74 | 49 |
| 4 | 1b | 2-CH3C6H4 | Me | 3.2 | 154 | 4c | 72 | 45 |
| 5 | 1c | 3-CH3C6H4 | Me | 3.2 | 154 | 4d | 75 | 47 |
| 6 | 1d | 4-CH3C6H4 | Me | 3.0 | 144 | 4e | 78 | 52 |
| 7 | 1e | 4-CH3OC6H4 | Me | 3.5 | 168 | 4f | 85 | 49 |
| 8 | 1f | 4-FC6H4 | Me | 3.2 | 154 | 4g | 82 | 51 |
| 9 | 1g | 3-BrC6H4 | Me | 3.0 | 144 | 4h | 87 | 58 |
| 10 | 1h | 2-NO2C6H4 | Me | 3.2 | 154 | 4i | 65 | 41 |
| 11 | 1i | 3-NO2C6H4 | Me | 3.3 | 158 | 4j | 67 | 41 |
| 12 | 1j | 4-NO2C6H4 | Me | 3.2 | 154 | 4k | 64 | 43 |
| 13 | 1j | 4-NO2C6H4 | Et | 3.0 | 144 | 4l | 66 | 44 |
| 14 | 1k | 3-Py | Me | 3.3 | 158 | 4m | 52 | 32 |
The developed process is scalable. Thus, 30 min stirring of benzaldehyde, methylcyanoacetate and dimethylmalonate (50 mmol of each) in the presence of NaBr–NaOAc (5 mmol of each) in methanol at rt followed by passing electricity (3.5 F mol−1, until complete conversion of starting material controlled by GLC) at 0 °C afforded 13.8 g (87%) of 4a in 8 h 50 min (Table 2, entry 2).
In the NMR spectra of 4a–m only a single set of signals was present, indicating stereoselective formation of a single isomer in the electrocatalytic process. The structure of 4a was previously confirmed by a single-crystal X-ray diffraction study.10b An appropriate mechanism of the chemical-electrocatalytic domino process is outlined in Scheme 5.
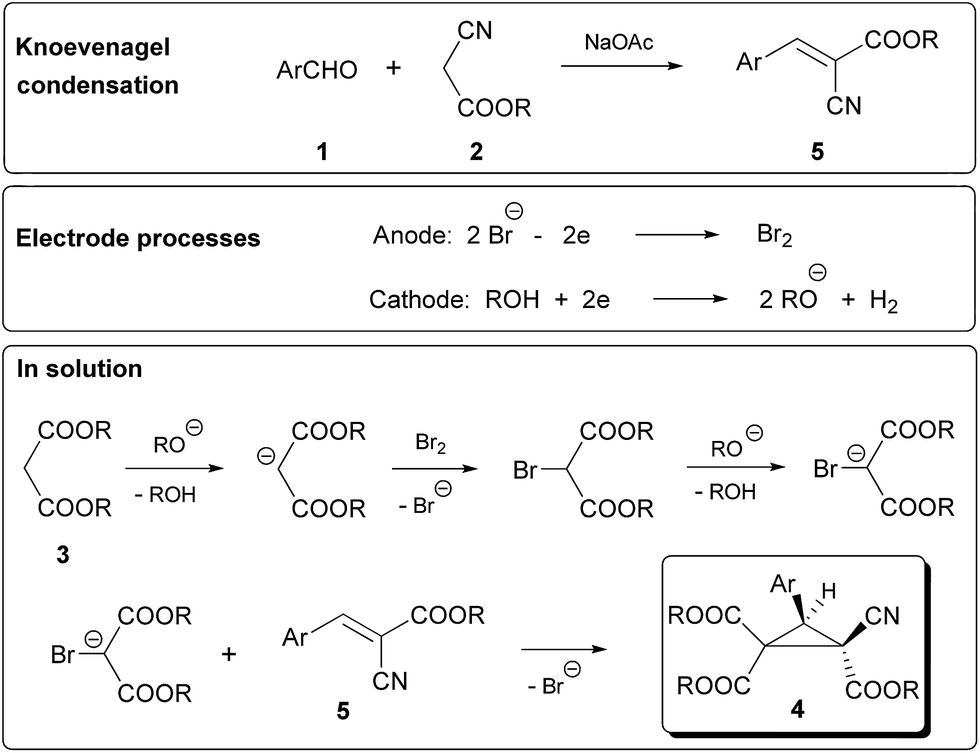 | ||
| Scheme 5 Mechanism of the electrocatalytic stereoselective multicomponent synthesis of cyclopropanes 4. | ||
Knoevenagel condensation of alkyl cyanoacetate and aromatic aldehyde is catalyzed by NaOAc, and afforded alkyl arylidenecyanoacetate 5.15a Reactions at electrodes are standard for the applied mediatory system and lead to the formation of bromine at the anode and the deprotonation of alcohol at the cathode with the evolution of hydrogen. The reaction in solution between an alkoxide ion and malonate leads to the formation of a malonate anion. Bromination of the malonate anion,16 then the formation of the bromomalonate anion followed by the addition to olefin 5 stereoselectively give rise to trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylate 4.17
Conclusions
To summarize, we have performed one-pot electrocatalytic stereoselective domino synthesis of the trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylates from aromatic aldehydes (bearing both electron-donating and electron-withdrawing groups in all positions) alkyl cyanoacetates and dialkyl malonates in the presence of sodium bromide–sodium acetate as a double mediatory system in alcohol in a beaker. In double mediatory system, NaOAc is a catalyst of the chemical process (Knoevenagel condensation of aldehyde and methyl cyanoacetate), and NaBr is a mediator of the electrocatalytic transformation between arylidenecyanoacetate formed in situ and malonate under constant current mode in alcohol.Using classical organic chemistry, this transformation can only be accomplished as a three-step process comprising: (1) Knoevenagel condensation of an aldehyde and alkyl cyanoacetate with formation of an alkylidenecyanoacetate,15 (2) halogenation of the malonate16 and (3) addition of the halogenated malonate to the double bond of the alkylidenecyanoacetate followed by cyclization.17
This new electrocatalytic process is an efficient and convenient method for the synthesis of trialkyl(2R*,3R*)-3-aryl-2-cyanocyclopropane-1,1,2-tricarboxylates. The techniques for electrolysis and isolation of the desired compounds are simple and convenient to use both under laboratory conditions and in large-scale apparatus.
Acknowledgements
The reported study was funded by RFBR, according to the research project No. 15-33-20168.Notes and references
- (a) The chemistry of the cyclopropyl group, ed. Z. Rappoport, J. Wiley & Sons, New York, 1987 Search PubMed; (b) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (c) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051 RSC; (d) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804 RSC.
- For reviews see: (a) W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS; (b) W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625 CrossRef PubMed; (c) D. Y.-K. Chen, R. H. Pouwerb and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC.
- (a) D. W. Graham, W. T. Ashton, L. Barash, J. E. Brown, R. D. Brown, L. F. Canning, A. Chen, J. P. Springer and E. F. Rogers, J. Med. Chem., 1987, 30, 1074 CrossRef CAS PubMed; (b) J. Salaün and M. S. Baird, Curr. Med. Chem., 1995, 2, 511 Search PubMed; (c) D. L. Boger, T. V. Hughes and M. P. Hedrick, J. Org. Chem., 2001, 66, 2207 CrossRef CAS PubMed; (d) K. Yamaguchi, Y. Kazuta, K. Hirano, S. Yamada, A. Matsuda and S. Shuto, Bioorg. Med. Chem., 2008, 16, 8875 CrossRef CAS PubMed.
- L. A. Yanovskaya, V. A. Dombrovsky and A. K. Khusid, Tsiklopropani s funktsionalnimi gruppami. Sintez I primenenie. (Cyclopropanes with Functional Groups. Synthesis and Application), Nauka, Moscow, 1980 Search PubMed.
- (a) D. L. Roman, J. N. Talbot, R. A. Roof, R. K. Sunahara, J. R. Traynor and R. R. Neubig, Mol. Pharmacol., 2007, 71, 169 CrossRef CAS PubMed; (b) Boston biomedical research institute, Pat. Appl., WO2006/79057, 2006.
- M. Ohkita, S. Nishida and T. Tsuji, Recent advances in synthesis of cyclopropanes, Wiley & Sons, New York, 1995 Search PubMed.
- (a) B. Zwanenburg and M. de Kimpe, in Houben-Weyl Methods of Organic Chemistry, Thieme, Stuttgart, 1997, vol. E17, pp. 45–105 Search PubMed; (b) H. Xie, L. Zu, H. Li, J. Wang and W. Wang, J. Am. Chem. Soc., 2007, 129, 10886 CrossRef CAS PubMed; (c) M. Lombardo, E. Montroni, A. Quintavalla and C. Trombini, Adv. Synth. Catal., 2012, 354, 3428 CrossRef CAS; (d) J. Ito, D. Sakuma and Y. Nishii, Chem. Lett., 2015, 44, 297 CrossRef CAS; (e) Y. Zhu and Y. Gong, J. Org. Chem., 2015, 80, 490 CrossRef CAS PubMed.
- (a) J. Grimshow, Electrochemical Reactions and Mechanisms in Organic Chemistry, Elsevier, Amsterdam, 2000 Search PubMed; (b) Organic electrochemistry, ed. H. Lund, Marcel Dekker, New York, 4th edn, 2000 Search PubMed.
- (a) M. N. Elinson, E. O. Dorofeeva, A. N. Vereshchagin and G. I. Nikishin, Russ. Chem. Rev., 2015, 84, 485 CrossRef CAS; (b) Y. N. Ogibin, M. N. Elinson and G. I. Nikishin, Russ. Chem. Rev., 2009, 78, 89 CrossRef CAS.
- (a) M. N. Elinson, S. K. Feducovich, S. G. Bushuev, A. A. Zakharenkov, D. V. Pashchenko and G. I. Nikishin, Mendeleev Commun., 1998, 8, 15 CrossRef; (b) M. N. Elinson, S. K. Feducovich, Z. A. Starikova, A. N. Vereshchagin, P. A. Belyakov and G. I. Nikishin, Tetrahedron, 2006, 62, 3989 CrossRef CAS; (c) M. N. Elinson, S. K. Feducovich, A. N. Vereshchagin, A. S. Dorofeev, D. E. Dmitriev and G. I. Nikishin, Russ. Chem. Bull., 2003, 52, 2235 CrossRef CAS; (d) M. N. Elinson, S. K. Feducovich, T. A. Zaimovskaya, A. N. Vereshchagin, S. V. Gorbunov and G. I. Nikishin, Russ. Chem. Bull., 2005, 54, 1593 CrossRef CAS; (e) M. N. Elinson, S. K. Feducovich, S. G. Bushuev, D. V. Pashchenko and G. I. Nikishin, Russ. Chem. Bull., 1998, 47, 1133 CrossRef CAS.
- (a) E. O. Dorofeeva, M. N. Elinson, A. N. Vereshchagin, N. O. Stepanov, I. S. Bushmarinov, P. A. Belyakov, O. O. Sokolova and G. I. Nikishin, RSC Adv., 2012, 2, 4444 RSC; (b) M. N. Elinson, E. O. Dorofeeva, A. N. Vereshchagin, A. D. Korshunov and M. P. Egorov, Res. Chem. Intermed., 2015 DOI:10.1007/s11164-015-2142-y.
- (a) M. N. Elinson, S. K. Feducovich, T. L. Lizunova and G. I. Nikishin, Tetrahedron, 2000, 56, 3063 CrossRef CAS; (b) M. N. Elinson, E. O. Dorofeeva, A. N. Vereshchagin, R. F. Nasybullin and M. P. Egorov, Catal. Sci. Technol., 2015, 5, 2384 RSC.
- (a) M. N. Elinson, S. K. Feducovich, A. N. Vereshchagin, S. V. Gorbunov, P. A. Belyakov and G. I. Nikishin, Tetrahedron Lett., 2006, 47, 9129 CrossRef CAS; (b) A. N. Vereshchagin, M. N. Elinson, E. O. Dorofeeva, N. O. Stepanov, T. A. Zaimovskaya and G. I. Nikishin, Tetrahedron, 2013, 69, 1945 CrossRef CAS.
- A. J. Fatiadi, Synthesis, 1978, 165 CrossRef CAS.
- (a) M. N. Elinson, S. K. Feducovich, T. A. Zaimovskaya, A. N. Vereshchagin and G. I. Nikishin, Russ. Chem. Bull., 2005, 54, 673 CrossRef CAS; (b) Y. Wei, S. Zhang, S. Yin, C. Zhao, S. Luo and C.-T. Au, Catal. Commun., 2011, 12, 1333 CrossRef CAS.
- S. Torii, K. Uneyama and N. Yamasaki, Bull. Chem. Soc. Jpn., 1980, 53, 819 CrossRef CAS.
- G. V. Kryshtal, G. M. Zhdankina and S. G. Zlotin, Russ. Chem. Bull., 2011, 60, 2286 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19690f |
| This journal is © The Royal Society of Chemistry 2015 |