
Salen based Schiff bases to flame retard thermoplastic polyurethane mimicking operational strategies of thermosetting resin†
Anil D. Naik,
Gaëlle Fontaine,
Séverine Bellayer and
Serge Bourbigot*
ISP/UMET – UMR/CNRS 8207, Ecole Nationale Supérieure de Chimie de Lille (ENSCL), Avenue Dimitri Mendeleïev – Bât. C7a, BP 90108, 59652 Villeneuve d'Ascq Cedex, France. E-mail: serge.bourbigot@ensc-lille.fr
First published on 22nd May 2015
Abstract
A classical Schiff base N,N′-bis(4-hydroxysalicylidene)ethylenediamine (L2), a member of the Salen group that has been introduced as a non-phosphorus and non-halogen flame retardant in thermoplastic polyurethane (TPU) is found to operate in an intriguing way to fire retard the material. L2 blended with TPU significantly improves the inherent flammability of TPU without a synergist. A plausible mechanism including intermediates in the reinforcement of this elastomer towards flame retardancy is elaborated in line with the char forming abilities of this β-resorcylaldehyde based Schiff base. Conclusions were drawn based on the input from thermal, spectroscopic, microscopic and operando spectroscopic techniques. While its un-substituted counterpart, N,N′-bis(salicylidene)ethylenediamine (L1) lags behind in performance, its structural isomer N,N′-bis(5-hydroxysalicylidene)ethylenediamine (L3) is equally efficient but exhibits another mechanism of action. The performance of L2-TPU is found to be mainly in a condensed phase and is due to decoding of intrinsic cross-linking ability of the key building block of the additive i.e. resorcinol via structural transformation over a range of temperature. This results in the methylene bridged phenolic resin type material having high temperature stability. The process is also found to interfere with TPU unzipping process delaying its thermal degradation and favoring extensive cross-linking promoting char formation with insulative properties.
1. Introduction
Thermoplastic polyurethane (TPU) belongs to thermoplastic elastomers that combine the mechanical properties of vulcanized rubber with the processability of thermoplastic polymers and are considered to be the most versatile polymers.1,2 They are used in a wide variety of applications including adhesives, sealants, coatings, fibers, injection molded components, thermoplastic parts, industrial cables, energy exploration, automotive, transportation etc.1–5 On the downside the flammability of PU materials presents a threat to both the performance and integrity of the product. Further fire degraded products contribute to environmental pollution and health risks. Considering these facts coupled with regulatory issues, flame retardancy in PU has become a necessity as well as an obligation.6–9 To tackle these issues, flame retardants (FRs) are added to the PU system which may or may not be part of the PU macromolecular system. Depending upon the type of FR, they would exert varying influences on quenching, delaying, minimizing or retarding the burning process. Wide array of additives ranging from inorganic fillers to composites has been employed in this direction.1–6,10–23 Inorganic oxides, hydroxides and carbonates; organic and inorganic derivatives of boron, silicon, phosphorus and melamine; and expandable graphite to name some. On economic and efficiency scale, intumescence promoting FRs is proposed to be highly desirable. An intumescence is a protection mechanism of the FR involving the formation of an expanded foamy char. Thus formed carbonaceous char acts as physical barrier to heat, air and pyrolysis products protecting the underlying polymer.6–9 This constitutes condensed phase of action in flame retardancy. FRs are also capable of acting in gas phase by various mechanism.6–9 Recently Fontaine et al.4 introduced a non-halogen, non-phosphorus, and relatively simple organic framework of ‘Salen’ (Fig. 1a) and their metal chelates to the flame retardancy of thermoplastic polyurethane elastomer (Fig. 1b) triggering the entry of Schiff bases for high temperature applications. The nominal dosage of 10 wt% and absence of any synergist are creditable features which results in significant reduction in peak of heat release rate. | ||
| Fig. 1 (a) Salen based Schiff base additives (b) framework of thermoplastic polyurethane. | ||
In order to expand the applicability of this FR system it is necessary to understand the underlying physical and chemical aspects leading to flame retardancy, particularly by metal free ‘Salen’ systems. It is the main goal of this paper.
We have prepared three Schiff bases (L1–L3). L1 represents the simplest framework among them and assumed to have limited H-bonding network. L2 and L3 have hydroxy group at 4 and 5 position respectively on the aromatic group due to which they can expand the supramolecular interaction by intermolecular H-bonding. L1–L3 were blended in TPU and the resulting formulations were characterized by FTIR, 13C MAS NMR, TGA, DSC and microscopic techniques. Pyrolysis combustion flow calorimetry (PCFC), cone calorimetry and limiting oxygen index (LOI) are the evaluating tools for fire properties. The observed flame retardancy is broadly discussed examining condensed phase and gas phase mode of action to elucidate the mechanism of flame retardancy. Samples for the condensed phase analysis were collected from cone calorimetry experiment which represents different stages of degradation and studied by FTIR, 13C MAS NMR, SEM and EPMA. Gas phase analysis were studied by TGA coupled FTIR and py-GCMS. A comprehensive study is given in the following Section on how a structured Schiff base could reinforce TPU thermally, chemically and physically in contending fire risks.
2. Experimental
2.1. Materials
Thermoplastic polyurethane (Elastollan C85A) was kindly supplied by BASF. N,N′-Bis(salicylidene)ethylenediamine (L1) and N,N′-bis(4-hydroxysalicylidene)ethylenediamine (L2) and N,N′-bis(5-hydroxysalicylidene)ethylenediamine (L3) were synthesized based on earlier report.42.2. Formulations, processing and sampling
2.3. Instrumental
3. Results and discussion
3.1. Thermoplastic polyurethane, additives and formulations
Thermoplastic polyurethane elastomers (TPUs) are manufactured by reacting a diisocyanate, a macroglycol and a chain extender. The framework of TPU (Fig. 1b) consists of block copolymers of, soft segment (SS) and hard segments (HS) with a repeating unit of urethane linkage (–NHC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O–). The HS are produced by the reaction between the diisocyanate (e.g. 4,4′-methylene diphenyl diisocyanate (MDI)) and the chain extender (e.g. butanediol), and are responsible for rigidity, and are highly polar and present a relatively high melting temperature. HSs have high interchain interactions due to hydrogen bonding between the urethane groups. On the other hand, the SS consist of long polyester chains interconnecting two HS. This relatively non-polar segment impart flexibility and elastomeric properties to the TPU and present a relatively low softening temperature. The differences in polarity between hard and soft segments produce phase separation in the TPUs structure, giving micro-domains. TPUs will be elastic in the range of temperature between the glass transition temperature (Tg) generally located at about −60 °C and the softening temperature of the elastomeric domains (60–100 °C). Source of TPU in our study is Elastollan (C85A) which is a thermoplastic polyurethane elastomer containing polyester chains.5 Flame retardants (L1–L3) used in our study are classical Schiff bases and quite a large number of publications are available in the literature on their synthesis and properties.4 Their syntheses were performed in ethanol and are isolated as crystalline materials. Their thermal degradation and associated changes in physical and chemical properties are studied by us.24 L1 is found to have least thermal stability among all, melting at 128 °C followed by decomposition. Both L2 and L3 have better thermal stability, exhibit thermochromism and intriguing structural transformation associated with weight loss. Blending of L1–L3 in TPU was performed with a dosage of 10 wt% as reported before.4 Bright yellow color of L1 impart yellow color in L1-TPU, yellow-orange color of L2 to red color in L2-TPU and beige color of L3 to yellowish-brown to L3-TPU. We have particularly focused on resorcinol based ‘Salen’ additive (L2, Fig. 1a) in TPU as resorcinol is known for its extraordinary ability of cross-linking and radical chemistry.22,23 Thus present study is focused on L2-TPU with occasional reference to L1-TPU and L3-TPU.
O)O–). The HS are produced by the reaction between the diisocyanate (e.g. 4,4′-methylene diphenyl diisocyanate (MDI)) and the chain extender (e.g. butanediol), and are responsible for rigidity, and are highly polar and present a relatively high melting temperature. HSs have high interchain interactions due to hydrogen bonding between the urethane groups. On the other hand, the SS consist of long polyester chains interconnecting two HS. This relatively non-polar segment impart flexibility and elastomeric properties to the TPU and present a relatively low softening temperature. The differences in polarity between hard and soft segments produce phase separation in the TPUs structure, giving micro-domains. TPUs will be elastic in the range of temperature between the glass transition temperature (Tg) generally located at about −60 °C and the softening temperature of the elastomeric domains (60–100 °C). Source of TPU in our study is Elastollan (C85A) which is a thermoplastic polyurethane elastomer containing polyester chains.5 Flame retardants (L1–L3) used in our study are classical Schiff bases and quite a large number of publications are available in the literature on their synthesis and properties.4 Their syntheses were performed in ethanol and are isolated as crystalline materials. Their thermal degradation and associated changes in physical and chemical properties are studied by us.24 L1 is found to have least thermal stability among all, melting at 128 °C followed by decomposition. Both L2 and L3 have better thermal stability, exhibit thermochromism and intriguing structural transformation associated with weight loss. Blending of L1–L3 in TPU was performed with a dosage of 10 wt% as reported before.4 Bright yellow color of L1 impart yellow color in L1-TPU, yellow-orange color of L2 to red color in L2-TPU and beige color of L3 to yellowish-brown to L3-TPU. We have particularly focused on resorcinol based ‘Salen’ additive (L2, Fig. 1a) in TPU as resorcinol is known for its extraordinary ability of cross-linking and radical chemistry.22,23 Thus present study is focused on L2-TPU with occasional reference to L1-TPU and L3-TPU.
3.2. Spectroscopic characterization
Selected FTIR of TPU and formulations are compared in Fig. 2a. Spectral pattern are similar except following changes. The –NH doublet (3331, 3305 cm−1) in TPU is replaced by a broad peak at 3320 cm−1 in formulations due to disruption in H-bonding network caused by renewed H-bonding by additives. In the –CH absorption region sharp signals at 2922 and 2852 cm−1 bands disappear while their shoulder bands (2950, 2870 cm−1) are retained. The band at 1704 cm−1 (due to –CO stretching vibration of urethane), decreases in intensity but the –CO stretch of polyol signal (1727 cm−1) is unaffected.10–12,16 13C MAS NMR of TPU shows (Fig. 2b) following signals: carbonyl carbon at 154.4 ppm; aromatic carbons between 137–118 ppm; a doublet at 65 ppm and multiplet between 24.9–41.6 ppm belonging to –CH2 carbons. L3 bands (azomethine at 162.7 ppm, aromatic carbons between 110–155 ppm, –CH2 carbon at 57 ppm) are detected in L3-TPU but with slight change in intensity and position although L2 bands (azomethine at 176.1 ppm, aromatic carbons at 168.5–104 ppm; –CH2 at 48.2 ppm) are less resolved in L2-TPU. Based on the observed FTIR spectral change in the region of –NH and carbonyl group of urethane group it is assumed that, ‘Salen’ additives establish intra and interchain H-bonding interaction between urethane groups which bear both H-bonding acceptor and donor group. A tentative supramolecular interaction is shown in Fig. 3a focusing on HS region. Long polyester chain of soft segment with only H-acceptor groups is expected to form limited H-bonding with Salen molecules.
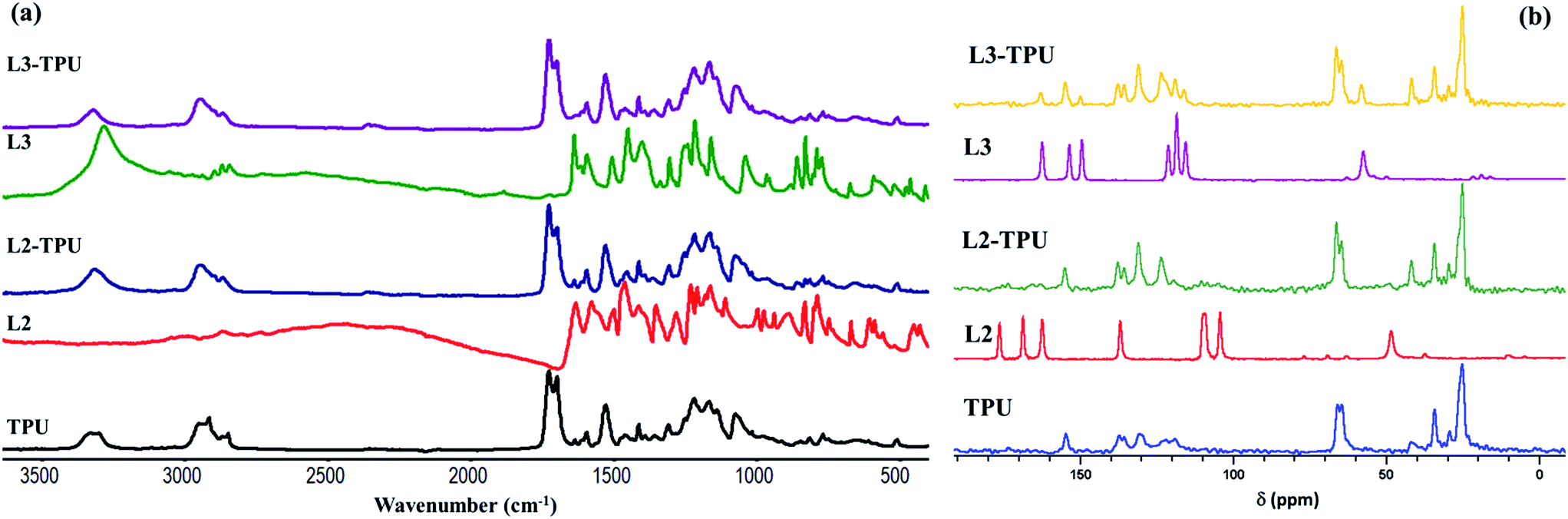 | ||
| Fig. 2 (a) FTIR spectra (b) 13C MAS NMR on TPU, selected additives and formulations. | ||
 | ||
| Fig. 3 (a) Possible interchain H-bonding (focused by arrows) by Schiff base L2 (or L3) between urethane group of TPU. Part of the PU macromolecular chain is shown. (b) Crystalline sample of L2 as observed under optical microscope. (c) Optical microscope image showing distribution of L2 in TPU. (d) Back scattering electron (BSE) image showing L2 additive as light colored aggregates in TPU matrix. | ||
3.3. Thermal properties of formulations
Thermal decomposition behavior of TPU, and formulations are shown in Fig. 4a. The initial thermal stability (until ∼260 °C) of TPU and additives filled TPU are in the order TPU > L2-TPU > L3-TPU > L1-TPU (Table 1). TPU is found to be stable until 250 °C and begin to lose weight gradually decomposing in two overlapping steps. The first major decomposition step is between 275–340 °C although there is sluggish weight loss prior to this. The second major step of decomposition is more abrupt than the first step and takes place until 374 °C. On the other hand, formulations (L2-TPU and L3-TPU) begin to lose weight early (around ∼220 °C) but subsequent decomposition steps are gradual extending the complete decomposition above 400 °C. Residue weights are in the order TPU-L1 < TPU < TPU-L2 = TPU-L3 suggesting char formation in L2-TPU and L3-TPU. It is intriguing that char formation takes place despite absence of an acid source. Although it has been found from our studies24 that L2 and L3 themselves are capable of forming char, in the present case an interaction between Salen and TPU matrix can be envisioned. Although the initial thermal stability of formulations (de-stabilizing effect) is less, subsequent decomposition steps are shifted to higher temperature. The indistinguishable two steps in TPU are also segregated in these formulations. The first weight loss during thermal degradation of TPU is due to the degradation of hard segment as a consequence of the relatively low thermal stability of urethane groups whereas the second weight loss has been associated to soft segment decomposition.2 | ||
| Fig. 4 (a) Thermal decomposition in TPU and formulations (b) DSC profile of TPU and formulations. | ||
| Materials | TGA | DSC | ||||
|---|---|---|---|---|---|---|
| Weight loss (%) at 262 °C | Onset of 2nd step | End of 2nd step (°C) | Residuea (%) (at 450 °C) | Heating (°C) | Cooling (°C) | |
| a Standard deviation given. | ||||||
| TPU | 1.5 | 330 | 374 | 6.0 ± 0.12 | 177, 213 | 81 |
| L1-TPU | 6.0 | 336 | 396 | 4.4 ± 0.13 | 79, 180, 209 | 84 |
| L2-TPU | 3.1 | 344 | 406 | 12.2 ± 0.11 | 83, 170, 208 | 72 |
| L3-TPU | 4.6 | 342 | 391 | 12.1 ± 0.12 | 76, 170, 203 | 74 |
In order to understand the interaction between TPU and additives, experimental and theoretical TGA curves were computed.25a Fig. S1† shows the TGA curves of additives,24 formulations, neat TPU and theoretical curves of the mixture. The difference between the experimental and theoretical TGA curves on formulations gives information on the reactivity of additives with TPU. If the experimental curve is above the theoretical one, the loss of weight is lower than expected, showing synergistic interaction of additives with polymer that leads to a thermal stabilization of the formulation. If the experimental curve is below the theoretical one, the reactivity of the polymer with additive leads to a thermal destabilization of the formulations.25a It can be seen from Fig. S1† that L2 and L3 in TPU stabilize the degrading matrix at higher temperature (above 290 °C). Considering high thermal instability of L1, there is gain in thermal stability in L1-TPU as judged from theoretical curve but fails at later stage due lack of char formation. The weight loss difference between the theoretical and the experimental TGA curve is also presented in Fig. S1.† In case of L2-TPU, the difference between the calculated and experimental weight loss becomes larger (>30%) over a wider temperature range (330–400 °C), an indication of char forming stage. L3-TPU although has ability to form char its efficiency is lower than L2-TPU.
Neat TPU in DSC (Fig. 4b) shows a broad endothermic signal around 177 °C due to disordering of HS crystallites.25b,26 The signal at 213 °C corresponds to melting of hard segment of TPU. In the cooling mode an exothermic peak is observed (∼81 °C) due to crystallization. This profile is nearly followed in formulations (Table 1) with slight change in peak position. The shift in crystallization temperature is prominent in L2 and L3 based formulations due their supramolecular associations of hydroxyl groups with the matrix. The broad peak observed during heating around 85 °C is due to softening temperature of TPU.
3.4. Material's response to simulated fire scenario
Flammability characteristic of TPU and flame retarded formulations are evaluated from rate of heat release (HRR) curves derived from two different techniques – pyrolysis combustion flow calorimetry (PCFC) and mass loss calorimetry (MCC) and are displayed in (Fig. 5a and b) with their associated parameters in Table 2. Neat TPU under cone calorimetry condition (35 kW m−2) burns easily and to the full extent resulting in small amount of char (pHRR, 426 kW m−2). On the other hand in formulations (L2-TPU and L3-TPU), bubbling is seen at the beginning which soon collapse to form protective char layer and swells by the blowing action of gases released underneath. These events influence ignition time and pHRR value. Order of efficiency is in the order L2-TPU ≈ L3-TPU > L1-TPU > TPU in terms of reduction in pHRR. Although both L2-TPU (pHRR, 266 kW m−2) and L3-TPU (pHRR, 279 kW m−2) perform well in terms of reduction of total heat release but it is L2-TPU which in addition significantly extend the ignition time (115 s).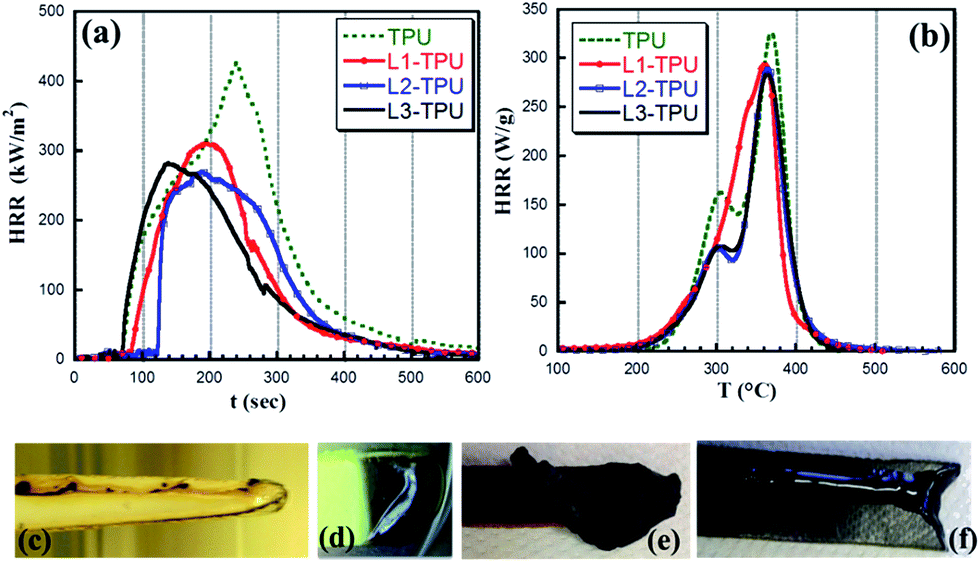 | ||
| Fig. 5 (a) HRR curve obtained from cone calorimetry. (b) HRR curve obtained from PCFC (c–f) physical response to flaming under LOI condition (c) neat TPU (d) L1-TPU (e) intumescence in L2-TPU (f) dripping in L3-TPU. | ||
| Materials | Mass loss calorimeter parameters | PCFC parameters | LOI (%) at 20 °C | ||||
|---|---|---|---|---|---|---|---|
| Time to ignition (s) | pHRR (kW m−2) | THR (MJ m−2) | pHRR reduction (%) | pHRR (W g−1) | THR (kJ g−1) | ||
| TPU | 60 ± 8.2 | 426 ± 12.0 | 83.8 ± 1.1 | — | 163 ± 5.4, 327 ± 7.5 | 25.7 ± 1.1 | 23 |
| L1-TPU | 65 ± 9.1 | 310 ± 13.2 | 55.1 ± 1.2 | 27 | 295 ± 9.4 | 24.9 ± 1.4 | 23 |
| L2-TPU | 115 ± 8.3 | 266 ± 14.2 | 53.2 ± 1.4 | 37 | 104 ± 6.1, 286 ± 7.2 | 23.1 ± 1.2 | 24 |
| L3-TPU | 60 ± 8.0 | 279 ± 13.1 | 54.6 ± 1.2 | 34 | 109 ± 6.0, 283 ± 8.2 | 22.7 ± 1.3 | 28 |
HRR curve (Fig. 5b) from PCFC for TPU shows two stages of heat release peaks (at 302 and 369 °C) the profile of which nearly matches to that of thermal degradation curve (from TGA) having two unequal degradation steps. Formulations based on L2 and L3 follow the same trend but with reduction in pHRR in the first step (up to 33%) whereas it is marginal (13%) in the second step. FTIR on residue collected at 329 °C (end of 1st degradation) confirms that it is indeed the HS that has been eliminated at this stage as ν(–CO) and ν(–NH) of urethane group disappears. Unlike L2-TPU and L3-TPU, L1-TPU deviates (see PCFC) from the trend and undergoes single stage decomposition likely to be due to high sublimation probability of L1.
PCFC recorded (Fig. S2†) on neat L2 and L3 shows pHRR in the temperature range that corresponds to their respective decomposition steps in TGA (Fig. S1†). The band in L2 (pHRR, 34 W g−1) and L3 (112 W g−1) means their individual contribution to pHRR but it is not so evident in their formulation counterparts except a small hump at the onset of HRR curve around 260 °C.
There are also differences in limiting oxygen index (LOI) results among these formulations (Fig. 5c–f). No improvement of LOI value is found in L1-TPU (LOI, 23%) which shows only dripping. The increase of LOI in L2-TPU is marginal (24%) wherein it performs by char formation (Fig. 5e) whereas the increase although significant (28%) in L3-TPU works mainly by dripping (Fig. 5f). Thus based on physical response in LOI and variation in HRR curve it is presumed that L1–L3 follow different mode of action. The convenient protocol to investigate the flame retardancy mechanism is to follow the chemical species generated/transformed/released in condensed and/or gas phases during the simulated fire scenario like in cone calorimetry. As HRR curve witness the decomposition pathway, analysis of samples at different stages of this curve (like beginning, peak, end of decomposition) gives valuable information to understand decomposition mechanism of the polymer/formulation. Operando techniques like TGA-FTIR and py-GCMS wherein volatile species are detected and analyzed should provide information on gas phase action.
3.5. Gas phase analysis
Neat TPU. As discussed in the previous Section TPU decomposes mainly in two overlapping steps. The volatile species evolved in these regions are detected by continuously monitoring by TGA coupled FTIR. Some selected FTIR are shown in Fig. 6. Until 23 min (230 °C) no significant species are detected. Thereafter slowly CO2 (2371, 2308, 670 cm−1) begin to evolve. Along with it hydrocarbons (2998–2874 cm−1), possibly species containing terminal –C–OH or C–O–C group (1079 and 912 cm−1) and hydroxyl species (bands above 3594 cm−1) are detected. Based on the reported20,21 data it can be assumed that these bands are mainly due to butanediol evolved in this region. Between 280 °C (28 min) to 320 °C (32 min) there is considerable increase of a band at 2279 cm−1 which is characteristic of –NCO group along with bands for butanediol. This corresponds to the beginning of the first major decomposition step and this is due to urethane dissociated product methanediphenyl diisocynate (MDI).2,14 As the thermal degradation enters second major degradation step (35 min and until 41 min, 412 °C), isocyanate begins to decrease at the expense of strong hydrocarbon and carbonyl (1760 cm−1) bands (possibly cyclopentanone, a cyclized product or linear esters fragments from soft segment of TPU) which considerably increases.
 | ||
| Fig. 6 (a) TGA weight loss derivative curve. (b) Selected FTIR of L2-TPU and neat TPU recorded during simultaneous TGA run. | ||
L2-TPU. The onset of degradation here begins slightly early but the detected species are essentially similar to TPU degradation product (until 31 min, 310 °C). This tentatively reveals that mode of onset of polymer dissociation is similar in both cases. In the following step, (31–36 min, 310–360 °C), signals for hydrocarbon and carbonyl group increases at the expense of CO2 much similar to neat TPU but differ with respect to their time of evolution. For example (Fig. 6b) the carbonyl species has already been distinguished at 32 min in L2-TPU whereas it appeared at 34 min in neat TPU. A more detailed scenario about evolution of species is presented in Fig. S1.† But there is significant difference with respect to isocyanate evolution which decreases considerably in L2-TPU. This could be due to –NCO species involving in crosslinking/dimerization process of char formation or there may be multiple modes of polymer dissociation which are discussed in later Section.2,8 It has to be noted that TPU can dissociate in several ways.2,14
Desorption studies. The temperature regions selected for this study is based on TGA pattern: 150–250 °C; 250–300 °C; 300–350 °C; and 350–450 °C. The first two temperature region corresponds to gradual minor weight loss steps that should reveal the mode of dissociation of TPU matrix. Analysis between 350–450 °C corresponds to major degradation steps and mainly concerned with both primary and secondary degradation products of dissociating fragments of HS/SS. Identified products are summarized in Table S1.† For neat TPU until 250 °C, trace amount of water, CO2, butanediol, methanediphenyl diisocyanate (MDI), tetrahydrofuran (THF), and a cyclic product 1,6-dioxacyclododecane-7,12-dione are identified.14 On the other hand, along with these species, 4,4′-diaminodiphenylmethane, 3-butenyl-4-hydroxybutyl adipate are detected in L2-TPU in the first step. Amine and olefin products are known to result from another degradation pathway of urethane group of TPU meaning operation of more than one type dissociation mechanisms in L2-TPU.
In the next step (250–300 °C), the dominant species in TPU are 1,4-butanediol and MDI. These products are signature of urethane dissociation mechanism involving release of isocyanate and alcohol. It is found that amount of these two main degradation products are significantly reduced in the formulations (Fig. 7b and c). This observation was also made in TGA-FTIR. THF and cyclopentanone are also detected in both cases at this stage which are formed from dehydration-cyclization of (1,4-butanediol and adipic acid components) 1,6-dioxacyclododecane.14 It is also found that products identified in the next two steps are similar among TPU and formulations and are much complicated due to numerous break-down, cyclized products in all formulations. Fragments of additives like resorcinol, benzoquinone and salicylaldehyde are also identified (in <250 °C range) in case of L2-TPU, L3-TPU and L1-TPU respectively. Although ethylenediamine and its deammonation product pyrazine are identified during the desorption experiment in neat L2 they are not traced in formulation.24
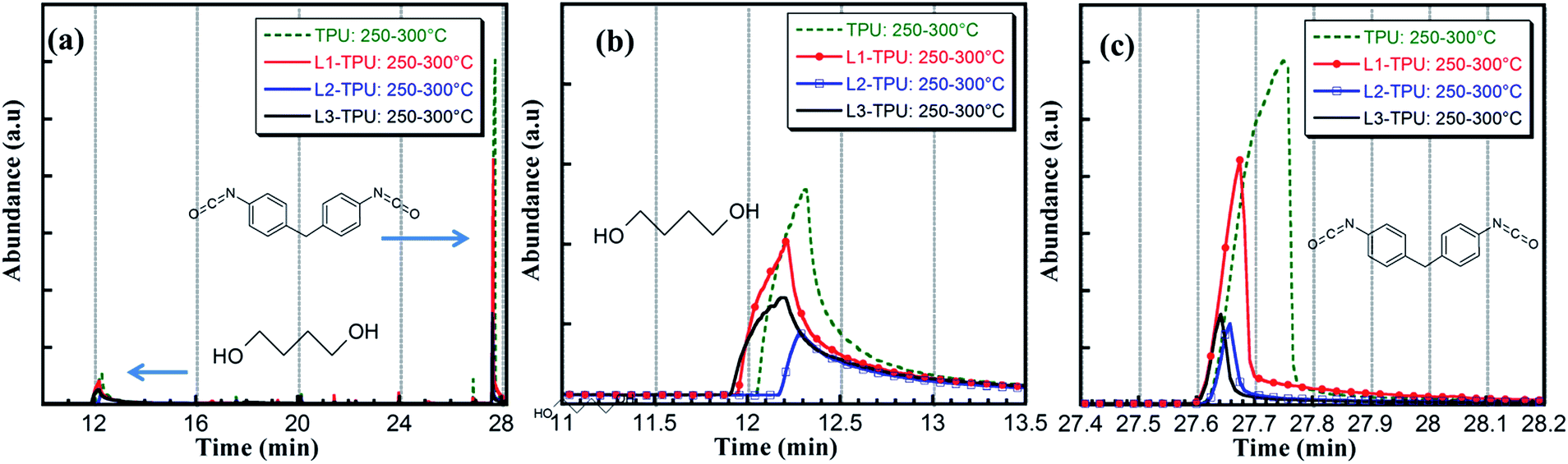 | ||
| Fig. 7 pyGCMS desorption experiment between 250 and 350 °C focusing on evolution of butanediol and MDI. | ||
Pyrolysis studies (at 600 °C). Pyrolysis at 600 °C which corresponds to completion of degradation in formulations provides additional information. Although it is not the scenario of cone calorimetry, the material is subjected directly to high temperature like in some fire scenario (e.g. open flame impinging the material). Some of the products identified (H2O, CO2, THF, cyclopentanone) at 600 °C are similar in TPU and L2-TPU whereas more of MDI, hydrocarbons, short chain ester fragment of 1,3-butenyl pentanoate (m/z, 156) and cyclized molecule like 1,6-dioxacyclododecane are found in TPU. Larger ester fragments like adipic acid 4-butenylester (m/z, 200) and 3-dibutenyl adipate (m/z, 254) are found to be dominant in L2-TPU. Scheme 1 depicts an overview of tentative degradation products in TPU and L2-TPU. Product formation and detection is influenced by heating rate, column temperature, polarity of eluting molecules, interference with other products, adsorption on column, gas-phase interaction, and secondary degradations. Despite this a good correlation is found from TGA-FTIR and pyGCMS results.
 | ||
| Scheme 1 Thermal degradation products in TPU and L2-TPU as evaluated from pyGCMS and supported TGA-FTIR in gas phase. For complete list of products refer Table S1.† | ||
3.6. Condensed phase analysis by 13C MAS NMR and FTIR studies
We have seen in mass loss calorimetry experiment that formulations swells and consequently forms char under heat flux. TPU itself being least contributor to intumescence/char, the observed intumescence/char is expected to be from additives or influence of additives on HS/SS of TPU. The exact way of its contribution could be evaluated from condensed phase analysis. As explained in Experimental section, samples for condensed phase analysis were collected based on HRR curve which represents different stages of degradation. Analysis of samples at the onset, peak, descending and end of decomposition normally should provide useful inputs to reveal the degradation mechanism. FTIR and 13C MAS NMR are routine technique to probe these degradation steps.Since additive signals due to its smaller dosage are not distinctly seen in MAS 13C NMR or FTIR and some of the signals are overlapped by dominant TPU signals tracking the degradation mechanism difficult. Despite this following noticeable changes are seen particularly in samples collected at peak of degradation and at later stages. As an example a comparative figure is given (Fig. 8) showing TPU and L2-TPU at different stages of degradation. In L2-TPU-peak, the region between 115–140 ppm characteristic of aromatic carbons in neat TPU becomes considerably broader (stretches between 100–150 ppm) with variation in intensity. Indeed aliphatic region also has seen some changes. There is an additional band around 40 ppm appeared as a shoulder on the degrading aliphatic fragments from TPU (Fig. 8). Both these changes (in aromatic and aliphatic regions) are thought to be brought about by additives. Since both the regions are overlapped by still remaining degrading TPU components no conclusive evidence on fate of additive is drawn-out. Since decomposition of neat TPU also stretches over a period of temperature the crucial information in the initial stages of additive structure-functioning escape detection. Sample collected at the descending part of the HRR curve also provides limited information being similar to L2-TPU-peak. Formation of char is confirmed in the residue with a broad signal around 125 ppm. The peak at 175 ppm might be due to some carbonyl species that may have origin from oxidation of methylene chain or some cyclized carbonyl species.
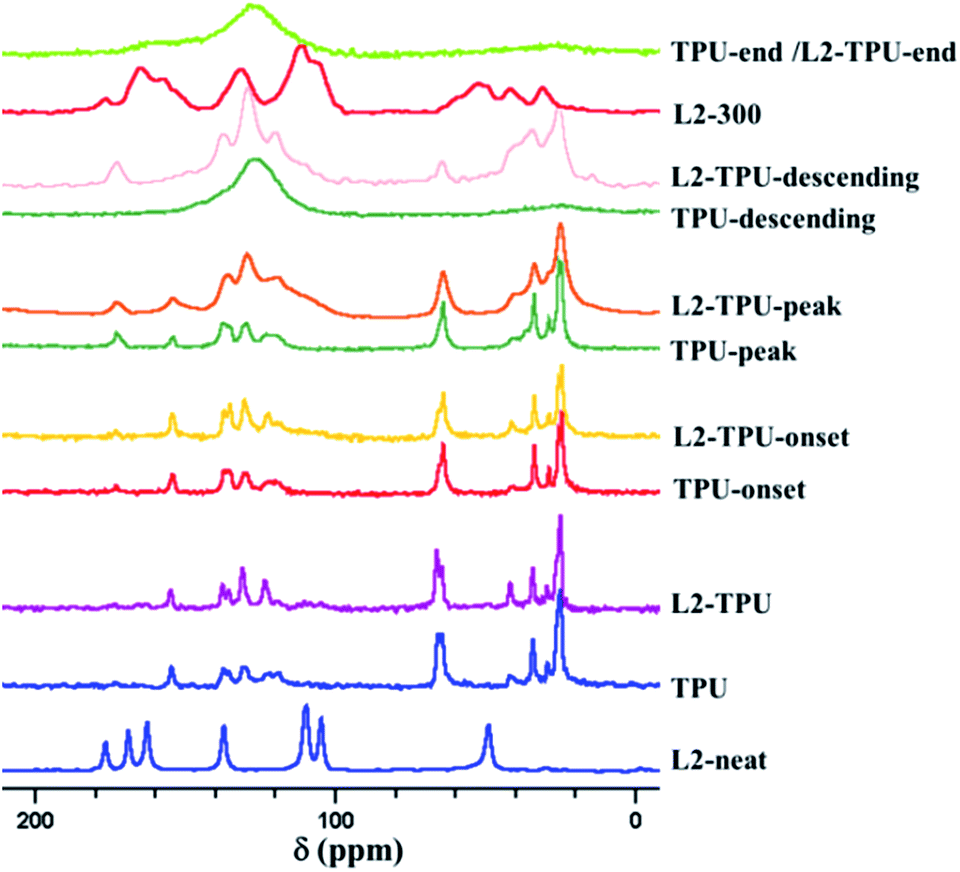 | ||
| Fig. 8 13C MAS NMR on TPU and L2-TPU at different degradation steps (onset, peak, descending and end refers to samples collected from mass loss calorimetry based on HRR curve which are at different stages of decomposition; L2-300 is a stage24 in the thermal decomposition of L2). | ||
Selected FTIR spectra of L2-TPU degradation are given in Fig. 9. Only expanded region is shown as region above 2700 cm−1 is not quite well resolved. Spectrum of L2-TPU is not shown as it closely resembles L2-TPU-onset.
 | ||
| Fig. 9 Expanded part of FTIR on samples (L2-TPU) collected from mass loss calorimetry (onset, peak, and descending refers to samples collected based on HRR curve which are at different stages of degradation. Arrow indicates new bands in L2-TPU-descending). L2-resin formation around 240 °C (L2-240) is also shown for comparison.24 | ||
It can be seen that in L2-TPU-peak it is carbonyl stretching (1704 cm−1) of urethane group that undergoes reduction in intensity confirming our earlier observation that HS is the preferred choice of cleavage.11,12,16 FTIR spectrum of L2-TPU-descending shows several new bands at 1661 m, 1592 s, 1509 s, multiplets around 1452 m, 1246 m, and 813br cm−1. The first four signals are assigned to the formation of new aromatic structure possibly involving additive framework. This was concluded because as the other aromatic contributor MDP (from TPU) decompose completely by this stage. The signal at 1452 cm−1 together with signal at 40 ppm in 13C MAS NMR indicates formation of new aliphatic fragments.
It is also possible that both new aromatic and aliphatic signals are arising from the same framework and likely from chemical modification of additive as most of the TPU framework is destroyed at this stage. This is evident from decrease or disappearance of several bands in FTIR spectrum. The molecules at this stage are possibly one among several species down the chain of reactive chemicals, a contributor to cross-linker and char former. In order to retrieve these masked species and their precursors it is necessary to compare with the thermal behavior of L2 which was investigated by us.24 It was found that L2 upon thermal treatment undergoes poly-condensation via covalent cross-linking forming hyperbranched cross-linked resins much similar to phenolic resins.27–34 The active molecule involved in this process is assumed to be resorcinol from the breakdown product of L2. Methylene bridge formation is conclusively confirmed by FTIR and solid state NMR which is in close alignment with that observed in the present case. These resorcinol based resins favor the formation of intumescence and char.
It was also found that the poly-condensation sites differ in L2 and L3 which involve novolac and resol type resin formation respectively.24 Formation of phenolic resin are also observed in L3-TPU as evidenced by FTIR and MAS NMR (Fig. S4 and S5†). In this respect it is worth mentioning a recent study of self-polymerisation of resole type prepolymer in blocked polyurethane which forms interpenetrating polymer network of blocked polyurethane and phenolic resin.36
L1, the unsubstituted saline molecule used in the present study although still hold promise because of its limited performance has high inclination for sublimation and lower thermal stability could not hold up to the expectation. Moreover, salicylaldehyde, a breakdown product of L1 is not a solid unlike resorcinol or hydroquinone and vaporizes easily.
3.7. Texture studies
Optical microscopic images on residue (on surface) were displayed in Fig. 10. Residue from TPU shows a hexagonal cellular structure which are loosely held. Char surface of L2-TPU shows slightly elongate cellular structure with thick walls occasionally raptured creating pores. But this char formation is peculiar in the sense it is multilayered, strong, but less-flexible. L3-TPU shows more elongated cellular structure with slightly shiny surface. Each cell bear numerous folding, flexible, single layered and moderately strong. It can be recalled that L2 residue possess nitrogen in variable amount.24 This is partially attributed to in situ formation of benzoxazine type resins. Considering the advantages of polybenzoxazines over traditional phenolic resins these observations are significant as also noted in reported poly(urethane-benzoxazine) films as novel polyurethane/phenolic resin composites.35 However EPMA (not shown) on L2-TPU-800 did not show any evidence of presence of nitrogen thus concluding that char is mainly composed of phenolic resin and no traces of benzoxazine type resin. | ||
| Fig. 10 Optical microscope images on residues (surface) collected in cone calorimetry experiments (a) TPU (b) L2-TPU (c) L3-TPU (d) SEM image on residue of L2-TPU showing scattered aggregates of resins. | ||
3.8. Mapping the degradation mechanism of additive blended TPU
In thermal decomposition (thermolysis) of TPU, the weakest point of the polyurethane chain is the urethane group.15 Thermal decomposition of TPU proceeds mainly through 2 steps. The first step is the degradation of hard segment followed by the decomposition of soft segment. There are three main pathways for the initial degradation of the urethane linkage (Scheme 2). Depolymerization into isocyanate and alcohol; dissociation into primary amine, olefin, and CO2; formation of secondary amine with elimination of CO2. Additives often influence these paths and polymer may adopt any of these routes or more likely combination of them. From FTIR we found that Schiff base (L2 and L3) establish H-bonding interactions with urethane segment (Fig. 3a) in L2-TPU and L3-TPU and thus assumed to have influence on TPU dissociation process. | ||
| Scheme 2 Possible thermal degradation routes of urethane segment of polyurethane. | ||
Although there is early onset of TPU break down (see TGA) in formulations the subsequent process is gradual stretching the complete degradation of TPU. We have seen in the previous Section and in our earlier work24 how resorcinol or hydroquinone component of L2 or L3 respectively can contribute to char formation. Resorcinol or 2,4-dihydroxybenzaldehyde alone due its high sublimating tendency at higher temperature fails to build-up resin/char. Stabilizing such hydroxyphenols by structuring into Schiff bases is an alternative approach which can influence their sublimation rate, control release, resin formation, and thermal stability. Recent work shows that in the derived form like in resorcinol bis(diphenyl phosphate), it was found to be an efficient flame retardant.37 Resorcinol or benzoquinone are known to be radical scavengers that could be released from L2 and L3 respectively and may act both in gas phase and condensed phase.22,23,38 The additive L2 is found to release (from pyGCMS) small amount of resorcinol in the gas phase during the curing process which may fractionally contribute to the radical quenching chemistry of gas phase action of flame retardancy. As TPU itself is thought to contribute to the oxidation by producing either OH radicals2 or water which may act as oxidizing agent, any free resorcinol trapped in condensed may also act as radical scavenger. On the other hand its sublimation causes blowing action which partly made char surface to swell increasing the gap between burning and unburnt polymer. Additives promote both segments of TPU to contribute to char formation either actively or passively. From TGA-FTIR, pyGCMS and analysis of residue from PCFC by FTIR (∼329 °C) indicate that HS of neat TPU decomposes first followed by SS. MDI and butanediol results from HS while a cyclized species mainly 1,6-dioxacyclododecane results from SS. Neat TPU has high inclination for formation of cyclized product from SS segment which degrades to smaller fragments like cyclopentanone, butanediol, pentanoic acid and 1,3-butenyl pentanoate. L2-TPU does not seem to favor cyclic products but utilizes SS. It is reported that ester segment of the TPU chain can bind to phenolic resin22 during its resin formation for cross-linking. Thus L2-TPU at higher temperature is found to release larger fragments like, adipic acid, 4-butenylester and 3-dibutenyladipate (Scheme 1) which are considerably less in neat TPU. These species pass through stages like formation of diene, polyene, polyaromatics before forming char (Scheme 3).2
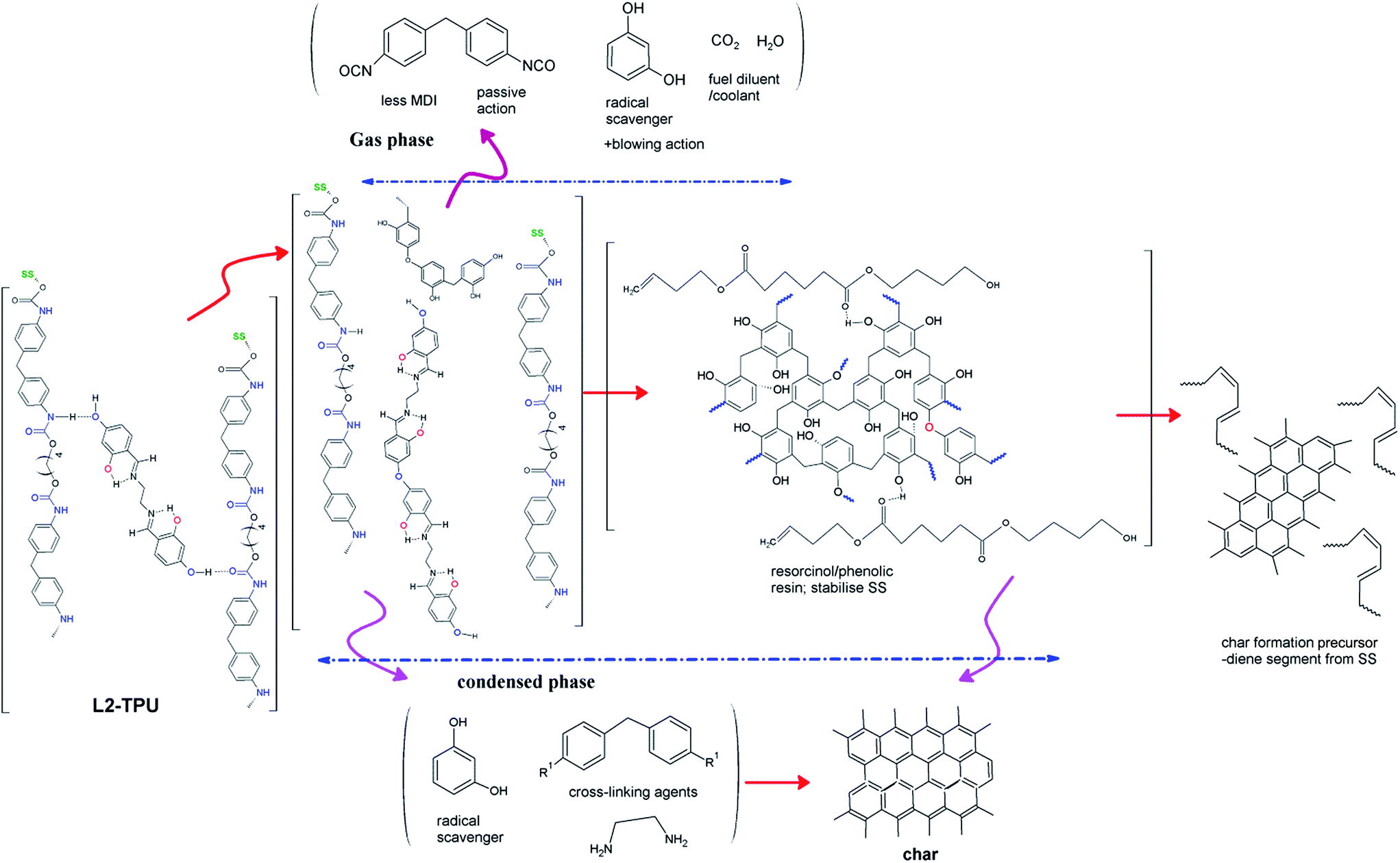 | ||
| Scheme 3 A simplified tentative degradation mechanism showing active and passive species responsible for flame retardancy in L2 blended TPU (R1 = –NCO or –NH2 group; SS = soft segment). | ||
Isocyanate production in TPU degradation is not considered favorable due to environmental issues.20 On the other hand its high reactivity and cross-linking ability are positive aspects to exploit it towards char formation in the condensed phase if suitable reactants are made available. From TGA-FTIR and pyGCMS it was tentatively estimated that diisocyanate (MDI) evolution in the gas phase is considerably lowered in formulations possibly reducing the pHRR of PCFC curve. Several reasons are reported for such decrease in the evolution of isocyanate and discussed in the following Section.2,16,20
The interference of additive in the urethane dissociation stage (Scheme 2) may modify the usual retropolymerisation process decreasing MDI production. The –OH groups of free resorcinol from disjointing L2 or L3 may reacts with released highly reactive diisocyanates. This leads to isocyanate-derived cross-links which are more robust and more resistive to shrinkage like in case of carbon aerogels.22 Phenolic hydroxyls can also react with isocyanates to form carbamates. The isocyanates formed may be dimerised to carbodiimide or undergo trimerization leading to isocyanurate are also reported.2 These species may react with remaining urethane groups or polar groups on polyester chain to form a cross-linked structure. We have seen that during cross-linking or curing of resorcinol from Schiff base, a highly branched interconnected resorcinol/phenolic resin is formed by polycondensation reaction between the phenolic prepolymer units.24 This cross-linking/resin formation density found in neat L2 not necessarily be the same in TPU matrix as TPU matrix may reduce cross-link density of this resin allowing additional reaction between resin and ester fragment of hard segment similar to that taking place in resorcinol–formaldehyde (RF) resins in polyester.22 Nevertheless the major flame retardancy action in this Schiff base flame retarded TPU is in the condensed phase by intumescence via resin intermediate which acts as physical defensive barrier.
This tightly cured bonding network of aromatic phenolic resin slows down heat and mass transfer between gaseous and condensed phases and thus retards the degradation and acts as heat shield. A key characteristic of such thermoset phenolic resin is its ability to withstand high temperature. Cured phenolic resin also provides the rigidity necessary to maintain structural integrity and dimensional stability of char which in its absence leads to feeble char. Thus Salen Schiff base that mimic the thermosetting behavior of resorcinol/phenol resin plays a pivotal role in this flame retardancy of action.
4. Conclusions
Schiff bases are the most versatile molecules among organic compounds finding diverse application like catalysis, medicine, molecular sensors, as ligands in coordination chemistry, magnetic materials to name some but rarely leaped into high temperature applications. This was partially due to the perception about ‘Salen’ molecules as a ‘soft’ molecular framework. Our investigation reveals for the first time that such molecules can be used for high temperature applications. This can be further improved by modifying the framework of the ‘Salen’ molecules and can be structure tuned to improve the performances. For example, L2/L3 which bear hydroxyl substitution is found to perform better than their unsubstituted counterpart, L1 and thus seemingly outperform L1-TPU. The observed flame retardancy of L2-TPU is mainly credited to the char forming ability of L2 as it endorses phenolic resin formation thus upcoming of new generation intumescence promoting FRs. The remote possibility of radical chemistry of flame quenching by L2 or L3 in the gas phase also cannot be excluded. It is worth to mention that intumescence observed in the present case works without any acid source or any synergist. The scope for this type of additives is enormous as there is large scope for redesigning this molecule and further stabilizing by known flame retardant active cations (Al, Zn) or anions (phosphate, borate) by encapsulation.Acknowledgements
Authors would like to thank Mr Bertrand Doumert for his assistance during MAS NMR recording.References
- S. Duquesne, M. Le Bras, S. Bourbigot, R. Delobel, G. Camino, B. Eling, C. Lindsay, T. Roels and H. Vezin, J. Appl. Polym. Sci., 2001, 82, 3262 CrossRef CAS PubMed.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068 CrossRef CAS PubMed.
- D. Tabuani, F. Bellucci, A. Terenzi and G. Gamino, Polym. Degrad. Stab., 2012, 97, 2594 CrossRef CAS PubMed.
- G. Fontaine, T. Turf and S. Bourbigot, Fire and Polymers V, ACS symposium, 2009, vol. 1013, ch. 20, p. 329 Search PubMed.
- http://www2.basf.us/urethanechemicals/tpu/pdf/C85A.pdf.
- S. Bourbigot, M. Le Bras and J. Troitzsch, Fundamentals: introduction, in Flammability handbook, ed. J. Troitzsch, Hanser Verlag, Munich, 2003, pp. 3–7 Search PubMed.
- C. A. Wilkie and A. B. Morgan, Fire Retardancy of Polymeric Materials, CRC Press, 2nd edn, 2009, ISBN: 978-1-4200-8399-6 Search PubMed.
- B. Schartel, Materials, 2010, 3, 4710 CrossRef CAS PubMed.
- Fire retardancy of polymers: new strategies and mechanisms, ed. T. R. Hull and B. Kandola, RSC Publishing, 2008, ISBN: 978-0-85404-149-7 Search PubMed.
- D. Rosu, N. Tudorachi and L. Rosu, J. Anal. Appl. Pyrolysis, 2010, 89, 152 CrossRef CAS PubMed.
- J. M. Cervantes-Uc, J. I. Moo Espinosa, J. V. Cauich-Rodríguez, A. Ávila-Ortega, H. Vázquez-Torres, A. Marcos-Fernández and J. San Román, Polym. Degrad. Stab., 2009, 94(10), 1666 CrossRef CAS PubMed.
- M. Herrera, G. Matuschek and A. Kettrup, Polym. Degrad. Stab., 2002, 78, 323 CrossRef CAS.
- A. Konig, U. Fehrenbacher, T. Hirth and E. Kroke, J. Cell. Plast., 2008, 44, 464 Search PubMed.
- B.-H. Kim, K. Yoon and D. Cheul Moon, J. Anal. Appl. Pyrolysis, 2012, 98, 236 CrossRef CAS PubMed.
- J. Simon, F. Baria, A. Kelemen-Haller, F. Farkas and M. Kraxner, Chromotographia, 1988, 25, 99 CAS.
- M. Berta, C. Lindsay, G. Pans and G. Camino, Polym. Degrad. Stab., 2006, 91, 1179 CrossRef CAS PubMed.
- W. P. Yang, C. W. Macosko and S. T. Wellinghoff, Polymer, 1986, 27, 1235 CrossRef CAS.
- A. Toldy, G. Harakaly, B. Szolnoki, E. Zimonyi and G. Marosi, Polym. Degrad. Stab., 2012, 97, 2524 CrossRef CAS PubMed.
- G. Trovati, E. Ap Sanches, S. Claro Neto, Y. P. Mascarenhas and G. O. Chierice, J. Appl. Polym. Sci., 2010, 115, 263 CrossRef CAS PubMed.
- X. Chen, L. Huo, C. Jiao and S. Li, J. Anal. Appl. Pyrolysis, 2013, 100, 186 CrossRef CAS PubMed.
- P.-S. Wang, Polym. Degrad. Stab., 1999, 66, 307 CrossRef CAS.
- R. B. Durairaj, Resorcinol: Chemistry, Technology and Applications, Springer, 2005, ISBN: 3540251421 Search PubMed.
- V. Thavasi, R. Phillip, A. Bettens and L. P. Leong, J. Phys. Chem. A, 2009, 113(13), 3068 CrossRef CAS PubMed.
- A. D. Naik, G. Fontaine, S. Bellayer and S. Bourbigot, in preparation.
- (a) M. Jimenez, S. Duquesne and S. Bourbigot, Thermochim. Acta, 2006, 449, 16 CrossRef CAS PubMed; (b) A. Frick and A. Rochman, Polym. Test., 2004, 23, 413 CrossRef CAS PubMed.
- Y. Zhu, J. Hu, K.-F. Choi, K.-W. Yeung, Q. Meng and S. Chen, J. Appl. Polym. Sci., 2008, 107, 599 CrossRef CAS PubMed.
- L. Costa, L. Rossi di Montelera, G. Camino, E. D. Weil and E. M. Pearce, Polym. Degrad. Stab., 1997, 56, 23 CrossRef CAS.
- S. Mulik, C. Sotiriou-Leventis and N. Leventis, Chem. Mater., 2008, 20(22), 6985 CrossRef CAS.
- L. B. Manfredi, D. Puglia, J. M. Kenny and A. Vazquez, J. Appl. Polym. Sci., 2007, 104, 3082 CrossRef CAS PubMed.
- M.-F. Grenier-Loustalot, S. Larroque and P. Grenier, Polymer, 1996, 37, 639 CrossRef CAS.
- S. Lin-Gibson, Cresol novolac/epoxy networks: synthesis, properties and processability, PhD thesis, 2001, http://vtechworks.lib.vt.edu/handle/10919/27296.
- W. Hesse and J. Lang, Phenolic resins, Ullmann's Encyclopedia of Industrial Chemistry, 2000, vol. 26, p. 583 Search PubMed.
- E. Fitzer and W. Schafer, Carbon, 1970, 8, 353 CrossRef CAS.
- S. Zeng, L. Guo, L. Zhang, F. Cui, J. Zhou, Z. Gao, Y. Chen and J. Shi, Macromol. Chem. Phys., 2010, 211, 845 CrossRef CAS PubMed.
- T. Takeichi, Y. Guo and T. Agag, J. Polym. Sci., 2000, 38, 4165 CrossRef CAS.
- Y.-Y. Sun and C.-H. Chen, Polym. Eng. Sci., 2011, 51, 285 CAS.
- K. H. Pawlowski and B. Schartel, Polym. Int., 2007, 56(11), 1404 CrossRef CAS PubMed.
- D. Mikulski, R. Górniak and M. Molski, Eur. J. Med. Chem., 2010, 45, 1015 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Identification of pyrolysates (Table S1); experimental and theoretical TGA curves (Fig. S1); PCFC (Fig. S2); TGA-FTIR (Fig. S3); 13C MAS NMR on L3 (Fig. S4); FTIR on L3 (Fig. S5). See DOI: 10.1039/c5ra06242j |
| This journal is © The Royal Society of Chemistry 2015 |