
Chitosan nanoparticle carrier based on surface molecularly imprinted polymers for the recognition and separation of proteins†
Cenjin Zhanga,
Yuzhi Wang*a,
Junxia Guoa,
Yanjin Liua and
Yigang Zhoub
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: wyzss@hnu.edu.cn; Fax: +86-731-88821848; Tel: +86-731-88821903
bDepartment of Microbiology, College of Basic Medicine, Central South University, Changsha, 410083, P. R. China
First published on 1st December 2015
Abstract
This paper discusses the construction of surface molecularly imprinted polymers (MIPs) based on the modified chitosan (CS) nanoparticle carrier to recognize and separate bovine serum albumin (BSA, pI 4.9, MW 69.0 kDa) in aqueous solution. Functional biopolymer CS was selected as the supporting material. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group was introduced to CS by glycidyl methacrylate (GMA) to form ethylene-modified chitosan (GMA-CS), so that the imprinting polymerization could be initiated onto GMA-CS. Both bi-functional monomers MIP1 composed of acrylamide (AAm) and acrylic acid (AA) and single functional monomer MIP2 polymerized only by AA were prepared. Transmission electron microscopy (TEM) and field emission scanning electron microscopy (FESEM) were used to characterize the micro-morphology of MIP1 and MIP2, respectively. Static adsorption experiments showed that the adsorption capacity of MIP2 for BSA was much higher than MIP1, so we chose MIP2 for further research. The results showed that the MIP2 could accomplish adsorption equilibrium within only 2 h under an initial BSA concentration of 1.0 mg mL−1 and gave an imprint factor of 2.35. The theoretical maximum adsorption capacity (Qmax) was determined by the Langmuir model, which turned out to be 373.13 mg g−1. The selectivity of the MIP2 was evaluated by direct adsorption of a single reference protein and mixed proteins. The UV measurement and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis results indicated that the MIP2 had a relatively higher adsorption capacity with good recognition and binding selectivity for BSA, which made it possible to remove the template protein from different sample solutions. After three adsorption–desorption cycles, MIP2 could still maintain 66.5% of absorption capacity. It was also found that the buffer pH had a great influence on the adsorption capacity of MIP2, indicating electrostatic interactions played an important role in the absorption and recognition process.
C group was introduced to CS by glycidyl methacrylate (GMA) to form ethylene-modified chitosan (GMA-CS), so that the imprinting polymerization could be initiated onto GMA-CS. Both bi-functional monomers MIP1 composed of acrylamide (AAm) and acrylic acid (AA) and single functional monomer MIP2 polymerized only by AA were prepared. Transmission electron microscopy (TEM) and field emission scanning electron microscopy (FESEM) were used to characterize the micro-morphology of MIP1 and MIP2, respectively. Static adsorption experiments showed that the adsorption capacity of MIP2 for BSA was much higher than MIP1, so we chose MIP2 for further research. The results showed that the MIP2 could accomplish adsorption equilibrium within only 2 h under an initial BSA concentration of 1.0 mg mL−1 and gave an imprint factor of 2.35. The theoretical maximum adsorption capacity (Qmax) was determined by the Langmuir model, which turned out to be 373.13 mg g−1. The selectivity of the MIP2 was evaluated by direct adsorption of a single reference protein and mixed proteins. The UV measurement and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis results indicated that the MIP2 had a relatively higher adsorption capacity with good recognition and binding selectivity for BSA, which made it possible to remove the template protein from different sample solutions. After three adsorption–desorption cycles, MIP2 could still maintain 66.5% of absorption capacity. It was also found that the buffer pH had a great influence on the adsorption capacity of MIP2, indicating electrostatic interactions played an important role in the absorption and recognition process.
1. Introduction
Molecule imprinting is a powerful method to prepare tailor-made synthetic polymers capable of molecular recognition by copolymerization of template molecule-functional monomer complexes and crosslinkers. After the removal of the template molecule from the resulting polymer network, specific binding cavities used as recognition sites for the template molecule are left which are composed of functional monomer residues assembled to fit the template molecule in terms of their size, shape, and exposed chemical functionality.1 Because of its excellent advantages like specific recognition, high stability, low cost and reusability, MIP material can be applied in many fields of sensors,2,3 solid-phase extraction,4 separation,5,6 catalysis,7 water treatment8 and drug design.9 Despite the attractive features of MIP material, it has been largely limited to small molecules. The technique of protein imprinting to specifically recognize the target protein still remains a challenge, because proteins have their own characteristics of large molecular size, high flexibility of spatial conformation and complex surface structures.10 The protein imprinted polymers can be used as cell scaffold materials, antibodies and enzymes, which can substitute for natural biological structures.11–13 Therefore, it is necessary to study in depth the preparation of protein imprinted polymers.However, due to the thick polymeric network, the MIPs prepared by the conventional bulky polymerization technique had some disadvantages especially for protein imprinting. For example, the polymeric network restricted the template molecule from removal and rebinding, which resulted in materials with poor site accessibility and low rebinding capacity for the target molecules.14,15 Because the protein template molecules were easily entrapped in the matrices, the elution was difficult, the diffusion barrier for the template molecules was higher, the rate of mass transfer was lower, and the template molecules were not easy to bind with recognition sites.16,17 To overcome these drawbacks effectively and enhance the imprinted efficiency, the surface molecularly imprinted method in which the imprinted polymerization system was formed on a support substrate material surface18,19 even combined with nanotechnology20,21 have been proposed in place of the traditional bulky imprinting methods. For surface molecular imprinting, carbon nanotubes,22 magnetic nanoparticles,23 silica particles24 and quantum dots25 have already been used as solid support materials.
CS is a type of natural alkaline polysaccharose, which is a linear biopolymer consisting of β-(1,4)-2-acetamido-β-D-glucose units with excellent features such as biodegradability, biocompatibility, nontoxicity, nonantigenicity, abundant source, hydrophilicity and low-cost nature.26 It can initiate a lot of chemical reactions which contain plenty of reactive hydroxyl and amino functional groups. So chitosan can be modified by acylation, alkylation, etherification, esterification, halogenation, etc. These controllable chemical modifications produce derivatives with different structures and chemical properties.27–31 Owing to its advantageous nature, chitosan and its derivatives have drawn wide attention in the biomedical field. Many researchers have made several attempts to apply chitosan to the molecular imprinting technique. Wei32 et al. synthesized a metal ion imprinted chitosan resin, which could considerably enhance the adsorption capacity and selectivity of the metal ion. Guo33 et al. have prepared hemoglobin protein molecularly imprinted polymers, using acrylamide as the functional monomer, and cross-linked chitosan beads as the supporting matrix. The obtained MIP showed a much higher adsorption capacity for hemoglobin than the NIP with the same chemical composition.
In this work, we attempted to employ the functional biopolymer CS as the supporting substrate material to improve the adsorption capacity of surface imprinting because CS has plenty of amino and hydroxyl groups which might self-assemble onto the template protein by hydrogen bonding, hydrophobic interactions and van der Waals forces. The CC group was introduced to CS from GMA so that imprinting polymerization could occur on the modified GMA-CS carrier, which could form the uniform and nano-sized production particles to offer a high surface area for the adsorption of the template. Both bi-functional monomers MIP1 composed of AAm with AA and single functional monomer MIP2 polymerized only by AA were prepared. After the template protein BSA was removed by 10% (w/v) SDS-10% (v/v) acetic acid (HAc) eluent solution, specific binding sites that matched the template were obtained on MIPs. The resulting MIPs could effectively solve the problem of difficult elution, which was also conducive for the template protein rebinding process. MIP1 was characterized by TEM. MIP2 was characterized by FESEM, Fourier transform infrared spectroscopy (FT-IR) and thermogravimetric analysis (TG). The recognition performance of MIP2 for BSA was further investigated by adsorption capacity, imprinting efficiency and specific selectivity through static adsorption tests.
2. Experimental section
2.1 Instrumentation
A UV-2450 UV-vis spectrophotometer (Shimadzu, Japan) was used to determine the absorbance of protein. IR spectra were recorded on a FT-IR spectrometer (Perkin Elmer, USA). TG curves of samples were acquired by thermal gravimetric analyzer (Netzsch, Germany). A Virtis freeze drier (YiKang Experimental Equipment Co. Ltd, China) was employed to obtain the freeze-dried polymers. After being dried, the samples were imaged under a JEM-3010 transmission electron microscope (JEOL Company, Japan) or a Hitachi S-4700 scanning electron microscope (Hitachi Company, Japan). QYC 200 incubator shaker (FuMa Experimental Equipment Co. Ltd, China) was used for the static adsorption process. An RM 220 ultrapure water instrument (LiDe Experimental Equipment Co. Ltd, China) was used throughout the experiments.2.2 Chemicals and reagents
N,N-Methylene-bis-acrylamide (MBAA), chitosan, glycidyl methacrylate was supplied by Aladdin chemistry Co., Ltd. (Shanghai, China). Acrylic acid, acetone, acetic acid, ammonium persulfate (APS), acrylamide, N,N,N′,N′-tetramethylenediamine (TEMED), hydrochloric acid (HCl), tris(hydroxymethyl)aminomethane (Tris), bovine serum albumin (BSA, MW 69 kDa, pI 4.9), bovine hemoglobin (BHb, MW 65 kDa, pI 6.9), ovalbumin (OVA, pI 4.7, MW 43.0 kDa) were all purchased from Guoyao chemical reagents company Co., Ltd. (Beijing, China). Sodium dodecyl sulfate was purchased from FuCheng Chemical Reagent (Tianjin, China). Tris–HCl buffer solution (pH 7.0, 10 mM) was used as the working medium. All chemicals used were of analytical grade and used directly without further purification. Ultrapure water (18.25 MΩ cm−1) used throughout the experiment was obtained from the laboratory purification system.2.3 Preparation of functional chitosan
Functional CS carrier was prepared by the introduction of CC from GMA following a procedure reported by Guan et al.34 The synthesis process was shown in Fig. 1. In this experiment, 1 g of CS was resolved in 50 mL of 2% (v/v) HAc aqueous solution. The system was stirred for 1 h at room temperature and deoxygenated by purging with nitrogen for 5 minutes, then 5.3 mL of GMA was added dropwise to the above aqueous solution of CS at the mole rate of vinyl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amino = 6:1. The reaction system was left to stand for 24 h at room temperature under nitrogen atmosphere protection and magnetic stirring. After reaction, a large amount of acetone was poured into the product solution to form functional CS precipitate. The precipitate was filtered and washed by acetone three times to remove unreacted GMA. Finally, the product was dried by a Virtis freeze drier to obtain ethylene-modified chitosan (GMA-CS).
:amino = 6:1. The reaction system was left to stand for 24 h at room temperature under nitrogen atmosphere protection and magnetic stirring. After reaction, a large amount of acetone was poured into the product solution to form functional CS precipitate. The precipitate was filtered and washed by acetone three times to remove unreacted GMA. Finally, the product was dried by a Virtis freeze drier to obtain ethylene-modified chitosan (GMA-CS).
 | ||
| Fig. 1 Reaction diagram of GMA-chitosan. | ||
2.4 Preparation of imprinted and non-imprinted polymers
In this experiment, both bi-functional monomers MIP1 and single functional monomer MIP2 were prepared. Highly schematic representations of the preparation processes are shown in Fig. 2. At first, 100 mg of GMA-CS carrier was dispersed in 3 mL of Tris–HCl buffer solution (pH = 7.0, 10 mM) with shaking for 30 minutes. Then, 40 mg of template protein BSA, 30 mg of crosslinkers MBAA, 90 mg of main functional monomer AAm combined with 60 μL of second functional monomer AA for MIP1 or 150 μL of single functional monomer AA for MIP2 were added into the GMA-CS solutions, with stirring for 2 h to obtain the completely self-assembled complexes on GMA-CS carriers. After the prepolymerization, the solutions were deoxygenated by purging with nitrogen for 8 minutes, then 40 μL of 20% wt initiator APS and 10 μL of TEMED were added. The reaction systems were purged with nitrogen for another 5 minutes and sealed immediately. The polymerization was carried out for 24 h at room temperature with shaking. Afterwards, the obtained polymers were first washed with a solution of SDS (10%, w/v) and HAc (10%, v/v) to remove the template protein several times until no BSA could be detected by a UV-vis spectrophotometer at 278 nm and then washed with ultrapure water to remove SDS. Finally, the imprinted polymers were obtained after drying by a Virtis freeze drier. The control non-imprinted polymers were also prepared and treated in the same way except for the addition of the template BSA.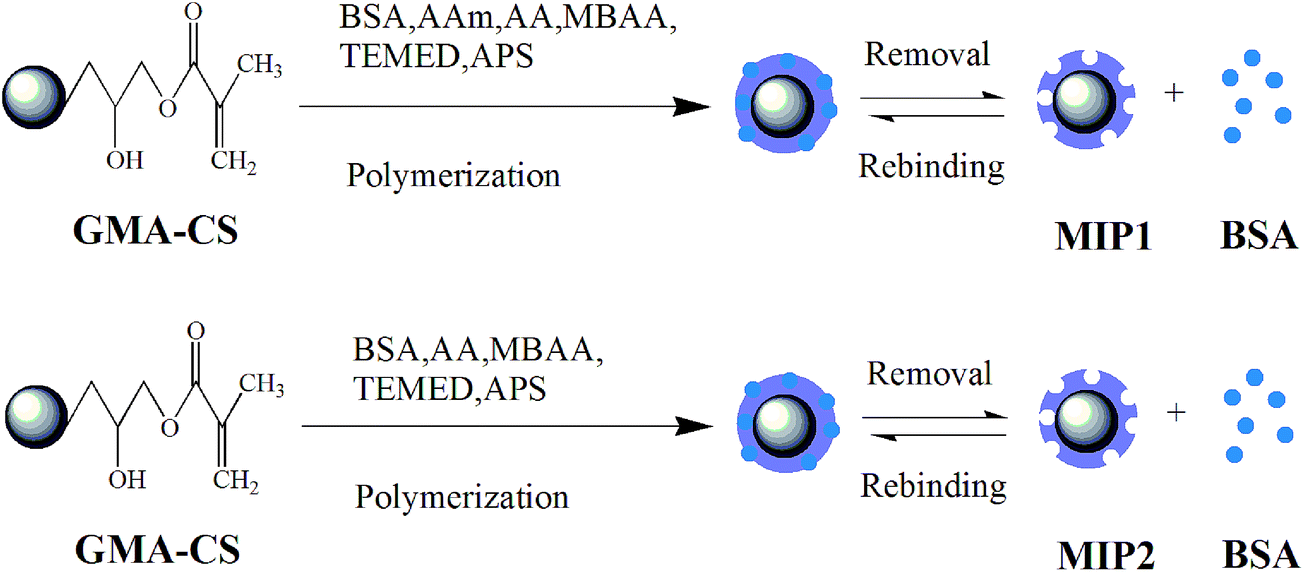 | ||
| Fig. 2 Schematic representation of the molecular imprinting procedures for MIP1 and MIP. | ||
2.5 Protein adsorption experiments
The adsorption performance of the MIPs were studied by adsorption experiments, including adsorption kinetics experiments, adsorption isothermal experiments, selectivity experiments and reusability experiments. The prepared hydrogels were first swollen in the Tris–HCl buffer (pH = 7.0, 10 mM) to gain equilibrium prior to use. Adsorption experiments were conducted by incubating 5.0 mg of MIP or NIP in 4.0 mL of a certain initial concentration protein Tris–HCl buffer solution (pH = 7.0, 10 mM). All the adsorption experiments were conducted in the incubator shaker (200 rpm) under room temperature. After adsorption equilibrium, samples were centrifuged at 5000 rpm for 6 minutes. The concentration of protein in the supernatant was measured by detecting the absorbance at 278 nm for BSA and OVA and 404 nm for BHb using a UV-2450 UV-vis spectrophotometer. As an example, the UV-vis spectra of BSA was shown in Fig. 3. The capacity of protein adsorbed by the MIP or NIP was calculated by the following formula:
 | (1) |
 | ||
| Fig. 3 UV-vis spectrum of BSA in pure water. | ||
The specific recognition characteristic of the MIP was evaluated by the imprinting factor α which was defined by following formula:
| α = QMIP/QNIP | (2) |
In the adsorption kinetics experiments, the initial concentration of BSA was 1.0 mg mL−1. The concentration of BSA at different times was measured using a UV-vis spectrophotometer at a wavelength of 278 nm. Adsorption isothermal experiments were performed by adsorption of a series of different initial concentrations of BSA solutions (0.2–1.6 mg mL−1) for 2 h.
In the selectivity experiments, BHb (1.0 mg mL−1) and OVA (1.0 mg mL−1) were chosen as the reference proteins to prove the selectivity of MIP towards the template protein BSA (1.0 mg mL−1). The concentration of each protein was measured by the UV-vis spectrophotometer at their own maximum absorption wavelength. For the mixed adsorption experiments, OVA was chosen as the competitive protein. The adsorption was performed with a protein mixture (containing 1.0 mg mL−1 of OVA and 1.0 mg mL−1 of BSA). 10 μL of the mixed solution, before and after the adsorption, was extracted for SDS-PAGE analysis with a 12% polyacrylamide separation gel. In the reusability experiment, the MIP was subjected to an adsorption–desorption cyclic operation three times. SDS-HAc was used for elution to completely remove the BSA adsorbed on MIP to free the imprinted sites and make MIP regain the ability to adsorb protein.
In the adsorption experiments, the adsorption capacities of MIP1 and MIP2 for BSA at the initial protein concentration of 1.0 mg mL−1 after an adsorption time of 7 h were compared. The result showed that the adsorption capacity of MIP2 was 369.93 mg g−1, which was much higher than that of MIP1 whose adsorption capacity value was 50.25 mg g−1. The difference of adsorption capacity between MIP1 and MIP2 might be related to the thickness of the polymer layer. In the synthetic process of MIP2, 150 μL of AA was used as functional monomer. While in MIP1, 90 mg of AAm and 60 μL of AA was used as bi-functional monomer, which could increase the thickness of the polymer layer. And then blocked the site accessibility and led to the decrease in the binding amount of BSA. So we selected MIP2 and NIP2 for all subsequent experiments.
2.6 Characterization of polymers
The surface morphologies of GMA-CS, MIP2 and NIP2 were determined by SEM. TEM was mainly used to confirm nano-sized GMA-CS carrier particles, besides, MIP1 and NIP1 were also characterized by TEM. The CS, GMA-CS, MIP2 and NIP2 were characterized by an FT-IR spectrometer in the range of 4000–500 cm−1 to verify the successful synthesis of MIP2 on the surface of the GMA-CS carrier. TG curves of GMA-CS, MIP2 and NIP2 were acquired to further verify the relative composition of the prepared materials.3. Results and discussion
3.1 Characterization of the prepared materials
SEM images of GMA-CS (a), MIP2 (b) and NIP2 (c) are shown in Fig. 4 to characterize their surface morphology and microstructure. As seen from the images, the surface morphology of the prepared materials were different from each other. The surface of GMA-CS carrier was very smooth with a compact and dense microstructure. After grafting with AA, MIP2 and NIP2 with an extra polymer layer appeared with a rough surface morphology in SEM images. In addition, when carefully compared MIP2 with NIP2, it could be found that although the porosity could not be obviously seen on the surface of MIP2, the MIP2 had a relatively rougher surface, indicating the success of the polymerization in the presence of the template protein. | ||
| Fig. 4 SEM images of GMA-CS (a), MIP2 (b), NIP2 (c) and TEM images of GMA-CS (d), NIP1 (e) and MIP1 (f). | ||
TEM images of GMA-CS (d), NIP1 (e) and MIP1 (f) are shown in Fig. 4 to characterize their microscopic size and shape. A uniform and regular spherical particle structure could be observed from all three of the TEM images, which verified the successful formation of the desired material shape. It could be clearly observed that the average diameters of GMA-CS, NIP1 and MIP1 were different from each other. Compared with the GMA-CS whose diameter ranged from 30 to 40 nm, the prepared NIP1 and MIP1 had a much bigger size with a diameter ranging from 110 to 120 nm and 150 to 160 nm, respectively. This meant the modified polymer layer was successfully grafted onto the surface of the GMA-CS carrier during polymerization. After the preparation of MIP1 and NIP1 was successfully proven by TEM images, and the much higher absorption capacity of MIP2 for BSA when compared with MIP1, we only selected MIP2 and NIP2 for further characterization.
In order to further determine the chemical structure of the synthetic products, FT-IR spectra of GMA-CS (a), CS (b), NIP2 (c) and MIP2 (d) are shown in Fig. 5A. As can be seen from the figure, compared with CS, GMA-CS showed a new peak at 1637 cm−1 which could be ascribed to CC. In our work, CC was successfully introduced to CS by GMA. The FT-IR spectra of NIP and MIP were similar to each other in terms of pattern profile, and they both had some new peaks when compared with GMA-CS. The new characteristic peak of 1728 cm−1 and other two observed new peaks at 3430 cm−1 and 3150 cm−1 could be ascribed to the CO and O–H bond of the –COOH group from AA. This FT-IR spectrum verified that the functional monomer had been successfully grafted onto the surface of GMA-CS after imprinting polymerization.
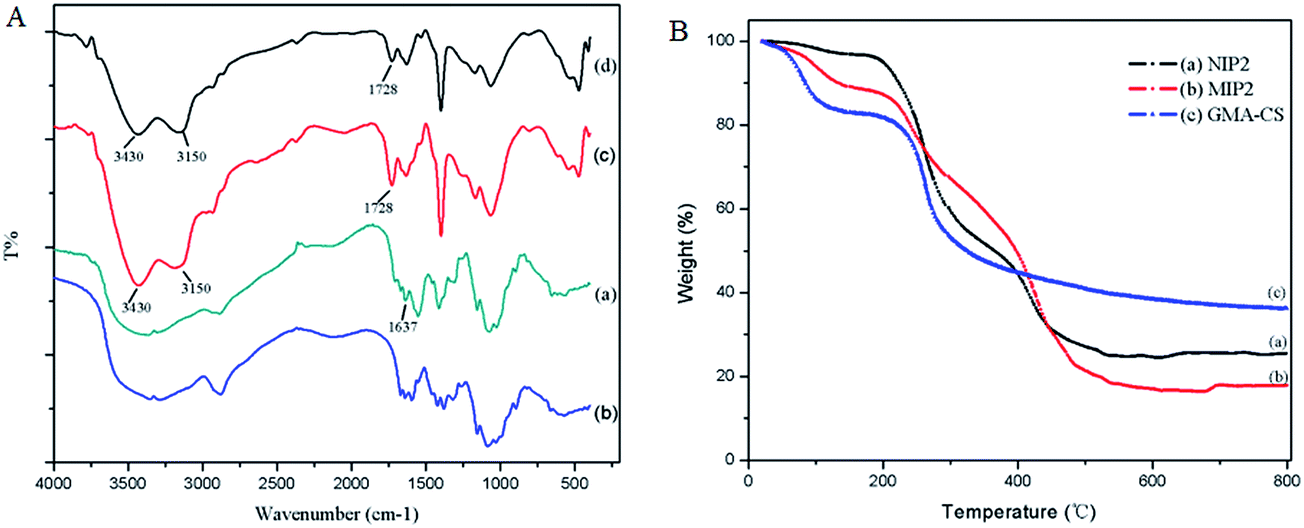 | ||
| Fig. 5 (A) FT-IR spectra of GMA-CS (a), CS (b), NIP2 (c) and MIP2 (d), and (B) TG curves of (a) NIP2, (b) MIP2 and (c) GMA-CS. | ||
Thermogravimetric analysis was performed to further explore the thermal stability and structure characteristic of the prepared products. TG curves of (a) NIP2, (b) MIP2 and (c) GMA-CS are shown in Fig. 5B. From the figure, we see that the TG curves of NIP2, MIP2 and GMA-CS are quite different from each other with temperatures ranging from 0 °C to 800 °C. When the temperature reached 800 °C, there was a weight loss of GMA-CS, NIP2 and MIP2 of 36%, 25% and 17% of their initial weights, respectively. This illustrated that the thermal stability of the polyacrylic acid outer layer was weaker than the carrier material GMA-CS which was also a type of polymer. But in the temperature range from 100 °C to 200 °C, the degree of weight loss ranked as GMA-CS > MIP2 > NIP2. Considering the weight loss of the above temperature range was mainly caused by water evaporation, we could speculate that GMA-CS carrier was a kind of material with a great water adsorption capacity and the water adsorption capacity of MIP2 and NIP2 decreased when a layer of polyacrylic acid was modified on the surface of the GMA-CS carrier. Besides, the greater water adsorption capacity of MIP2 than NIP2 might be explained by the existence of an imprinted cavity on MIP2.
3.2 Swelling measurement
As an important feature of hydrogels material, the saturated swelling ratio was investigated in swelling studies. The saturated swelling ratio (S) was calculated from the following equation:
 | (3) |
In our study, the results of the saturated swelling ratio of GMA-CS, MIP2 and NIP2 are shown in Table 1. It can be seen that the saturated swelling ratio of MIP and NIP corresponded to 3.66 and 3.14, which was much lower than that of GMA-CS (13.79). This was because there were plenty of hydrophilic amino and carboxyl functional groups on the surface of CS, which could adsorb a lot of water. However, the diffusion and penetration resistance of water molecules would increase when the CS was coated with polymer, which could decrease the water absorbency of hydrogel materials. Therefore, the results proved the successful grafting of PAA on the surface of GMA-CS.
| Samples | GMA-CS | MIP2 | NIP2 |
| Saturated swelling ratio | 13.79 | 3.66 | 3.14 |
3.3 Adsorption kinetics
In the evaluation of adsorption kinetics, the adsorption capacities of the MIP2 and NIP2 were tested as a function of time. The kinetic curves are shown in Fig. 6a. From the figure, we could see that the adsorption capacities of both MIP2 and NIP2 for BSA increased as time goes by and they could adsorb template protein quickly and easily owing to the low mass transfer resistance of thin shell. The adsorption equilibrium times for MIP2 and NIP2 were 2 h and 4 h, respectively. In addition, MIP2 clearly adsorbed more template protein compared to NIP2 under the same conditions. This result also validates the ability of MIP2 to recognize the template and confirmed the fact that recognition sites with the specific shape and the orientation of functional groups were successfully formed in the imprinted polymer network during the imprinting process. The relatively higher adsorption capacity of MIP2 compared with NIP2 could be attributed to the electrostatic interaction and hydrogen bonding interaction between the –COOH group of MIP2 and the template protein BSA. However, the NIP had no recognition sites for BSA, the adsorption capacity was mainly from non-specific adsorption.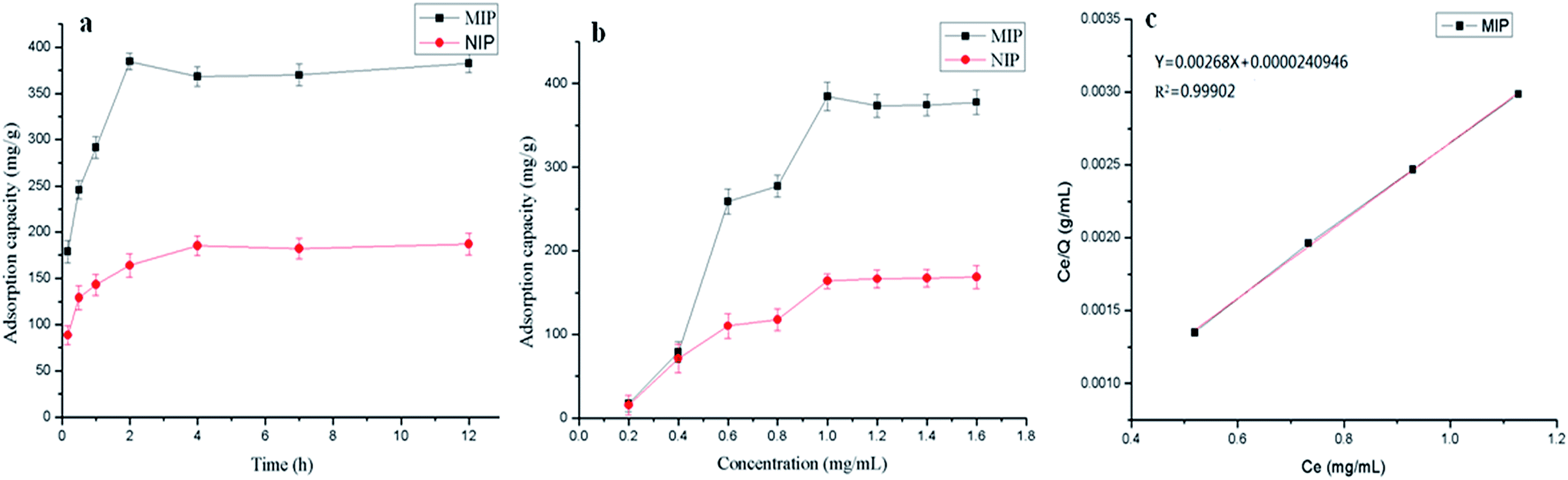 | ||
| Fig. 6 (a) Adsorption kinetic curves of the BSA on MIP2 and NIP2, (b) adsorption isotherms of the BSA on MIP2 and NIP2, and (c) Langmuir adsorption thermodynamics model of MIP2 for the curve of Ce/Q versus Ce. | ||
3.4 Adsorption isotherms
To investigate the adsorption capacities of both MIP2 and NIP2 for BSA, the adsorption isotherms were tested under room temperature with different initial concentrations of BSA solution and the results can be seen in Fig. 6b. As the isotherm curves showed, the adsorption trend of NIP2 was similar to MIP2. They both increased with the increase of the initial BSA concentration from 0.2 mg mL−1 to 1.0 mg mL−1. Then these curves became flat and reached thermodynamic equilibrium in the concentration region above 1.0 mg mL−1. So the concentration of 1.0 mg mL−1 was chosen as the optimal concentration condition for the following experiments. It also can be found that the adsorption capacities of MIP2 and NIP2 were almost the same in the low concentration region of 0.2 mg mL−1 to 0.4 mg mL−1, which indicated the existence of serious non-specific adsorption and the limited amount of proteins were completely absorbed by non-specific adsorption before imprinted sites were used. MIP2 showed a much higher adsorption capacity than NIP2 when the BSA initial concentration increased to 0.6 mg mL−1 or more, and a good imprinting effect could be obtained with an imprinting factor α of 2.35 at the BSA initial concentration of 1.0 mg mL−1, which also suggested that the specific recognition sites were successfully formed on MIP2 by the imprinting template molecule BSA during the polymerization process.The adsorption thermodynamic data of MIP2 could be further analyzed according to the Langmuir isotherm model proposed by Irving Langmuir.35 The equation of the Langmuir model is listed as follows:
| Ce/Q = Ce/Qmax + 1/(KQmax) | (4) |
A highly linearized plot of Ce/Q versus Ce is presented in Fig. 6c. It can be seen that the Langmuir equation fitted well for the adsorption of BSA on MIP2 within the concentration range studied (correlation coefficient, r2 = 0.99902). The value of Qmax was equal to the reciprocal of the slope of the Langmuir equation simulated linear curve, which turned out to be 373.13 mg g−1. This result evidenced a relatively high adsorption affinity for our synthesized MIP2 towards BSA.
3.5 Selectivity adsorption
In order to study the selectivity of MIP2 towards BSA, reference proteins BHb and OVA were used in the selective adsorption experiment. This choice was based on the fact that BHb had the same molecular weight and OVA had the same isoelectric point as the template BSA. The adsorption capacities of MIP2 and NIP2 for these three proteins are illustrated in Fig. 7a. It could be seen from the figure that, MIP2 obviously displayed a higher adsorption capacity for template BSA than the other two reference proteins, indicating the high adsorption selectivity of MIP2 for BSA. Moreover, a comparison of the adsorption of the MIP2 and NIP2 for each protein substrate suggested that MIP2 had no imprinting effect for BHb and OVA because both MIP2 and NIP2 nearly had the same adsorption capacity for them. | ||
| Fig. 7 (a) Selective adsorption between reference proteins and BSA on MIP2 and NIP2, and (b) influence of adsorption times on BSA adsorption capacity to the MIP2. | ||
The selectivity factor β was used to evaluate the specific selectivity and was defined by the following formula:
| β = αBSA/αRP | (5) |
| Protein | QMIP (mg g−1) | QNIP (mg g−1) | Imprinting factor α | Selectivity factor β |
|---|---|---|---|---|
| BSA | 384.59 | 163.96 | 2.35 | — |
| BHb | 155.94 | 153.42 | 1.02 | 2.30 |
| OVA | 231.31 | 187.75 | 1.23 | 1.91 |
The selectivity of MIP2 for BSA in a mixture of proteins was also studied. In this test, OVA was selected as the competitive protein. The results of the SDS-PAGE analysis are shown in Fig. S1.† From the degree of change in the band width and color of gel electrophoresis, we could find that BSA was the most adsorbed protein by MIP2. Although both BSA and OVA were adsorbed by the NIP2, the adsorption capacities were much lower than those of MIP2. So good selectivity was observed.
The preferred adsorption and selective recognition of MIP2 for template BSA might be due to the presence of imprinted recognition sites formed during the imprinting process, which only matched with template BSA in shape, size and arrangement of functional groups. However, the different spatial structure of the reference proteins did not match the imprinted sites and their access to the imprinted binding cavities may be limited by the steric hindrance of the polymer chains. Thus the adsorption capacity of MIP2 for the reference proteins were relatively lower than that for BSA, and mainly came from physical adsorption associated with nonspecific interactions. As for NIP2, the nonspecific adsorption was the dominant driving force because of the lack of imprinted recognition sites. Therefore, the adsorption capacity of NIP2 for BSA was much lower than that for MIP2.
3.6 Regeneration feature
In order to investigate the reusable property, MIP2 was regenerated by an eluent of SDS (10%, w/v) and HAc (10%, v/v) after the BSA adsorption process. In this work, MIP2 was repeatedly used three times, and the adsorption capacity of MIP2 for each adsorption experiment was measured and shown in Fig. 7b. After a one time adsorption–desorption cycle, the adsorption capacity of MIP2 for BSA was about 82.93% relative to that in the first adsorption experiment, and maintained at 66.45% in the third adsorption experiment. The lost adsorption capacity of MIP2 after repeated adsorption may be due to damage of some of the imprinted binding sites for BSA in the template removal operation process. Although ideal reuse efficiency of MIP2 was not achieved in this study, MIP2 still maintained a certain degree of capacity adsorption for BSA, indicating the imprinted cavities could regain the ability to adsorb template protein BSA and MIP2 could be used repeatedly several times.3.7 Effect of buffer pH on the adsorption of BSA
In order to study the mechanism for static adsorption of BSA onto MIP2 and verify the important role played by electrostatic interactions during the adsorption process, a series of Tris–HCl buffer solutions with different pH values were prepared as the working medium to test the effect of buffer pH on the adsorption capacity of MIP2 and NIP2 for BSA. The different pH values of Tris–HCl buffer could change the nature and the amount of electric charge on the protein, leading to various natures and extents of electrostatic interaction between the protein and imprinted cavities on MIP2 during adsorption. If the electrostatic interaction was the main driving force in the adsorption process, the adsorption capacity of MIP2 or NIP2 for BSA under different pH environments would be significantly different from each other. The results presented in Table 3 show this was the case. This might be due to the functional monomer acrylic acid ionizing and forming negatively charged carboxylate ions –COO− on the polymer side chain in aqueous solution after polymerization, which could interact with different parts of the protein by electrostatic interactions.| Buffer pH | 2 | 4 | 6 | 7 | 8.5 | 10 |
|---|---|---|---|---|---|---|
| QMIP mg g−1 | 276.04 | 437.18 | 301.63 | 384.59 | 0 | 0 |
| QNIP mg g−1 | 58.57 | 231.63 | 152.08 | 163.96 | 0 | 0 |
| Imprinting factor α | 4.71 | 1.89 | 1.98 | 2.35 | 0 | 0 |
When the pH of the buffer was higher than 8.5, the BSA protein molecule carried a lot of negative charge and formed strong electrostatic repulsion interactions with –COO− in MIP2, leading to a zero adsorption capacity of MIP2 for BSA. However, the adsorption capacity of MIP2 for BSA at a buffer pH value of 7 was higher than that at a buffer pH value of 6. This might be because 7 was also the pH value chosen for the imprinting polymerization process, the conformation of adsorption substrate BSA has a similar charge distribution and morphology as the imprinted template BSA which could better match the imprinted cavity when the pH value in the adsorption process was the same as the pH value in the imprinting polymerization process. The maximum adsorption capacity of MIP2 appeared at the buffer pH value of 4. Taking into account the BSA isoelectric point of 4.9, the BSA should carry some positive charge at buffer pH value of 4 and could form electrostatic attraction interactions with –COO− in MIP2, which could greatly improve the affinity between BSA and MIP2. When the pH value of the buffer further reduced to 2, the adsorption capacity of MIP2 for BSA was relatively low within the whole test pH range. It might be because too low a pH value of the environment would inhibit the ionization of –COOH in MIP2 and reduce electrostatic attraction interaction between BSA and MIP2. Moreover, there was a large number of hydrogen ions H+ in the buffer at a pH value of 2, which would compete with BSA for the electrostatic interaction sites in MIP2 and further reduce the affinity between BSA and MIP2, resulting in a low adsorption for MIP2 towards BSA. Meanwhile, NIP2 behaved similarly to MIP2 in the same series of buffers, which further validated the above speculation and interpretation because both the imprinted cavities of MIP2 and the polymer shell of NIP2 were filled with a lot of ionizable carboxyl groups which had a great tendency to form electrostatic interactions.
Although MIP2 had the highest adsorption capacity at the buffer pH value of 4, considering the protein is unstable and easily loses its biological activity in acidic medium, we still chose the buffer with the pH value of 7 as the working medium in the batch adsorption experiments.
4. Conclusions
In our work, a new kind of surface molecular imprinted polymer was successfully synthesized to recognize the template protein BSA based on the supporting material GMA-CS carrier. AA was used as functional monomer and MBAA was adopted as the cross-linker. The CC group was introduced to CS by GMA to facilitate imprinting polymerization. The results of the batch adsorption experiments demonstrated that MIP2 could successfully recognize BSA. MIP2 had a much higher adsorption capacity for BSA when the testing concentration was above 0.6 mg mL−1 and offered a faster adsorption kinetic rate than NIP2. The equilibrium adsorption isotherms of BSA on MIP2 could be well fitted by the Langmuir adsorption model and gave a theoretical maximum adsorption capacity of 373.13 mg g−1. At the same time, the selectivity of the MIP2 for BSA was verified by direct adsorption of single reference protein and competitive adsorption of mixed proteins. The results of all the experiments suggested that specific imprinted recognition sites which were complementary to BSA were created during the imprinting polymerization step. In addition, MIP2 could be reused after three adsorption–desorption cycles with a reuse efficiency of 66.45%. The effect of buffer pH on the adsorption of BSA was also determined, and the result indicated that electrostatic interaction between BSA and MIP2 was the main driving force in adsorption. All the above mentioned advantageous features such as easy preparation, fast mass transfer rate, a satisfactory adsorption capacity and specific recognition capability to the template protein BSA proved that this method and MIP2 material provides a promising potential for the practical application in the field of protein rapid separation and enrichment for the near future.
Abbreviations
| AA | Acrylic acid |
| AAm | Acrylamide |
| APS | Ammonium persulfate |
| BSA | Bovine serum albumin |
| BHb | Bovine hemoglobin |
| CS | Chitosan |
| FESEM | Field emission scanning electron microscope |
| FT-IR | Fourier transform infrared spectroscopy |
| GMA | Glycidyl methacrylate |
| GMA-CS | Ethylene-modified chitosan |
| Diacetic | acid |
| HCl | Hydrochloric acid |
| Lyz | Lysozyme |
| MBAA | N,N-Methylene-bis-acrylamide |
| MIPs | Molecularly imprinted polymers |
| NIP | Non-imprinted polymer |
| OVA | Ovalbumin |
| Qmax | Theoretical maximum adsorption capacity |
| SDS | Sodium dodecyl sulfate |
| SDS-PAGE | Sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TEM | Transmission electron microscopy |
| TEMED | N,N,N′,N′-Tetramethylenediamine |
| TG | Thermogravimetric analysis |
| Tris | Tris(hydroxymethyl)aminomethane |
Acknowledgements
The authors greatly appreciate the financial supports by the National Natural Science Foundation of China (No. 21175040; No. 21375035; No. J1210040) and the Foundation for Innovative Research Groups of NSFC (Grant 21221003).References
- N. Masqué, R. M. Marcé, F. Borrull, P. A. G. Cormack and D. C. Sherrington, Anal. Chem., 2000, 72, 4122–4126 CrossRef.
- J. Niu, Z. H. Liu, L. Fu, F. Shi, H. W. Ma, Y. Ozaki and X. Zhang, Langmuir, 2008, 24, 11988–11994 CrossRef CAS.
- J. W. Zhang, Y. L. Liu, G. L. Wu, M. Schönhoff and X. Zhang, Langmuir, 2011, 27, 10370–10375 CrossRef CAS PubMed.
- F. Augusto, E. Carasek, R. G. Silva, S. R. Rivellino, A. D. Batista and E. Martendal, J. Chromatogr. A, 2010, 1217, 2533–2542 CrossRef CAS PubMed.
- J. P. Wang, A. G. Cormack, D. C. Sherrington and E. Khoshdel, Angew. Chem., Int. Ed. Engl., 2003, 42, 5336–5338 CrossRef CAS PubMed.
- H. Kubo, T. N. Player, S. Shinoda, H. Tsukube, H. Nariai and T. Takeuchi, Anal. Chim. Acta, 2004, 504, 137–140 CrossRef CAS.
- C. D. Anderson, K. J. Shea and S. D. Rychnovsky, Org. Lett., 2005, 7, 4879–4882 CrossRef CAS PubMed.
- T. Kobayashi, P. S. Reddy, M. Ohta, M. Abe and N. Fujii, Chem. Mater., 2002, 14, 2499–2505 CrossRef CAS.
- C. J. Allender, C. Richardson, B. Woodhouse, C. M. Heard and K. R. Brain, Int. J. Pharm., 2000, 195, 39–43 CrossRef CAS PubMed.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O’Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS.
- S. M. Reddy, Q. T. Phan and H. El-Sharif, Biomacromolecules, 2012, 13, 3959–3965 CrossRef CAS PubMed.
- E. L. Holthoff and F. V. Bright, Anal. Chim. Acta, 2007, 594, 147–161 CrossRef CAS PubMed.
- J. Gao, H. Tian and Y. Wang, Biomaterials, 2012, 33, 3344–3352 CrossRef CAS PubMed.
- Y. Jin, M. Jiang, Y. Shi, Y. Lin, K. Peng and B. Dai, Anal. Chim. Acta, 2008, 612, 105–113 CrossRef CAS PubMed.
- Q. Z. Feng, L. X. Zhao, W. Yan, J. M. Lin and Z. X. Zheng, J. Hazard. Mater., 2009, 167, 282–288 CrossRef CAS PubMed.
- C. Y. Huang, M. J. Syu, Y. S. Chang, C. H. Chang, T. C. Chou and B. D. Liu, Biosens. Bioelectron., 2007, 22, 1694–1699 CrossRef CAS PubMed.
- J. L. Urraca, M. C. Moreno-Bondi, G. Orellana, B. Sellergren and A. Hall, Anal. Chem., 2007, 79, 4915–4923 CrossRef CAS.
- T. Shiomi, M. Matsui, F. Mizukami and K. Sakaguchi, Biomaterials, 2005, 26, 5564–5571 CrossRef CAS PubMed.
- X. P. Jia, M. L. Xu and Y. Z. Wang, Analyst, 2013, 138, 651–658 RSC.
- C. J. Tan and Y. W. Tong, Langmuir, 2007, 23, 2722–2730 CrossRef CAS.
- C. J. Tan, S. Wangrangsimakul and R. Bai, Chem. Mater., 2008, 20, 118–127 CrossRef CAS.
- R. Liu, M. Sha, S. S. Jiang, J. Luo and X. Y. Liu, Talanta, 2014, 120, 76–83 CrossRef CAS.
- Q. Q. Gai, F. Qu, Z. J. Liu, R. J. Dai and Y. K. Zhang, J. Chromatogr., A, 2010, 1217, 5035–5042 CrossRef CAS PubMed.
- W. Cheng, Z. Liu and Y. Wang, Talanta, 2013, 116, 396–402 CrossRef CAS.
- W. Zhang, X. W. He, Y. Chen, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2011, 26, 2553–2558 CrossRef CAS PubMed.
- F. L. Mi, Y. C. Tan, H. F. Liang and H. W. Sung, Biomaterials, 2002, 23, 181–191 CrossRef CAS PubMed.
- Z. Y. Wang, Z. J. Jiang and X. Q. Hu, Chin. J. Appl. Chem., 2002, 19, 1002–1004 CAS.
- F. Li, Chem. Ind. Eng., 2002, 19, 281–285 CAS.
- H. Y. Hao, Polym. Mater.: Sci. Eng., 2006, 22, 223–226 CAS.
- A. B. Huang, New Chem. Mater., 2010, 38, 17–20 CAS.
- D. H. Li, Chin. J. Biochem. Pharm., 2002, 23, 124–126 CAS.
- T. T. Wei, H. X. Jing and D. W. Xia, J. Chem. Technol. Biotechnol., 2001, 76, 191–195 CrossRef.
- T. Y. Guo, Y. Q. Xia and G. J. Hao, Carbohydr. Polym., 2005, 62, 214–221 CrossRef CAS.
- H. M. Guan and Y. J. Tong, Sci. Technol. Energ. Mater., 2009, 9, 2038–2041 Search PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1916, 38, 2221–2295 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14088a |
| This journal is © The Royal Society of Chemistry 2015 |