DOI:
10.1039/C5RA19326E
(Paper)
RSC Adv., 2015,
5, 106787-106794
Effect of annealing temperature on the structure of carbon encapsulated Fe3O4 nanospheres†
Received
19th September 2015
, Accepted 9th December 2015
First published on 10th December 2015
Abstract
Carbon-encapsulated Fe3O4 nanospheres were synthesized by a one-step solvothermal method. The effect of annealing temperature on the structure of carbon-encapsulated Fe3O4 nanospheres was investigated. The morphologies and microstructures of all the products were characterized by field emission scanning electron microscopy, high resolution transmission electron microscopy, X-ray diffraction, N2 adsorption–desorption isotherms, thermogravimetry and vibrating sample magnetometry. The results show that the as-obtained carbon-encapsulated Fe3O4 nanospheres possess a core–shell structure with a size of about 120 nm. The thickness of the carbon layer, specific surface area and pore volume are 20 nm, 33 m2 g−1 and 0.06 cm3 g−1, respectively. With the increase of annealing temperature from 300 to 500 °C for 1 h, the carbon layer of the carbon-encapsulated Fe3O4 nanospheres becomes thinner, and the specific surface area and pore volume become larger. After annealing at 500 °C, the carbon layer thickness, specific surface area and pore volume of the carbon-encapsulated Fe3O4 nanospheres are 13 nm, 191 m2 g−1 and 0.14 cm3 g−1, respectively. After annealing at 600 °C, the carbon-encapsulated Fe3O4 nanospheres change to a yolk–shell structure. When annealed at 700 °C, the carbon layer is destroyed and some Fe nanoparticles are exposed. A plausible formation mechanism for yolk–shell Fe3O4@C nanospheres is proposed.
1. Introduction
In recent years, core–shell nanoparticles with a magnetic response have attracted intensive interest owing to their remarkable chemical and physical properties, which endow them with a large variety of applications in the fields of catalyst supports,1 microwave absorbents,2–4 Li-ion batteries,5 waste water treatment,6,7 bioseparation and enrichment,8,9 and drug delivery.10 For these applications, carbon encapsulated Fe3O4 (designated as Fe3O4@C) nanoparticles are the most promising candidates because carbon has been proved to have chemical inertness, low toxicity, thermal and mechanical stability, electrical conductivity and biocompatibilty, as well as significant synergetic or complementary behavior.2
Up to date, several strategies have been developed to synthesize Fe3O4@C nanoparticles, such as hydrothermal and solvothermal methods,11–13 co-carbonization technique,14 atomic layer deposition,15 and phase transition.16 Among these methods, hydrothermal and solvothermal methods have received considerable attention for their outstanding advantages including mild reaction condition, environmental friendliness and low cost. Xuan and co-workers17 designed a facile method for carbon-encapsulated magnetic composites using glucose as both carbon precursor and reducing agent under hydrothermal conditions, where the amount of glucose had a great influence on the morphology of the products. Wang et al.13 chose ferrocene as both iron source and carbon source, and hydrogen peroxide as oxidant to fabricate Fe3O4@C core–shell microspheres under solvothermal conditions. The Fe3O4@C range in size from 100 to 250 nm, and demonstrated a significant difference for optical properties. However, the Fe3O4@C nanoparticles, fabricated by these methods have poor thermal stability and carbonation degree because of low synthesis temperature. In addition, they have low specific surface area for lack of pore structures on the surface. These shortcomings put some limits on the in application in lithium ion battery, catalyst support and microwave absorption. Heat treatment can improve thermal stability and carbonization degree, and introduce pores on the surface of Fe3O4@C nanoparticles. At present, there are many reports for preparing and modifying the Fe3O4@C by hydrothermal and solvothermal methods combined with heat treatment. Gong Cheng et al.18 fabricated Fe3O4 particles with solvothermal approach, and coated polydopamine on the surface of Fe3O4. Then they did heat treatment at 550 and 700 °C for 1 hour, getting core–shell structure Fe3O4@C and hollow magnetic carbon microspheres respectively. Lu Li et al.19 obtained monodisperse Fe3O4@C core–shell spheres, chains, and rings by annealing Fe2O3@poly(acrylic acid) at 400 °C for 3 hours, 600 °C for 5 hours and 600 °C for 7 hours, respectively. Obviously, annealing temperature has great effect on the structure and morphology of Fe3O4@C. Furthermore, as is well known, oxidation–reduction reaction may occur between carbon and Fe3O4 under high temperature conditions.2 So it is necessary to investigate the effect of heat treatment temperature on the structure of Fe3O4@C nanoparticles.
In this paper, we synthesized Fe3O4@C nanospheres by one-step solvothermal method and investigated the morphological and structural evolution during annealing. A series of investigations using X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), nitrogen adsorption, thermogravimetry (TG), differential scanning calorimetry (DSC) and vibrating sample magnetometry (VSM) were carried out to characterize the annealed Fe3O4@C nanospheres. The Fe3O4@C nanospheres with yolk–shell structure formed at 600 °C. The structural evolution mechanism of Fe3O4@C nanospheres from core–shell to yolk–shell structure was proposed, which can be explained by synergistic growth of Fe3O4 nanoparticles and pyrolysis of carbon layer. Compared with the selective etching method,20,21 which is the common method to prepare the yolk–shell structure of Fe3O4@C, the method of heat treatment is simpler and easier. The unique yolk–shell structured Fe3O4@C nanospheres have large specific surface areas, low density and hollow spaces, which are preferable in applications such as drug delivery,22 lithium-ion batteries20 and nanoreactors.23 Moreover, the systematical structure investigation and mechanism establishment are beneficial for understanding the development of structures of nanospheres during annealing, and also helpful for other researchers to prepare the Fe3O4@C structure for various application fields.
2. Experimental
2.1. Materials
Ferrocene, hydrogen peroxide (H2O2, 30%, wt) and acetone were purchased from Dongli Chemical Reagent Factory, Tianjin, China. All the reagents are analytically pure. Argon was supplied by Feiyue gas Co Ltd.
2.2. Preparation of Fe3O4 @C nanospheres
Fe3O4@C nanospheres were prepared using the modified method as described previously.13 Typically, 1.2 g of ferrocene was dissolved in 40 mL of acetone with ultrasonic irradiation, followed by addition of 2 mL H2O2 into above solution, and sonication for 10 minutes successively. The mixture solution was transferred into 100 mL Teflon lined stainless autoclave. Then the mixture was heated to 200 °C and kept at 200 °C for 48 h and then cooled to room temperature naturally. The products were collected via an external magnetic field and rinsed sequentially with acetone, ethanol and deionized water. The obtained samples were dried at 60 °C in oven overnight.
2.3. Annealing
The annealed Fe3O4@C nanospheres were obtained from the thermal treatment of the as-obtained Fe3O4@C (denoted as Fe3O4@C-0) nanospheres in an argon flow of 200 mL min−1 at 300, 400, 500, 600 and 700 °C for 60 minutes with the heating rate 50 °C min−1 in a cylindrical furnace with a horizontal quartz glass tube. And we also discussed the effect of heating rate (50, 25 and 10 °C min−1) on the Fe3O4@C nanospheres at final temperature 500 °C. The results (Fig. S1 in ESI†) show that the heating rate discussed in our work has little effect on the morphology and structure of Fe3O4@C nanospheres. Therefore, we chose heating rate 50 °C min−1 for further experiment. The samples annealed at different temperature are accordingly named as Fe3O4@C-3, Fe3O4@C-4, Fe3O4@C-5, Fe3O4@C-6, and Fe3O4@C-7, respectively.
2.4. Materials characterization
The morphology and structure of the magnetic nanospheres were observed by FESEM (JEOL, JSM-6700, operated at 10 kV), TEM (JEOL, JEM-2100, operated at an acceleration voltage of 60 kV). DSC (Netzsch STA 449F3) was used to investigate the phase transformation of Fe3O4 to Fe0 and Fe3C. TGA was carried out on a Netzsch TG 209 F3 instrument from 100 to 900 °C with a heating rate of 10 °C min−1 under air atmosphere to evaluate the carbon content. The magnetic properties of the nanospheres were assessed with a quantum design VSM at 298 K and the saturation magnetization is normalized to the mass of test sample. The powder XRD patterns were collected on a Japan Rigaku D/MAX-γA X-ray diffractometer equipped with Cu Kα radiation (λ = 1.5406 Å) over the 2θ range of 10–70°. Specific surface area and pore size distribution were measured with Quadrasorb SI (USA). Before sorption measurement, the samples were degassed at 150 °C for 12 h. Specific surface areas were calculated according to the Brunauer–Emmett–Teller (BET) equation, and pore size distributions and pore volume were computed using density functional theory (DFT) (assuming cylindrical pores).
3. Results and discussion
3.1. Crystalline phase analysis
It is well known that there exist possible interactions between carbon species and iron oxides under high temperature conditions, where carbon species can induce a carbothermic reduction to produce metal Fe, while iron oxides or Fe may promote the graphitization degree of carbon materials.24 Therefore, it is of primary importance to determine the crystalline phases of samples after annealing. As shown in Fig. 1, Fe3O4@C-0, Fe3O4@C-3, Fe3O4@C-4, Fe3O4@C-5 and Fe3O4@C-6 composites exhibit XRD peaks at ∼30.0°, 35.4°, 43.0°, 53.4°, 57.0°, 62.5° (JCPD card no. 88-0866), which can be unambiguously assigned to the spinel phase of Fe3O4, and no other impurity is detected, except for a broad peak assigned to amorphous carbon at 21.6°, indicating Fe3O4 phase can be still well preserved during the carbonization process, which is in accordance with the following TEM results. These results suggest that chemical interactions between carbon species and iron oxides do not occur under current conditions (up to 600 °C, Ar atmosphere). It is worth noting that during thermal treatment at 700 °C under argon, the carbothermic reduction will be induced drastically, the diffraction peaks of Fe3O4 disappear. Diffraction peaks at around 45.1° confirm the formation of zero-valent iron and Fe3C.25
 |
| | Fig. 1 XRD patterns of original Fe3O4@C and annealed Fe3O4@C at different temperature. | |
3.2. Morphology and structure analysis
The FESEM images shown in Fig. 2 display the general morphology of as-obtained Fe3O4@C and annealed Fe3O4@C obtained at different heat treatment conditions. From Fig. 2(a), it can be seen that as-obtained Fe3O4@C composites are solid nanospheres with an average size around 120 nm. After annealed at 300, 400 and 500 °C for 1 h under Ar, as shown in Fig. 2(b)–(d), the annealed Fe3O4@C products still keep the spheric morphology, in spite of a slight shrink in size. Besides, they present a rougher surface (as shown in the inset in Fig. 2(d)) compared with the as-obtained Fe3O4@C, which should be ascribed to the pyrolysis of carbon layer. With further increased heat treatment temperature, the Fe3O4@C nanospheres become hollow, collapsed or even broken. Fig. 2(e) clearly shows that carbon layers of some Fe3O4@C nanospheres are broken, which indicates that the Fe3O4@C-6 nanospheres have hollow structures after annealed at 600 °C for 1 h. At the temperature of 700 °C, a part of nanospheres are cracked or even broken, and a few particles probably run out of carbon layer on their surfaces.
 |
| | Fig. 2 FESEM images of Fe3O4@C as-obtained (a) and annealed at 300 (b), 400 (c), 500 (d), 600 (e), and 700 °C (f) for 1 h. The inset in d shows a high magnification SEM image of the sample. | |
Fig. 3 gives TEM images of as-obtained Fe3O4@C and annealed Fe3O4@C samples. As shown in Fig. 3(a), as-obtained Fe3O4@C nanospheres exhibit solid texture, in accordance with FESEM observation. Fe3O4@C nanospheres show a typical core–shell structure, in which a 20 nm thick amorphous carbon layer covers the inner magnetite cores composed of multiple 12 nm Fe3O4 nanoparticles.13 After annealed at 300, 400 and 500 °C for 1 hour, as shown in Fig. 3(b), (d), (f) and (h), the thickness of carbon layer decreases from 20 nm to 18, 15 and 13 nm, respectively, and the core–shell structure is still maintained. Besides, we can observe that Fe3O4 nanoparticles agglomerate to form larger nanoparticles, leaving some vacancies between carbon shell and magnetic core (arrow in Fig. 3(g)). On the other hand, the yolk–shell structure, which has a distinctive core@void@shell configuration,26 is observed for the Fe3O4@C composite nanospheres annealed under inert atmosphere at 600 °C for 1 hour. TEM image in Fig. 3(i) indicates that the Fe3O4@C-6 nanospheres retain the spherical morphology of the as-obtained Fe3O4@C, while contain freely movable Fe3O4 cores (arrow A in Fig. 3(i)). The void space between the core and shell further confirms the hollow nature of the composite nanospheres. It is noteworthy, from the TEM image, all the small individual nanoparticles formed as a part of Fe3O4 cores during the solvothermal process are aggregate to form a larger spherical piece during the annealing process, as shown by arrow B in Fig. 3(i). With annealing at 700 °C, the carbothermic reduction is induced between carbon layer and Fe3O4, producing Fe and Fe3C, which breaking the carbon layer of the hollow Fe3O4@C nanospheres, and leaving some of the magnetic nanoparticles in core and aggregated to bigger spherical pieces.
 |
| | Fig. 3 TEM images of Fe3O4@C, as-obtained (a and b) and annealed at 300 (c and d), 400 (e and f), 500 (g and h), 600 (i) and 700 °C (j) for 1 h. | |
3.3. Specific surface area and pore size analysis
The specific surface area and pore structure of as-obtained Fe3O4@C and annealed Fe3O4@C were assessed on the basis of N2 adsorption/desorption measurement, as shown in Fig. 4. As-obtained Fe3O4@C exhibits a low adsorption capacity for nitrogen, demonstrating small amount of pores in the structure. However, annealed Fe3O4@C samples possess higher adsorption capacity for nitrogen, which is due to the presence of more porous structures. As-obtained Fe3O4@C and annealed Fe3O4@C reveal typical type-IV isotherm curves (according to IUPAC classification), which is characteristic of mesoporous materials. As indicated by the increase in the nitrogen adsorption amount at low pressures (P/P0 < 0.05), micropores are also presented in the materials.27 Fig. 4(b) shows the pore-size distributions calculated by the Density Functional Theory (DFT). It is clearly seen that Fe3O4@C-0 contains only micropores centered at 1.84 nm, and Fe3O4@C-3 possesses mesopores centered at 2.00 nm. For higher annealing temperature, Fe3O4@C-4 and Fe3O4@C-5 possess both micropores centered at 1.68 and 0.67 nm and a little mesopores. These phenomena suggested a large number of pores are created and the pore volume is enlarged with increasing carbonation temperature.
 |
| | Fig. 4 (a) N2 adsorption–desorption isotherms and (b) the pore size distribution curve obtained through the DFT method. | |
Table 1 describes the relevant porous data of as-obtained Fe3O4@C and annealed Fe3O4@C at different temperatures. The specific surface area of Fe3O4@C was calculated using the BET equation, and pore size and volume were determined by DFT analysis. It can be seen that the specific surface area of Fe3O4@C-3, Fe3O4@C-4 and Fe3O4@C-5 progressively increases from 30 to 87 and 191 m2 g−1, respectively, with the annealing temperature gradually increasing from 300 to 500 °C. At the same time, the pore volume correspondingly increases from 0.07 to 0.11 and 0.14 cm3 g−1. This implies that higher annealing temperature lead to more micropores through pyrolysis of carbonaceous matters and aggregation of Fe3O4 nanoparticles. It should be noted that, further increasing the annealing temperature to 600 °C increases the pore volumes and pore size, but slightly decreases the specific surface area. This phenomenon may originate from the expansion of some micropores into mesopores at high temperature. This is in agreement with the pore size distribution curves shown in Fig. 4(b).
Table 1 Specific surface area and pore parameters of as-obtained Fe3O4@C and annealed Fe3O4@Ca
| Sample |
SBET (m2 g−1) |
Pore size (nm) |
Pore volume (cm3 g−1) |
| SBET: specific surface area calculated by the BET method. Pore size and pore volume are calculated by the DFT method. |
| Fe3O4@C-0 |
33 |
1.84 |
0.06 |
| Fe3O4@C-3 |
30 |
2.00 |
0.07 |
| Fe3O4@C-4 |
87 |
1.68 |
0.11 |
| Fe3O4@C-5 |
191 |
0.67 |
0.14 |
| Fe3O4@C-6 |
172 |
1.43 |
0.15 |
3.4. Thermal stability and carbon content analysis
Fig. 5 displays the TG-DSC profiles of as-obtained Fe3O4@C in Ar atmosphere. The weight loss from room temperature to 200 °C is evoked by water evaporation and possible weak organic group removing. The weight loss from 200 °C to 450 °C may correspond to pyrolysis and carbonization of oligomer on the carbon shell. As the temperature increases to 640 °C, a step weight loss occurs, demonstrating that carbothermic reduction is induced between carbon layer and Fe3O4. As can be seen from DSC curve, two conspicuous exothermic peaks can be observed at 647 and 662 °C. The peak at 647 °C could be assigned to the reduction of Fe3O4 to Fe0. The other exothermic peak centering at 662 °C could be ascribed to the formation of Fe3C.21 The phase transformation of Fe3O4 to Fe0 and Fe3C at 647 °C and 662 °C provides a supporting explanation to results. The very high heat treatment temperature causes the destruction of the structure of Fe3O4@C. Accordingly, the structure of the Fe3O4@C turns out to be stable and ideal when the annealing temperature is below 600 °C. The TG-DSC analysis under Ar demonstrates the mechanism of the effect of temperature on the structure of Fe3O4@C. That is, the thickness of the carbon layer reduces and the size of Fe3O4 nanoparticles grows with increasing annealing temperature. At 600 °C the yolk–shell structured Fe3O4@C is obtained. As the temperature increases to 700 °C, the carbon layer destruction is caused by carbothermic reduction.
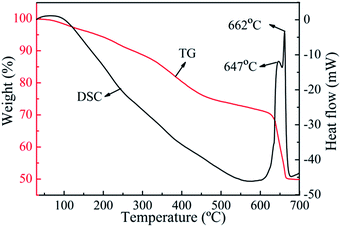 |
| | Fig. 5 TG and DSC curves of as-obtained Fe3O4@C in inert atmosphere (10 °C min−1). | |
TG analysis was performed in air to further quantitatively analyze the component evolution of the Fe3O4@C nanospheres. From the TG curves in Fig. 6(a), it can be seen that main weight-loss is due to the oxidation of the composite, i.e., the carbonaceous materials are oxidized to CO2 and Fe3O4 is oxidized to Fe2O3 under air atmosphere. Thermal stability is improved and carbon content is decreased with increasing annealing temperature. As carbon components can be completely burned in air, the final product is only Fe2O3. Therefore, the amount of carbonaceous matter in these composites can be calculated by2 the following eqn (1):
| |
 | (1) |
where wt%
R is the remaining weight percentage after combustion, and
M indicates the molecular weight of the compound. As deduced from
eqn (1), the weight percentages of carbonaceous matter in Fe
3O
4@C-0, Fe
3O
4@C-3, Fe
3O
4@C-4, Fe
3O
4@C-5, and Fe
3O
4@C-6 is 52.07%, 45.19%, 40.68%, 38.06% and 36.53%, respectively.
Fig. 6(b) shows the relationship between carbonaceous matter content and annealing temperature, the carbonaceous matter content presents a linear decrease with increasing annealing temperature.
 |
| | Fig. 6 (a) TGA curves of Fe3O4@C-0, Fe3O4@C-3, Fe3O4@C-4, Fe3O4@C-5 and Fe3O4@C-6. (b) The relationship between carbonaceous matter and annealing temperatures. | |
3.5. Magnetic properties
The magnetic hysteresis loops of as-obtained and annealed Fe3O4@C nanospheres under different temperatures are presented in Fig. 7(a). The saturation magnetization (Ms) of the six samples is 17.2, 23.5, 27.5, 32.2, and 35.5 emu g−1, respectively, suggesting that all of these materials could be easily separated from solution by applying an external magnetic field. The less the carbonaceous matter content is, the stronger the Ms value of the composites becomes. The inset of Fig. 7(a) depicts an enlarged section of the magnetization plot of Fe3O4@C-0 near the zero magnetic field. The corresponding remanent magnetization and coercivity are 2.52 emu g−1 and 35 Oe, respectively, displaying a quasi-superparamagnetic characteristic. The values of coercivity and remanence magnetization of other samples are summarized in Table 2. It is found that the remanence (Mr) and coercivity (Hc) of all the samples are very low. The five samples exhibit superparamagnetic behavior at room temperature, demonstrating that annealing temperature has little effect on their superparamagnetic property. From the relationship between Ms value and Fe3O4 nanoparticles content (Fig. 7(b)), a linear increase in Ms can be found with increasing Fe3O4 content in composites, which can be easily understood by the fact that annealing temperature does not affect the magnetic nature of the composites, but does increase the content of Fe3O4 nanoparticles. These results again verify that Fe3O4 can be well maintained after annealing.
 |
| | Fig. 7 (a) Magnetic hysteresis curves of Fe3O4@C-0, Fe3O4@C-3, Fe3O4@C-4, Fe3O4@C-5 and Fe3O4@C-6. The inset in panel (a) is the magnified view of magnetic hysteresis curves of Fe3O4@C-0 in the region of low magnetic field. (b) The relationship between saturation magnetization and Fe3O4 content in different Fe3O4@C composites. | |
Table 2 Magnetic properties of as-obtained Fe3O4@C and annealed Fe3O4@C at different temperatures
| Sample |
Magnetic properties |
| Ms (emu g−1) |
Hc (Oe) |
Mr (emu g−1) |
| Fe3O4@C-0 |
17.2 |
35.02 |
2.52 |
| Fe3O4@C-3 |
23.5 |
6.19 |
1.15 |
| Fe3O4@C-4 |
27.5 |
16.15 |
0.19 |
| Fe3O4@C-5 |
32.2 |
11.01 |
2.23 |
| Fe3O4@C-6 |
35.5 |
43.33 |
1.52 |
3.6. Formation mechanism of yolk–shell Fe3O4@C
The evolution mechanism of core–shell to yolk–shell Fe3O4@C nanospheres during the carbonization process is schematically explained in Fig. 8. Two possible growth processes result in forming Fe3O4@C nanospheres with yolk–shell structure.
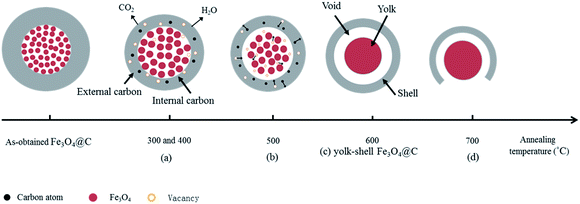 |
| | Fig. 8 Plausible mechanism for the formation of yolk–shell Fe3O4@C nanospheres. | |
At first, the formation of void between carbon layer and Fe3O4 core. There are two reasons for the formation of the void between the carbon layer and the Fe3O4 core. (1) Heat treatment can effectively promote the agglomeration of Fe3O4 nanoparticles, to facilitate the grain growth.28–30 Herein, at the annealing temperature higher than 300 °C, Fe3O4 nanoparticles agglomerate and grow to larger nanoparticles in the process of carbonization treatment,31 leaving small vacancies between carbon shell and Fe3O4, which can be observed in TEM image (arrow in Fig. 3(g)) and 8(b) and (c). As the annealing temperature increases to 600 °C, Fe3O4 nanoparticles further aggregate to form dense spherical structure, which leads to the decrease in the size of the Fe3O4 core particle and formation of void between carbon layer and Fe3O4 core, as shown in Fig. 3(i) and 8(c). (2) The low degree of graphitization of the carbon, as confirmed by XRD analysis, and rich oxygen functional groups13 in the Fe3O4@C nanospheres provide the basis for pyrolysis of carbon layer at annealing temperature. The curvature of Fe3O4@C becomes larger from outside to inside, the interior stress of Fe3O4@C is greater than their external stress, leading to easier breaking of C–C bonds of inner shells of Fe3O4@C.32 Besides, Fe3O4 can promote oligomer pyrolysis and carbonization at high temperature. Inner carbon layer decomposition releases H2O and CO2 and leaves many cavities inside the Fe3O4@C, which also plays a role in formation of void between carbon layer and Fe3O4 core.
Then, enlargement of the distance between core and shell to form yolk–shell structure. According to previous report,33 metal iron or iron oxides may promote the graphitization degree of carbon materials, even to generate crystalline graphite. Du et al.2 verified that Fe3O4 promotes the graphitization degree of carbon materials at 650 °C. With increasing carbonization temperature and prolonging carbonization time, the graphitization degree of the carbon layer near the Fe3O4 core (that is, the internal surface of carbon layer, denoted as internal carbon layer) is higher than that of Fe3O4@C surface (that is, the external surface of carbon layer, denoted as external carbon layer). So the thermal stability of internal carbon layer is greater than that of external carbon layer, and consequently, the pyrolysis degree of the external carbon layer is greater than that of the internal carbon layer. As shown in Fig. 8(a), the external carbon layer decomposes and releases H2O and CO2, and leaves many small vacancies at the surface layer, leading to the decreased size of Fe3O4@C. Meanwhile, the difference of pyrolysis speed in the external and internal carbon layer creates the carbon concentration gradient with continuous pyrolysis of carbon,34 that is, external carbon layer has lower carbon concentration than the internal carbon layer, resulting in the outward diffusion of carbon atoms and the inward diffusion of the related vacancies in carbon layer, as shown in Fig. 8(b). Through the continuous combination of the vacancies between the Fe3O4 core and carbon shell, the void between the carbon layer and the Fe3O4 core is further expanded, finally forming yolk–shell structures (Fig. 8(c)).
However, at further increased annealing temperature, the carbon layer of the hollow Fe3O4@C composite nanospheres is broken, Fe3O4 core is partly exposed to the air, as shown in Fig. 8(d). Or even carbothermic reaction is induced between carbon layer and Fe3O4 core to produce Fe and Fe3C.
Yolk–shell structured nanomaterial usually owns a movable core and hollow space between its core and shell. The uniquely constructed nanomaterials are promising functional nanomaterials owing to their favorable features such as low density, large specific area, big void space and tailorability and functionality in the cores as well as hollow shells.21,35 These excellent properties make the yolk–shell structured nanomaterials performed outstanding in the fields of catalysis,36 drug delivery,37 lithium-ion batteries20 and nanoreactors.38 For example, in the field of catalysis, the movable core provides more exposed active sites and the shell can protect the core from aggregation and divulgement. Besides, in the fields of lithium-ion batteries and drug release, the large voids between core and shell can provide available space for core materials.35
4. Conclusions
In summary, the effect of annealing temperature on the morphology and structure of Fe3O4@C nanospheres was systematically investigated. At the annealing temperature up to 600 °C, chemical reaction between carbon shell and Fe3O4 nanoparticles is not induced. When Fe3O4@C nanospheres are annealed below 500 °C, the core–shell structure can be well remained. The carbon layer reduces gradually with increasing annealing temperature. Meanwhile, a large number of micropores and a fraction of mesopores are created on the surface of Fe3O4@C, leading to gradual increase in surface area and pore volume. As annealing temperature rises, the carbon content linearly decreases and saturation magnetization linearly increases. At annealing temperature of 600 °C, yolk–shell structure of Fe3O4@C can be obtained. Meanwhile, the possible evolution mechanism of core–shell to yolk–shell Fe3O4@C nanospheres, including the formation of void, and expansion of void into hollow yolk–shell structure, can be explained through the synergistic effect of carbon pyrolysis and Fe3O4 nanoparticles agglomeration under high annealing temperature. The annealed core–shell and yolk–shell Fe3O4@C nanospheres with good thermal stability, excellent magnetic response and high surface area can be widely used in microwave absorption, lithium ion battery, and catalysis. At treatment temperature of 700 °C, the oxidation–reduction reaction is induced between carbon layer and Fe3O4, forming metallic iron and Fe3C. Heat treatment temperature plays an important role on the structure and morphology of Fe3O4@C. These findings have important theoretical and practical significance for the further application of carbon encapsulated metal magnetic materials.
Acknowledgements
We appreciate the financial support from National Natural Science Foundation of China (21176169), Shanxi Provincial Key Innovative Research Team in Science and Technology (2015013002-10), International Science & Technology Cooperation Program of China (2012DFR50460), Postgraduate Innovation Program of Shanxi Province (2015SY12) and Special/Youth Foundation of Taiyuan University of Technology (2014TD015).
Notes and references
- Y. Zhao, Y. Yeh, R. Liu, J. You and F. Qu, Solid State Sci., 2015, 45, 9–14 CrossRef CAS.
- Y. Du, W. Liu, R. Qiang, Y. Wang, X. Han, J. Ma and P. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 12997–13006 CAS.
- T. Zhang, D. Huang, Y. Yang, F. Kang and J. Gu, Mater. Sci. Eng., B, 2013, 178, 1–9 CrossRef CAS.
- W. Li, B. Lv, L. Wang, G. Li and Y. Xu, RSC Adv., 2014, 4, 55738–55744 RSC.
- D. Wang, L. Jiang, X. Wei and C. Song, Mater. Lett., 2015, 138, 164–166 CrossRef CAS.
- Y. Cui, S. Liu, K. Wei, Y. Liu and Z. Hu, Microchim. Acta, 2015, 182, 1337–1344 CrossRef CAS.
- P. Qiao, B. Zhao and Z. Nan, Mater. Sci. Eng., B, 2013, 178, 1476–1482 CrossRef CAS.
- X. Sun, X. Liu, J. Feng, Y. Li, C. Deng and G. Duan, Anal. Chim. Acta, 2015, 880, 67–76 CrossRef CAS PubMed.
- R. Gao, X. Mu, J. Zhang and Y. Tang, J. Mater. Chem. B, 2014, 2, 783–792 RSC.
- L. Chen, L. Li, H. Zhang, W. Liu, Y. Yang, X. Liu and B. Xu, RSC Adv., 2014, 4, 46806–46812 RSC.
- S. Zhang, H. Niu, Z. Hu, Y. Cai and Y. Shi, J. Chromatogr. A, 2010, 1217, 4757–4764 CrossRef CAS PubMed.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L.-J. Wan, Adv. Funct. Mater., 2008, 18, 3941–3946 CrossRef CAS.
- H. Wang, Q. W. Chen, Y. F. Yu, K. Cheng and Y. B. Sun, J. Phys. Chem. C, 2011, 115, 11427–11434 CAS.
- J. Zhang, J. Du, Y. Qian, Q. Yin and D. Zhang, Mater. Sci. Eng., B, 2010, 170, 51–57 CrossRef CAS.
- G. Z. Wang, Z. Gao, S. W. Tang, C. Q. Chen, F. F. Duan, S. C. Zhao, S. W. Lin, Y. H. Feng, L. Zhou and Y. Qin, ACS Nano, 2012, 6, 11009–11017 CrossRef CAS PubMed.
- S. Yuan, Z. Zhou and G. Li, CrystEngComm, 2011, 13, 4709 RSC.
- S. H. Xuan, L. Y. Hao, W. Q. Jiang, X. L. Gong, Y. Hu and Z. Y. Chen, Nanotechnology, 2007, 18, 6 CrossRef PubMed.
- G. Cheng, M. D. Zhou and S. Y. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 12719–12728 CAS.
- L. Li, T. Wang, L. Zhang, Z. Su, C. Wang and R. Wang, Chemistry, 2012, 18, 11417–11422 CrossRef CAS PubMed.
- Y. Zhao, J. Li, C. Wu, Y. Ding and L. Guan, ChemPlusChem, 2012, 77, 748–751 CrossRef CAS.
- Y. Shao, L. Zhou, C. Bao and J. Ma, Carbon, 2015, 89, 378–391 CrossRef CAS.
- Y. M. Zhou, H. B. Wang, M. Gong, Z. Y. Sun, K. Cheng, X. K. Kong, Z. Guo and Q. W. Chen, Dalton Trans., 2013, 42, 9906–9913 RSC.
- T. Yao, T. Cui, X. Fang, J. Yu, F. Cui and J. Wu, Chem. Eng. J., 2013, 225, 230–236 CrossRef CAS.
- Y. Du, W. Liu, R. Qiang, Y. Wang, X. Han, J. Ma and P. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 12997–13006 CAS.
- L. Wang, C. Tian, B. Wang, R. Wang, W. Zhou and H. Fu, Chem. Commun., 2008, 5411–5413 RSC.
- J. C. Park and H. Song, Nano Res., 2010, 4, 33–49 CrossRef.
- C. M. Ghimbeu, M. Sopronyi, F. Sima, L. Delmotte, C. Vaulot, C. Zlotea, V. Paul-Boncour and J. M. le Meins, Nanoscale, 2015, 7, 10111–10122 RSC.
- N. Zhao, S. Wu, C. He, Z. Wang, C. Shi, E. Liu and J. Li, Carbon, 2013, 57, 130–138 CrossRef CAS.
- Z. Zhou, W. Xie, S. Li, X. Jiang, D. He, S. Peng and F. Ma, J. Solid State Electrochem., 2015, 19, 1211–1215 CrossRef CAS.
- M. Lübke, N. M. Makwana, R. Gruar, C. Tighe, D. Brett, P. Shearing, Z. Liu and J. A. Darr, J. Power Sources, 2015, 291, 102–107 CrossRef.
- C. Zou, Y. Yao, N. Wei, Y. Gong, W. Fu, M. Wang, L. Jiang, X. Liao, G. Yin, Z. Huang and X. Chen, Composites, Part B, 2015, 77, 209–214 CrossRef CAS.
- Y. Z. Yang, X. G. Liu, Q. P. Luo, L. Jin and B. S. Xu, New Carbon Mater., 2010, 25, 431–437 CrossRef CAS.
- Z. H. Sun, L. F. Wang, P. P. Liu, S. C. Wang, B. Sun, D. Z. Jiang and F. S. Xiao, Adv. Mater., 2006, 18, 1968–1971 CrossRef CAS.
- D.-H. Nam, J. H. Lee, N. R. Kim, Y. Y. Lee, H. W. Yeon, S.-Y. Lee and Y.-C. Joo, Carbon, 2015, 82, 273–281 CrossRef CAS.
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. Lou, X. Xing and G. Q. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC.
- L. Chen, L. Li, T. Wang, L. Zhang, S. Xing, C. Wang and Z. Su, Nanoscale, 2014, 6, 6603–6608 RSC.
- L. Li, F. Tang, H. Liu, T. Liu, N. Hao, D. Chen, X. Teng and A. J. He, ACS Nano, 2010, 4, 6874–6882 CrossRef CAS PubMed.
- J. Lee, J. C. Park and H. Song, Adv. Mater., 2008, 20, 1523–1528 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra19326e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 








