
Development of asymmetrical near infrared squaraines with large Stokes shift†
Xiaoqian Liua,
Bokun Choc,
Li-Yan Chana,
Wei Lek Kwan*b and
Chi-Lik Ken Lee*a
aCentre for Biomedical and Life Sciences, Department of Technology, Innovation and Enterprise (TIE), Singapore Polytechnic, Singapore. E-mail: kenlee@sp.edu.sg
bEngineering Product Development, Singapore University of Technology and Design, Singapore. E-mail: kwanwl@sutd.edu.sg
cEnergetics Research Institute (EnRI), Nanyang Technological University, Singapore
First published on 7th December 2015
Abstract
A new strategy of obtaining large Stokes shift squaraine dyes is reported. Archetypal near infrared squaraines typically have very sharp absorption peaks and small Stokes shifts due to their very rigid ground and excited state molecular structures. TDDFT calculations revealed that large Stokes shift in squaraines can be reached by structural relaxation of the excited state. We achieved Stokes shifts of 90 nm by introducing a dibutyl-aniline side group and an electron withdrawing dicyano group to the squarate core. Wavefunction analysis indicates that that steric interactions and mesomeric effects in the ground and excited states of squaraines are crucial in determining the Stokes shift of the dye.
1. Introduction
In the last decade, there have been increasing interests in organic dyes, especially near infrared (NIR) dyes for diverse applications in light emitting diodes, field-effect transistors, organic photovoltaics, NIR-fluorescence imaging and photodynamic therapy.1,2 Their most notable features include strong absorption in the NIR region, tunable solubility in different solvents, and remarkable chemical and photostability. Great efforts have been made to develop more effective NIR dyes, including borondipyrromethenes (BODIPYs), pyrrolopyrrole cyanine and squaraines.3 Among these fluorophores, squaraine compounds are actively being investigated as high performance components with resonance stabilized zwitterionic structures.4Symmetric squaraines are the condensation products of one equivalent of squaric acid and two equivalents of suitable electron rich precursors.5 These dyes possess effective absorption in NIR region (>600 nm), narrow excitation and emission peaks with large molar extinction coefficients and quantum yields.6 However, compared to these popular and intensely studied “classical” squaraines, asymmetric derivatives are considerably less investigated. Asymmetrical squaraines in donor–acceptor–donor (D–A–D) types can provide unidirectional flow of electrons, which may affect charge transfer in the molecule, resulting in the change of the physical properties and improvement in the performance of organic solar cells and dye sensitized solar cells.7 More importantly, the structures with one site of functional group like –COOH can provide mono specific binding site to some bio-molecules, such as: oligonucleotides, which will act as probes for multiple detection applications.8
Large Stokes shift is desirable in fluorescent labeling applications of dyes, as it reduces self-quenching effects and interference from excitation source. However, despite the favorable characteristics of squaraines, the Stokes shifts are typically 20–30 nm, which limits its potential applications.
Recently, asymmetrical squaraine dyes with large Stokes shifts (∼90 nm) has been reported.2i,3k Shafeekh et al. has shown that the large Stokes shifts in their squaraines are due to the dipole moment inversion of the excited state and its interaction with the solvent.3k Since the effect is due to the redistribution of the charge density between the excited and ground states, it is highly sensitive to the local environment.
Archetypal near infrared squaraines have very rigid ground and excited state structures due to their large conjugated systems. As a result, squaraines typically have very sharp absorption peaks and small Stokes shifts. In this paper, we explore an alternative approach to increase the Stokes shift in squaraines by stabilizing the excited state through structural relaxation. We achieved a large Stokes shift of 90 nm by introducing a dibutyl-aniline side group and an electron withdrawing dicyano group to the squarate core. Wavefunction analysis indicates that steric interactions and mesomeric effects in the ground and excited states of squaraines is an important factor in determining the Stokes shift of the dye.
2. Results and discussion
Asymmetrical squaraines can be synthesized through a variety of multi-step procedures using electron rich precursors.9 In order to tune physical properties of asymmetrical squaraines, we choose a strategy that will facilitate structural diversification by first preparing semi-squaraine salts and subsequently attaching various donor moieties.10–13 Since squarylium dyes with tertiary arylamine groups are known to have better stability and solubility than those with heterocyclic end groups, and rotational relaxation is known the occur in the excited states of symmetrical arylamine-squaraines,5d new asymmetrical squaraine dyes containing the substituted aniline are firstly developed (Scheme 1).14 | ||
| Scheme 1 Multi steps synthesis of squaraine 3a–3c. | ||
The substituted aniline based semi-squaraine salt 1 is used as our first template and synthesized according to the steps reported in the literature.15 Different substituted benzothiazolium quaternary iodides 2a–2c are used in the reflux of 1-butanol and toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with salt 1 to achieve final product 3a–3c in moderate yields. Notably, the halide functionality for 3b and 3c can serve as diversity point for further structure tuning and optimization.16
:1) with salt 1 to achieve final product 3a–3c in moderate yields. Notably, the halide functionality for 3b and 3c can serve as diversity point for further structure tuning and optimization.16
To further investigate the influence on the structure difference of asymmetrical squaraines, another new series of compounds 6a–6c are synthesized based on semi squaraine salt 4 which contains benzothiazole moiety as a good electron donor and dicyanovinyl groups as a strong electron withdrawing group functionalized on the squarate core (Scheme 2).17 A dicyanovinyl group is added in 6a, in contrast to 3a, to investigate the influence of additional acceptor functionalities at the squarate core. Compound 6b and 6c are also successfully prepared, respectively, to tune the conjugation size in the structure. The yields for 6a–6c range from 8% to 45%.
 | ||
| Scheme 2 Multi steps synthesis of squaraine 4, 6a–6c. | ||
To gain insight into the relationship between molecular structure and physical properties, their absorption and fluorescence values are studied (Table 1; commercially available Cy5 and Cy5.5 standards were included for comparison).18 All of them have shown strong absorptions in red visible to NIR regions with high molar absorption coefficients (ε) up to 104 to 105 mol−1 cm−1 L−1 in DMSO. The introduction of dicyanovinyl group on the squarate core (6a) resulted in significant red shift for the absorption wavelength and a larger stokes shift of 90 nm compared to 3a.
| Dyesa | λmax (nm) | Δλb (nm) | ΔEc (eV) | Δλd (nm) | Øe |
|---|---|---|---|---|---|
| a All the measurements were carried out in the concentration of 10 μM (or less) dyes which dissolve in DMSO.b Δλ (nm) = Stokes shift (nm) in experiments.c Stokes shift presents in ΔE (eV).d Δλ (nm) = Stokes shift (nm) in simulations. CAM-B3LYP/6-311+G(d,p) TDDFT calculations with DMSO PCM solvation (ωB97xD/6-31G(d) PCM optimized structures).e Ø = quantum yield. | |||||
| 3a | 618 | 57 | 0.17 | 63 | 0.26 |
| 3b | 618 | 57 | 0.17 | — | 0.22 |
| 3c | 640 | 35 | 0.10 | — | 0.25 |
| 6a | 630 | 90 | 0.25 | 79 | 0.17 |
| Cy5 | 648 | 27 | 0.07 | — | 0.28 |
| 6b | 680 | 32 | 0.08 | 37 | 0.21 |
| 6c | 712 | 27 | 0.06 | 33 | 0.20 |
| Cy5.5 | 685 | 35 | 0.08 | — | 0.23 |
Large Stokes shift in organic fluorophores has been attributed to intramolecular charge transfer (ICT) excitation state or local excitation (LE) with competing steric and mesomeric interactions. Both phenomena can cause a significant geometric difference between ground state and excited state, resulting in a large Stokes shift. The dibutyl-aniline side chain of our squaraine dye is determined to be the deciding factor in causing a significant Stokes shift as both 3a and 6a has considerable larger Stokes shift than the other substrates despite scaffold similarity. Hence we will consider how both excitation mechanisms influence the N,N-dibutyl-aniline moiety during absorption and fluorescence.
ICT generates a pseudo-zwitterionic excited state S1 where a positive and negative charge resides at different regions of the fluorophore that stabilizes the charges respectively. This leads to a large structural change from the ground state S0 resulting in the large Stokes shift. A well-documented example is 4,4-dimethylaminobenzonitrile19 (DMABN, Fig. 1) and its para-alkylamine derivatives. Its close structural relation to our dibutyl-aniline side chain makes its excitation mechanism plausible for our substrate. Several charge transfer states of DMABN and their kinetic and thermodynamic feasibility has been investigated theoretically. These includes the twisted ICT, TICT model,19b where the amino group is in a perpendicular position relative to the benzene ring, the planar ICT, PICT19c which the amino group lies in the benzene plane, the wagged ICT, WICT19d which involves a rehybridization from planar sp2 to pyramidal sp3 of the amino nitrogen and lastly the rehybridized ICT, RICT19d involves a rehybridization of the cyano carbon atom from sp to sp2 entailing a bent cyano bond.
 | ||
| Fig. 1 DMABN ground state, LE S1 and possible ICT excited states. | ||
The ICT S1 is unobtainable by direct excitation and requires a kinetic transition from the LE S1.19e Due to its pseudo-zwitterionic properties, the ICT S1 will possess significantly higher dipole moment than the ground state S0 and LE S1, causing it to be more stable in polar solvents. Both of these phenomena will result in the observation of the dual fluorescence in the UV spectra produced by the LE S1 and ICT S1 with the ICT fluorescence peak increasing in magnitude with the solvent polarity. As dual fluorescence is absent in our squaraine dyes in both polar and non-polar solvents (Tables 2 and 3), this rules out the ICT mechanism in triggering the large Stokes shift observed.
| Dye | TDDFT method | Optimization method | Solvation | λabs (nm) | λem (nm) | Δλ (nm) |
|---|---|---|---|---|---|---|
| 3a | CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 520 | 598 | 78 |
| CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | PCM | 519 | 582 | 63 | |
| ωB97x-D/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 511 | 596 | 85 | |
| LC-BLYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 480 | 600 | 120 | |
| B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 572 | 606 | 34 | |
| PBE0/6-31+G(d) | ωB97x-D/6-31G(d) | SMD | 560 | 598 | 38 | |
| CAM-B3LYP/6-311+G(d,p) | PBE0/6-31+G(d) | SMD | 544 | 584 | 40 | |
| PBE0/6-311+G(d,p) | PBE0/6-31+G(d) | SMD | 570 | 594 | 24 | |
| CAM-B3LYP/6-311+G(d,p) | CAM-B3LYP/6-31+G(d) | PCM | 521 | 580 | 59 | |
| 6a | CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 520 | 609 | 89 |
| CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | PCM | 528 | 606 | 78 | |
| ωB97x-D/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 507 | 607 | 100 | |
| LC-BLYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 465 | 599 | 134 | |
| B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 595 | 632 | 37 | |
| PBE0/6-31+G(d) | ωB97x-D/6-31G(d) | SMD | 579 | 623 | 43 | |
| CAM-B3LYP/6-311+G(d,p) | PBE0/6-31+G(d) | SMD | 589 | 620 | 31 | |
| PBE0/6-311+G(d,p) | PBE0/6-31+G(d) | SMD | 546 | 608 | 63 | |
| CAM-B3LYP/6-311+G(d,p) | CAM-B3LYP/6-31+G(d) | PCM | 531 | 606 | 75 | |
| 6b | CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 576 | 614 | 39 |
| CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | PCM | 578 | 616 | 37 | |
| ωB97x-D/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 572 | 613 | 41 | |
| B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 604 | 634 | 30 | |
| PBE0/6-31+G(d) | ωB97x-D/6-31G(d) | SMD | 593 | 624 | 31 | |
| CAM-B3LYP/6-311+G(d,p) | CAM-B3LYP/6-31+G(d) | PCM | 579 | 6159 | 37 | |
| 6c | CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 596 | 631 | 35 |
| CAM-B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | PCM | 599 | 634 | 35 | |
| ωB97x-D/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 5918 | 628 | 37 | |
| B3LYP/6-311+G(d,p) | ωB97x-D/6-31G(d) | SMD | 622 | 651 | 29 | |
| PBE0/6-31+G(d) | ωB97x-D/6-31G(d) | SMD | 611 | 640 | 29 | |
| CAM-B3LYP/6-311+G(d,p) | CAM-B3LYP/6-31+G(d) | PCM | 601 | 633 | 32 |
| Dyes | Solvent | λmax | λem | Δλcal | Δλexp |
|
|---|---|---|---|---|---|---|
| 3a | DMSO | 519 | 582 | 63 | 65 | 13 |
| MeOH | 515 | 575 | 60 | 61 | 12 | |
| CHCl3 | 536 | 585 | 49 | 38 | 10 | |
| Hexane | 546 | 585 | 40 | 15 | 7.4 | |
| 6a | DMSO | 528 | 606 | 78 | 90 | 16 |
| MeOH | 525 | 600 | 75 | 80 | 15 | |
| CHCl3 | 557 | 617 | 60 | 67 | 12 | |
| Hexane | 580 | 631 | 51 | 26 | 9.3 | |
| 6b | DMSO | 578 | 616 | 38 | 32 | 6.1 |
| MeOH | 574 | 609 | 35 | 31 | 5.9 | |
| CHCl3 | 594 | 628 | 34 | 26 | 5.6 | |
| Hexane | 605 | 628 | 33 | 10 | 5.5 | |
| 6c | DMSO | 599 | 634 | 35 | 27 | 5.3 |
| MeOH | 593 | 626 | 33 | 25 | 5.3 | |
| CHCl3 | 615 | 648 | 33 | 28 | 5.1 | |
| Hexane | 627 | 640 | 13 | 16 | 5.3 |

In organic fluorophores, local excitation mechanism can cause significant Stokes shift due to the resonance effects being more dominant in the excited state stabilization while steric hindrance is more prominent in the ground state stabilization. This hierarchical change of stabilization factors during excitation creates a substantial geometric change in the process. Liu and coworkers20 exploited this feature in the local excitation mechanism by adding a rotatable substituent that can form conjugation with the fluorophore scaffold and customizing the steric hindrance on the substituent. The rotatable controlled steric hindrance on the substituent is strong enough to form a stabilized non-planar ground state structure but weak enough to be overridden by the mesomeric effect during the excited state formation to form a planar structure where the resonance is maximized to cause a substantial geometric change.
To understand the factors causing the large Stokes shift in 6a in comparison to its structurally analogues 3a and 6b and 6c, TDDFT calculations with implicit solvation in DMSO were utilized. CAM-B3LYP/6-311+G(d,p) TDDFT calculations with PCM21 solvation in DMSO gave the best qualitative trend with the calculated Stokes shift for 3a at 63 nm while that for 6a was 79 nm (Table 2). Initially a benchmark TDDFT studies at the 6-311+G(d,p) level with long range corrected functionals (CAM-B3LYP,22 ωB97X-D23 and LC-BLYP24) and popular DFT methods, B3LYP and PBE0 were computed with ωB97X-D/6-31G(d) optimized structures. SMD25 implicit solvation in DMSO was included in both TDDFT and optimization calculations (Table 2). Only long range corrected functionals were able to correctly determine the larger Stokes shift of 3a and 6a compare to 6b and 6c, demonstrating the importance of range correction in TDDFT calculations. To eliminate the possibility of the results being an artifact of ωB97X-D/6-31G(d) geometries, CAM-B3LYP/6-311+G(d,p) TDDFT calculations were accomplished with CAM-B3LYP/6-31+G(d) and PBE0/6-31+G(d) optimized structures to affirm that the qualitative trend of the Stoke shift (Table 2). The resulting Stokes shift reiterates the qualitative trend with respect to the experimental result.
The TDDFT calculations predicted that for 3a and 6a, 6b and 6c, both the excitation (S0 → S1) and fluorescence (S1 → S0) is caused primarily by the HOMO to LUMO transition. The π conjugation is predominant in the HOMO and LUMO of 3a, 6a, 6b and 6c and its excited states (ESI Fig. S1–S4†), predicting that both the excitation and fluorescence involves a π to π* transition. The orbital contributions from the ketone and dicyanovinyl functional group on the squarate core (Fig. 2) are absent in the LUMO while the squarate core has a significant contribution in the HOMO. This suggests that the squarate core have significant influence on the stabilization of the HOMO of the system. The involvement of the orbital of the dibutylamine N moiety in both the HOMO and LUMO suggest its significance in affecting both the S0 and S1 state in the LE mechanism.
 | ||
| Fig. 2 Isodensity plot of molecular orbitals (HOMO and LUMO) of 3a, 6a, 6b and 6c at ground state. | ||
The magnitude of the Stokes shift is a result of the magnitude of the geometric change between the absorption state and the fluorescence state geometry. The geometric and electron density change between the ground and excited state due to local excitation are studied to elucidate the factors behind the Stokes shift.
The deviation from planarity of the ground and excited state is used to evaluate the geometric change and assess the degree of optimization of the resonance effect. The dihedral angle deviation from planarity in the S0 and S1 state (θ, ESI Tables S2–S5†) is taken to compare the mesomeric effects where the geometric change in reflected in their dihedral deviation difference (Δθ, Table 4). The non-planarity of the dicyanovinyl functional group with respect to the squarate core and the phenyl ring of the N,N-dibutyl-aniline in 6a S0 caused the significant geometric change during excited state relaxation when mesomeric effects are maximized to form a near planar S1 state (Table 4 and Fig. 3). This is shown by the dihedral angle difference (θa, θa2 and θb, ESI Tables S2–S5†) of the mentioned functional group in S0 and S1 state. Despite the planarity of the π conjugated ring system in both 3a S0 and S1 state (Δθb), the significant Stokes shift is caused by the steric interaction between the N,N-dibutyl-amine functional group and the phenyl ring (Δθa, Δθa2). This impedes the mesomeric interaction of the amine with the phenyl ring and the π conjugated system in the ground state, preventing the amine from achieving a planar geometry. Both 6b and 6c S0 and S1 states have near planar geometries which do not experience much geometrical changes during excitation (Table 4) as shown in the small Stokes shift in Table 3.
| Dye | Solvent | Δra/Å | Δθab | Δθa2 | Δθb | Δθc | Δθd | Δθe |
|---|---|---|---|---|---|---|---|---|
| a The value of Δr in parenthesis refers to that of the S0 while the other value refers to that of the excited state.b Δθ is the dihedral angle between the S0 and S1 state's deviation from planarity. θ is calculated as the dihedral angle deviation from planarity where the values are taken form the modulus the difference of dihedral angle from 180° or 0° (i.e. 180° − |x| if x > 90° or |x| − 0 if x ≤ 90°), whichever is nearer to attaining a planar structure. The dihedral angles taken are shown in the figure to the left and also listed by its atomic label below its value in Table S2. Only for 3a and 6a will Δθa2 be applicable as it measures the dihedral angle of both t-butyl groups with respect to the phenyl ring. | ||||||||
| 3a | DMSO | 1.34 [3.13] | 4.1 | 8.8 | 0.0 | 0.1 | 0.5 | 1.3 |
| MeOH | 1.35 [3.03] | 4.7 | 8.1 | 0.1 | 0.1 | 0.5 | 0.7 | |
| CHCl3 | 1.51 [2.76] | 3.1 | 7.9 | 0.1 | 0.1 | 0.4 | 0.5 | |
| n-Hexane | 1.66 [2.40] | 1.8 | 6.2 | 0.3 | 0.5 | 0.6 | 0.3 | |
| 6a | DMSO | 1.33 [3.22] | 2.6 | 7.1 | 14.5 | 5.3 | 3.2 | 0.3 |
| MeOH | 1.34 [3.21] | 2.5 | 7.1 | 14.4 | 5.3 | 3.1 | 0.3 | |
| CHCl3 | 1.55 [2.92] | 4.8 | 3.2 | 6.5 | 3.9 | 0.8 | 0.3 | |
| n-Hexane | 1.76 [2.61] | 2.3 | 1.9 | 2.0 | 1.9 | 0.1 | 0.7 | |
| 6b | DMSO | 0.63 [1.28] | 0.2 | — | 2.8 | 0.3 | 0.6 | 1.6 |
| MeOH | 0.64 [1.25] | 0.4 | — | 0.9 | 0.5 | 0.3 | 1.0 | |
| CHCl3 | 0.75 [1.18] | 0.2 | — | 0.6 | 0.3 | 0.5 | 0.7 | |
| n-Hexane | 0.89 [1.10] | 0.2 | — | 0.1 | 0.3 | 0.5 | 0.4 | |
| 6c | DMSO | 0.69 [0.92] | 0.1 | — | 4.5 | 1.4 | 0.2 | 1.8 |
| MeOH | 0.71 [0.86] | 0.1 | — | 1.5 | 1.3 | 0.1 | 1.8 | |
| CHCl3 | 0.78 [0.85] | 0.2 | — | 1.5 | 1.3 | 0.2 | 1.4 | |
| n-Hexane | 0.90 [0.90] | 0.2 | — | 1.5 | 1.3 | 0.8 | 0.9 | |
 |
||||||||
 | ||
| Fig. 3 6a S0 and S1 RDG isosurface. Steric repulsion (red isosurface) between the N,N-butyl groups with the phenyl ring is increased significantly in the excited state as mesomeric effects override the steric influence. | ||
Cave et al.26 demonstrated that reorganization energy (eqn (1)) of the ground and excited state can be used as a qualitative relationship between the geometrical and energetic changes. From Table 3, the trend of the increasing Stokes shift with the corresponding increase in reorganization energy of the S1 state correlates with the respective magnitude of the geometrical change in Table 4.
The reorganization energy of the S1 state is given by the equation
 | (1) |
To account for the difference in steric repulsion experience by the S0 and S1 state, the NCI analysis27 is employed. The NCI index is based on a 2D plot of the reduced density gradient, s, and the electron density, ρ. The reduced density gradient, s, is derived from the electron density (ρ) of a system and its first derivative (eqn (2)).
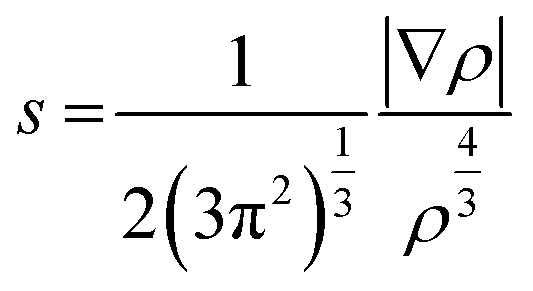 | (2) |
To differentiate between attractive and repulsive non-covalent interactions, the sign of the Laplacian of the density, ∇2ρ is used. The Laplacian is decomposed into a sum of contributions along the three principal axes of maximal variation. These components are the three eigenvalues λi of the electron-density Hessian (second derivative) matrix, such that ∇2ρ = λ1 + λ2 + λ3, (λ1 < λ2 < λ3). The second eigenvalue λ2 is used to differentiate the attractiveness or repulsiveness of an interaction. A positive sign λ2 > 0 (red isosurface) signifies steric repulsion while a negative sign λ2 < 0, (green to beige isosurface) shows attractive non-covalent interactions such as van der Waals or C–H–O interaction. This popular non covalent interaction evaluation method has been applied to both chemical and bio-system to analyse the essential intra and intermolecular interaction features.
The excited states of both 6a (Fig. 3) and 3a (ESI Fig. S6†) experiences higher steric interactions compared to its ground state as there is an observed increase in the red isosurface. This is a result of the hierarchical change of resonance interaction overriding the steric interactions in the excited state for both 3a and 6a, generating a more planar structure that experience more steric repulsion but maximizes the mesomeric effect. The higher geometric change in 6a.
An index described by Guido and coworkers, Δr (ref. 28) was introduced to characterize the amount of spatial rearrangement when the exchange correlation functional is inadequate to describe the charge transfer excitation. This is an issue for Tozer et al. Λ diagnostic index which in some cases is unable to differentiate the magnitude of short range charge transfer or local excitation. The hole–particle pair interactions using Δr is related to the average distance covered during the excitations correlated to the function of the excitation coefficients.
 | (3) |
| Kia = Xia + Yia | (4) |
The composition of Xia (excitation coefficient) and Yia (de-excitation coefficient) is made up of the molecular orbital overlap between the occupied orbitals φi and the virtual orbitals φa involved in the electronic transition. The Δr distance is related to the nature of the transition where the valence excitations and LE are characterized by short distances, while larger distances are associated with charge transfer excitation.
The Δr trend observed in Table 4 sees a higher CT characteristic in the excitation for both 3a and 6a with a larger Δr. This is consistent with the more significant geometrical change between the S0 and S1 in addition to the higher planarity of the excited structure where the optimized mesomeric interaction in S1 will stabilize a more polarized moiety. The smaller Δr value for 6b and 6c show the strong LE character of the excitation. The lack of NCI isosurface change between the S0 and S1 state shows the absence of competing mesomeric and steric induced geometrical change.
The solvent effects are reflected in Table 3 where the decrease in the solvent polarity by comparing DMSO to chloroform and to hexane results in the corresponding decrease in Stokes shift. This is caused by the decreasing stabilization of a more polarized and planar geometry in the excited state. This phenomenon is demonstrated in the increase of θ (ESI, Table Sx†) for all the squaraine dyes S1 states in CHCl3 and n-hexane solvents where they are unable to attain the more planar structure as achieved in the polar solvent DMSO. This result in a smaller geometric change between the solvated and excited state (Δθ, Table 4), hence a smaller Stokes shift (Table 3).
3. Conclusion
To conclude, six asymmetric squaraines were synthesized in a multistep manner through two semi-squaraine salts. Such an amenable strategy facilitates: (1) high value asymmetrical squaraines; (2) independent structure optimization to tune absorption and emission wavelengths within range from 600 nm to 720 nm. The quantum yields of these dyes are comparable to commercial available cyanine dyes. The unique large stokes shifts (up to 90 nm) are observed for this series of squaraines (especially for 6a) which can offer direct applications in multichannel molecular imaging with a clear and readable signal. TDDFT calculations indicate that the steric effect from N,N-dibutyl groups with the phenyl ring and the dicyanovinyl group on the squarate core contributes to the large geometric change, resulting in the large Stokes shift. All these findings help us to understand the relationships between structures and physical properties of squaraines.4. Computational details
All TDDT calculations were run in Gaussian 09 Rev B.01 (ref. 29) and molecular orbitals and electron density analysis were computed with Multiwfn.305. Experimental section
Materials and methods, details of synthesis and characterization of final compounds and all spectrums of absorption and emission for these dyes have been listed in the ESI.†Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by MOE-TIF grant (MOE2012-TIF-1-T-051) from the Ministry of Education and SP R&D (TIEFA) grant from Singapore Polytechnic.References
- (a) M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781 CrossRef CAS; (b) K. Y. Law, Chem. Rev., 1993, 93, 449 CrossRef CAS; (c) A. Ajayaghosh, Acc. Chem. Res., 2005, 38, 449 CrossRef CAS PubMed; (d) W. Shi and H. Ma, Chem. Commun., 2012, 48, 8732 RSC; (e) J. J. McEwen and K. J. Wallace, Chem. Commun., 2009, 6339 RSC; (f) S. L. Lam, X. Liu, F. Zhao, C. L. K. Lee and W. L. Kwan, Chem. Commun., 2013, 49, 4543 RSC; (g) F. Silvertri, M. D. Irwin, L. Beverina, A. Facchetti, G. A. Pagani and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 17640 CrossRef PubMed; (h) J. H. Yum, P. Waltre, S. Huber, D. Rentsch, T. Geiger, F. Nuesch, F. D. Angelis, M. Gratzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320 CrossRef CAS; (i) D. Demeter, T. Rousseau, P. Leriche, T. Cauchy, R. Po and J. Roncali, Adv. Funct. Mater., 2011, 21, 4379 CrossRef CAS.
- (a) B. Oswald, L. Patsenker, J. Duschl, H. Szmacinski, O. S. Wolfbeis and E. Terpetschnig, Bioconjugate Chem., 1999, 10, 925 CrossRef CAS; (b) B. Oswald, M. Gruber, M. Bohmer, F. Lehman, M. Probst and O. S. Wolfbeis, Photochem. Photobiol., 2001, 74, 237 CrossRef CAS PubMed; (c) L. D. Patsenker, A. L. Tatarets, Y. A. Povrozin and E. A. Terpetschnig, Bioanalytical reviews, 2011, 3, 115 CrossRef; (d) J. J. Lee, A. G. White, J. M. Baumes and B. D. Smith, Chem. Commun., 2010, 46, 1068 RSC; (e) E. M. Sevick-Muraca, J. P. Houston and M. Gurfinkel, Curr. Opin. Chem. Biol., 2002, 6, 642 CrossRef CAS PubMed; (f) M. S. Goncalves, Chem. Rev., 2009, 109, 190 CrossRef CAS PubMed; (g) R. R. Avirah, D. T. Jayaram, N. Adarsh and D. Ramaiah, Org. Biomol. Chem., 2012, 10, 911 RSC; (h) J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025 CrossRef CAS PubMed; (i) K. Funabiki, H. Mase, Y. Saito, A. Otsuka, A. Hibino, N. Tanaka, H. Miura, Y. Himori, T. Yoshida, Y. Kubota and M. Matsui, Org. Lett., 2012, 14, 1246 CrossRef CAS PubMed.
- (a) Y. Y. Huang, P. Mroz, T. Zhiyentayev, S. K. Sharma, T. Balasubramanian, C. Ruzie, M. Krayer, D. Fan, K. E. Borbas, E. Yang, H. L. Kee, C. Kirmaier, J. R. Diers, D. F. Bocian, D. Holten, J. S. Lindsey and M. R. Hamblin, J. Med. Chem., 2010, 53, 4018 CrossRef CAS PubMed; (b) S. H. Lim, C. Thivierge, P. N. Sliwinska, J. Y. Han, H. V. D. Bergh, G. Wagnières, K. Burgess and H. B. Lee, J. Med. Chem., 2010, 53, 2865 CrossRef CAS PubMed; (c) V. Rapozzi, L. Beverina, P. Salice, G. A. Pagani, M. Camerin and L. E. Xodo, J. Med. Chem., 2010, 53, 2188 CrossRef CAS PubMed; (d) M. Obata, S. Hirohara, R. Tanaka, I. Kinoshita, K. Ohkubo, S. Fukuzumi, M. Tanihara and S. Yano, J. Med. Chem., 2009, 52, 2747 CrossRef CAS PubMed; (e) T. Lu, P. Shao, I. Mathew, A. Sand and W. Sun, J. Am. Chem. Soc., 2008, 130, 15782 CrossRef CAS PubMed; (f) P. K. Frederiksen, S. P. McIlroy, C. B. Nielsen, L. Nikolajsen, E. Skovsen, M. Jørgensen, K. V. Mikkelsen and P. R. Ogilby, J. Am. Chem. Soc., 2005, 127, 255 CrossRef CAS PubMed; (g) A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619 CrossRef CAS PubMed; (h) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897 CrossRef CAS PubMed; (i) H. S. Choi, K. Nasr, S. Alyabyev, D. Feith, J. H. Lee, S. H. Kim, Y. Ashitate, H. Hyun, G. Patonay, L. Strekowski, M. Henary and J. V. Frangioni, Angew. Chem., Int. Ed., 2011, 6258 CrossRef CAS PubMed; (j) Y. Xu, B. Li, P. Han, S. Sun and Y. Pang, Analyst, 2013, 138, 1004 RSC; (k) K. M. Shafeekh, S. Das, C. Sissa and A. Painelli, J. Phys. Chem. B, 2013, 117, 8536 CrossRef CAS.
- (a) P. G. Jönsson and Å. Kvick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1827 CrossRef; (b) P. D. William, R. A. Jockusch and R. E. Williams, J. Am. Chem. Soc., 1997, 119, 11988 CrossRef; (c) H. S. Choi, K. Nasr, S. Alyabyev, D. Feith, J. H. Lee, S. H. Kim, Y. Ashitate, H. Hyun, G. Patonay, L. Strekowski, M. Henary and J. V. Frangioni, Angew. Chem., Int. Ed., 2011, 6258 CrossRef CAS PubMed; (d) W. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530 CrossRef CAS; (e) H. S. Choi and J. V. Frangioni, Mol. Imaging, 2010, 9, 291 CAS.
- (a) A. Treibe and K. Jacob, Angew. Chem., Int. Ed. Engl., 1965, 4, 694 CrossRef; (b) A. Treibs and K. Jacob, Justus Liebigs Ann. Chem., 1968, 712, 123 CrossRef CAS; (c) A. Treibs and K. Jacob, Justus Liebigs Ann. Chem., 1966, 699, 153 CrossRef CAS; (d) K. Y. Law, J. Phys. Chem., 1987, 91, 5184 CrossRef CAS.
- (a) C. Luo, Q. Zhou, B. Zhang and X. Wang, New J. Chem., 2011, 35, 45 RSC; (b) L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 2010, 1207 CrossRef; (c) A. Ajayaghosh, Acc. Chem. Res., 2005, 38, 449 CrossRef CAS PubMed; (d) A. Ajayaghosh, Chem. Soc. Rev., 2003, 32, 181 RSC; (e) M. V. Reddington, Bioconjugate Chem., 2007, 8, 2178 CrossRef PubMed; (f) V. Rapozzi, L. Beverina, P. Salice, G. A. Pagani, M. Camerin and I. E. Xodo, J. Med. Chem., 2010, 53, 2188 CrossRef CAS PubMed; (g) U. Mayerhoffer, M. Gasanger, M. Stolte, B. Fimmel and F. Wurthner, Chem.–Eur. J., 2013, 19, 218 CrossRef PubMed; (h) U. Mayerhoffer, B. Fimmel and F. Wurthner, Angew. Chem., Int. Ed., 2012, 51, 164 CrossRef PubMed.
- (a) J. Y. Li, C. Y. Chen, C. P. Lee, S. C. Chen, T. H. Lin, H. H. Tsai, K. C. Ho and C. G. Wu, Org. Lett., 2010, 12, 5454 CrossRef CAS PubMed; (b) L. Beverina, R. Ruffo, M. M. Salamone, E. Ronchi, M. Binda, D. Natali and M. Sampietro, J. Mater. Chem., 2012, 22, 6704 RSC.
- (a) Y. Q. Liu, A. Malkovskiy, Q. M. Wang and Y. Pang, Org. Biomol. Chem., 2011, 9, 2878 RSC; (b) L. I. Markova, V. L. Malinovskii, L. D. Patsenker and R. Haner, Org. Biomol. Chem., 2012, 10, 8944 RSC.
- (a) D. Keil and H. Hartmann, Dyes Pigm., 2001, 49, 161 CrossRef CAS; (b) S. Sreejith, P. Carol and A. Ajayaghosh, J. Mater. Chem., 2008, 18, 264 RSC; (c) J. Chen and R. F. Winter, Chem.–Eur. J., 2012, 18, 10733 CrossRef CAS PubMed; (d) D. E. Lynth, A. N. Kirkham, M. Chowdhury, E. S. Wane and J. Heptinstall, Dyes Pigm., 2012, 94, 393 CrossRef; (e) Y. Shi, R. B. Hill, J. H. Yum, A. Dualeh, S. Barlow, M. Gratzel, S. R. Marder and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2011, 50, 6619 CrossRef CAS PubMed.
- (a) S. S. Pandey, R. Watanable, N. Fujikawa, G. M. Shivashimpi, Y. Ogomi, Y. Yamaguchi and S. Hayase, Tetrahedron, 2013, 69, 2633 CrossRef CAS; (b) S. Kuster and T. Geiger, Dyes Pigm., 2012, 657 CrossRef CAS.
- M. C. Basheer, U. Santhosh, S. Alex, K. G. Thomas, C. H. Suresh and S. Das, Tetrahedron, 2007, 63, 1617 CrossRef CAS.
- (a) S. Kim, G. K. Mor, M. Paulose, O. K. Varghese, C. Baik and C. A. Grimes, Langmuir, 2010, 26, 13486 CrossRef CAS PubMed; (b) V. Hrobarikova, P. Hrobarik, P. Gajdos, I. Fitilis, M. Fakis, P. Persephonis and P. Zahradnik, J. Org. Chem., 2010, 75, 3053 CrossRef CAS PubMed.
- (a) S. Yasui, M. Matsuoka and T. Kitao, Dyes Pigm., 1988, 10, 13 CrossRef CAS; (b) Z.-S. Wang, F. Y. Li and C. H. Huang, J. Phys. Chem. B, 2001, 105, 9210 CrossRef CAS.
- (a) K.-Y. Law, Chem. Rev., 1993, 93, 449 CrossRef CAS; (b) K. A. Bello, S. N. Corns and J. Griffiths, J. Chem. Soc., Chem. Commun., 1993, 452 RSC; (c) H. E. Sprenger and W. Ziegenbein, Angew. Chem., Int. Ed. Engl., 1966, 5, 893 CrossRef PubMed.
- (a) L. H. Liu, K. Nakatani, R. Pansu, J. J. Vachon, P. Tauc and E. Ishow, Adv. Mater., 2007, 19, 433 CrossRef CAS; (b) V. Ramalingam, M. E. Domaradzki, S. Jang and R. S. Muthyala, Org. Lett., 2008, 15, 3315 CrossRef PubMed.
- (a) Y. Shi, R. B. Hill, J. H. Yum, A. Dualeh, S. Barlow, M. Gratzel, S. R. Marder and M. K. Nazeerudin, Angew. Chem., Int. Ed., 2011, 50, 6619 CrossRef CAS PubMed; (b) C. H. Chang, Y. C. Chen, C. Y. Hsu, H. H. Chou and J. T. Lin, Org. Lett., 2012, 14, 4726 CrossRef CAS PubMed; (c) J. Chen and R. F. Winter, Chem.–Eur. J., 2012, 18, 10733 CrossRef CAS PubMed; (d) S. Kuster and T. Geiger, Dyes Pigm., 2012, 95, 657 CrossRef CAS.
- (a) U. Mayerhoffer, M. Gsanger, M. Stolte, B. Fimmel and F. Wurthner, Chem.–Eur. J., 2013, 19, 218 CrossRef PubMed; (b) S. Kim, G. K. Mor, M. Paulose, O. K. Varghese, C. Baik and C. A. Grimes, Langmuir, 2010, 26, 13486 CrossRef CAS PubMed.
- M. Klessinger, Chem. Unserer Zeit, 1978, 12, 1 CrossRef CAS.
- (a) I. Gómez, M. Reguero, M. Boggio-Pasqua and M. A. Robb, J. Am. Chem. Soc., 2005, 127, 7119 CrossRef PubMed; (b) K. Rotkiewicz, K. H. Grellmann and Z. R. Grabowski, Chem. Phys. Lett., 1973, 21, 212 Search PubMed; (c) K. A. Zachariasse, S. I. Druzhinin, W. Bosch and R. Machinek, J. Am. Chem. Soc., 2004, 126, 1705 CrossRef CAS PubMed; (d) A.-D. Gorse and M. Pesquer, J. Phys. Chem., 1995, 99, 4039 CrossRef CAS; (e) W. Schuddeboom, S. A. Jonker and J. M. Warman, J. Phys. Chem., 1992, 96, 10809 CrossRef CAS.
- X. Liu, Z. Xu and J. M. Cole, J. Phys. Chem. C, 2013, 117, 16584 CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- R. J. Cave, K. Burke and E. W. Castner Jr, J. Phys. Chem. A, 2002, 106, 9294 CrossRef CAS.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed; (b) J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef CAS PubMed; (c) J. M. V. Franco, A. E. Ferao, G. Schnakenburg and R. Streubel, Chem. Commun., 2013, 49, 9648 RSC.
- C. A. Guido, P. Cortona, B. Mennucci and C. Adamo, J. Chem. Theory Comput., 2013, 9, 3118 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision A.2), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra18998e |
| This journal is © The Royal Society of Chemistry 2015 |