DOI:
10.1039/C5RA05472A
(Paper)
RSC Adv., 2015,
5, 48368-48381
Chloroquinoline–acetamide hybrids: a promising series of potential antiprotozoal agents†
Received
27th March 2015
, Accepted 15th May 2015
First published on 15th May 2015
Abstract
In an endeavour to develop efficacious antiprotozoal agents 2-[4-(7-chloroquinolin-4-yl)piperazin-1-yl]acetamide derivatives were synthesized and screened in vitro against the HM1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IMSS strain of E. histolytica and 3D7 strain of P. falciparum. Among the twenty-seven synthesized compounds, eleven evinced propitious anti-amoebic activity with IC50 values ranging from 0.41 to 1.80 μM) lower than the standard drug metronidazole (IC50 1.80 μM). All the compounds inhibited the in vitro growth of P. falciparum (IC50 range: 0.30–33.52 μM). Compounds A22 and A25 were found to be the most active antimalarial derivatives, and compound A16 the most active in inhibiting β-haematin formation; however compound A25 displayed the more favourable safety profile. The crystal structure for the compounds A7, A8, A12 and A21 was also determined. The molecular docking of crystal resolved inhibitors with PfDHFR allowed identification of stabilizing interactions within enzyme active sites. These compounds affirm the potential for further derivatives to enhance antiprotozoal activity whilst retaining their safety profile.
:IMSS strain of E. histolytica and 3D7 strain of P. falciparum. Among the twenty-seven synthesized compounds, eleven evinced propitious anti-amoebic activity with IC50 values ranging from 0.41 to 1.80 μM) lower than the standard drug metronidazole (IC50 1.80 μM). All the compounds inhibited the in vitro growth of P. falciparum (IC50 range: 0.30–33.52 μM). Compounds A22 and A25 were found to be the most active antimalarial derivatives, and compound A16 the most active in inhibiting β-haematin formation; however compound A25 displayed the more favourable safety profile. The crystal structure for the compounds A7, A8, A12 and A21 was also determined. The molecular docking of crystal resolved inhibitors with PfDHFR allowed identification of stabilizing interactions within enzyme active sites. These compounds affirm the potential for further derivatives to enhance antiprotozoal activity whilst retaining their safety profile.
1. Introduction
Two protozoal diseases, namely, amoebiasis and malaria continue to result in morbidity and mortality particularly in tropical countries. Amoebiasis is a common problem in areas with poor sanitary conditions and health education. Meanwhile over 200 million episodes of malaria occur annually, with approximately 600000 deaths every year mainly among children and pregnant women in sub-Saharan Africa.1–4 The main challenge in malaria control is that the malaria parasite has developed clinically significant resistance to many classes of drugs,5,6 and in recent years to artemisinin-derived antimalarials.7,8 To improve the efficacy and delay the onset of resistance, artemisinin combination chemotherapy (ACT) is a promising strategy in which an artemisinin derivative is used together with a conventional antimalarial drug.9 ‘Covalent biotherapy’ a recent rational approach involves linking two molecules with individual intrinsic activity into a single agent, thus developing hybrid molecule with dual functionality.10,11 The quinoline nucleus has proven to be a source of drug development for such protozoal infections. Chloroquine is a common drug used for both Entamoeba and Plasmodium infections.12 Many natural drugs isolated from the plant Cinchona officinalis namely quinine, quinidine and quinidinone bearing a quinoline ring are also active against amoebiasis.13 Chloroquine and amodiaquine are two 7-chlorquinoline-based drugs effective against malaria, while iodoquinol is used as a standard drug bearing a quinoline ring also used in the treatment of amoebiasis.14 Therefore quinoline-based heterocycles have a momentous role in drug design against protozoal diseases.15 In our previous research, some 2-quinoline hydrazones, 2-chloroquinoline chalcones and 7-chloroquinoline sulphonamides have displayed good antiprotozoal activity.16,17 Chloroquinoline derivatives of similar type have also been previously described in the literature as interesting antimalarial agents.18
Dihydrofolate reductase (DHFR) is one of the key enzymes in the folate pathway and is requisite for the maintenance of proper intracellular concentrations of tetrahydrofolate, a coenzyme involved in the biosynthesis of purines, pyrimidines, and some amino acids.19 It remains an attractive target for selective drug design of novel anticancer, antimalarial and antibacterial agents.20 It is proposed that 4-aminochloroquinoline will promote haem binding, piperazine is a tertiary amine which increases the hydrophilicity and the amide group will act as dihydrofolate reductase (DHFR) inhibitor.21
In the present study, a series of novel chloroquinoline based hybrid molecules bearing a core active 4-aminochloroquinoline moiety and substituted acetamide linked together with a piperazine have been designed using the strategy of linking two scaffolds in a single molecule and are expected to show an enhancement/amelioration of antiamoebic as well as antimalarial activity.
2. Results and discussion
2.1. Chemistry
The synthetic pathway leading to formation of 2-[4-(7-chloroquinolin-4-yl)piperazine-1-yl]acetamide hybrids (A1–A27) is depicted in Scheme 1. The chloro-4-piperazin-1-yl-quinoline was synthesized by aromatic nucleophilic substitution of piperazine on the commercially available 4,7-dichloroquinoline under refluxing conditions with good yield.17 Different chloroacetamides (1–27) were synthesized by substitution of the amine with chloro acetyl chloride.22 The formation of the final compounds (A1–A27) was achieved by reacting 7-chloro-4-piperazin-1-yl-quinoline with different chloroacetamides (1–27) using potassium carbonate as a base, potassium iodide as catalyst and dimethylformamide as solvent under refluxing conditions. All the products were soluble in polar solvents and re-crystallized from dichloromethane/hexane system. All the compounds are stable in solid states at room temperature. The NMR spectra of all the compounds were recorded in CDCl3 that also favours the proposed structures. The 1H NMR of the aromatic chloroacetamides (1–15) showed a broad singlet for –NH in the range δ 7.87–9.27 ppm whereas it appeared upfield for the aliphatic chloroacetamides (23–27) δ 5.20–7.33 ppm. The IR spectrum of the final compounds (A1–A27) exhibited characteristic absorption band due to carbonyl group in the region 1634–1720 cm−1. The structure of the compounds was further confirmed by the 1H NMR spectral data. A broad singlet in the range δ 8.60–11.79 ppm and a doublet δ 8.71–8.74 ppm due to –NH was observed for the compounds A1–A15 and A23–A27 respectively. The signals due to the aliphatic and aromatic proton appeared in the expected region. All the structures were further confirmed by 13C NMR and mass spectral studies and the purity of the compounds was confirmed by elemental analysis and data was found in accordance with ±0.3%.
 |
| | Scheme 1 Synthesis of substituted chloro acetamides (1–27) and 2-[4-(7-chloro-quinolin-4-yl)-piperazin-1-yl]acetamides (A1–A27). | |
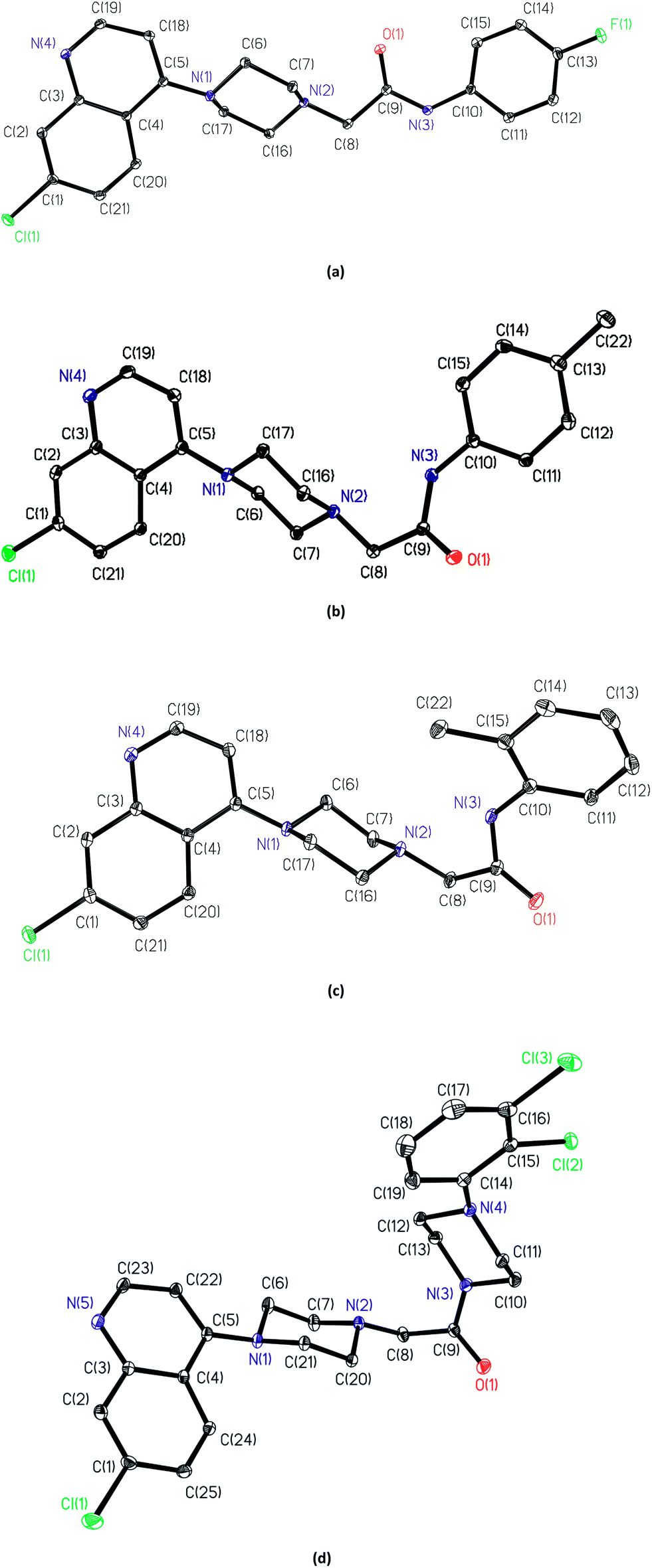
2.2. Single crystal structure
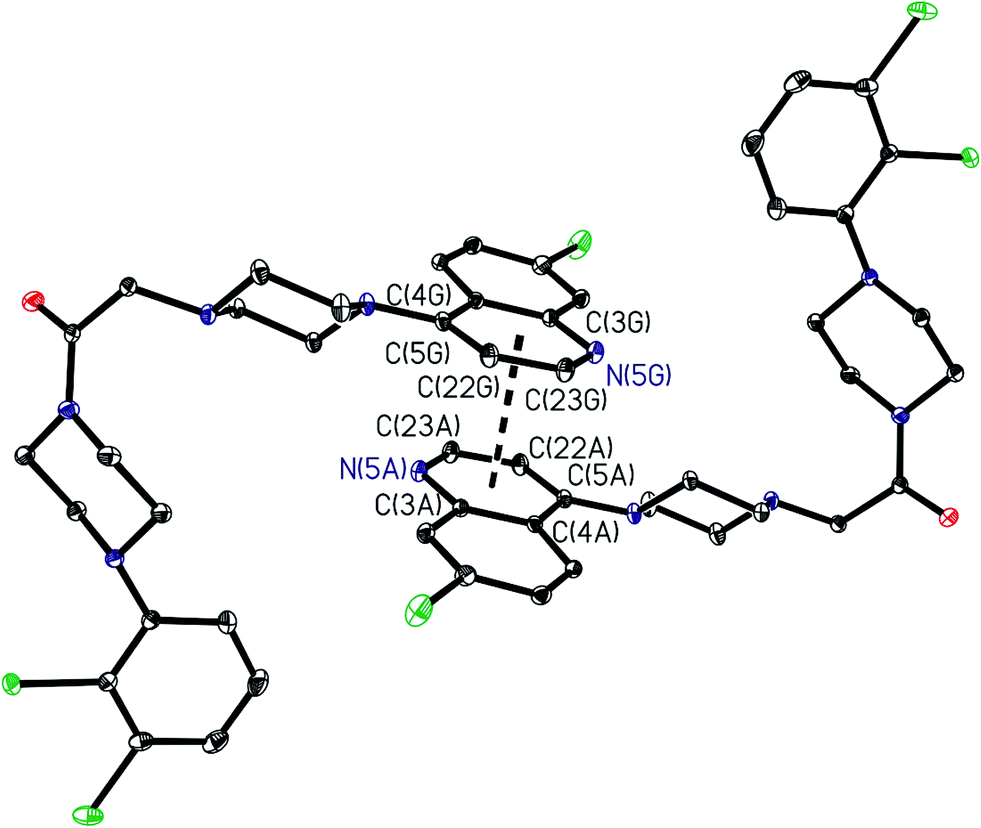
Single crystal structures of A7, A8, A12 and A21. The asymmetric unit of A7 contains the piperazine derivative and one water molecule; while the asymmetric units of A8, A12 and A21, only contains the piperazine derivative. The piperazine rings are in a chair form and hence the molecules have an extended conformation. Both ring N atoms [N(1) and N(2)] in each compound are bonded in a configuration which are distinctly pyramidals, with the sum of the C–N–C angles being 340.01(9)° and 330.45(9)° for A7, 344.07(11)° and 328.96(11)° for A8, 340.05(9)° and 333.38(10)° for A12 and 341.50(8)° and 332.61(8)° for A21.23 Bond lengths and angles are given in ESI.†In A7, the presence of intermolecular hydrogen bonds between the water molecule and nitrogen atoms of the piperazine derivative, determined the configuration around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. In A8 and A12 intramolecular hydrogen bonds between N(2) and N(3) atoms established the configuration of this part of the molecule (Fig. 1). In the solid state the compounds A7 and A21 form dimers through π–π interactions between chloroquinoline groups (Fig. 2 and 3), through phenyl rings in A7 and through pyridine rings in A21. The distances between centroids are: dc1–c2 = 3.754 Å [c1 (C1I–C2I–C3I–C4I–C20I–C21I), c2 (C1E–C2E–C3E–C4E–C20E–C21E) and repeated for other centroids] in A7, and dc3–c4 = 3.591 Å [c3 (C3A–C4A–C5A–C22A–C23A–N5A), c4 (C3G–C4G–C5G–C22G–C23G–N5G) and repeated for other centroids]. Compounds A8 and A12 do not show these π–π interactions.
O bond. In A8 and A12 intramolecular hydrogen bonds between N(2) and N(3) atoms established the configuration of this part of the molecule (Fig. 1). In the solid state the compounds A7 and A21 form dimers through π–π interactions between chloroquinoline groups (Fig. 2 and 3), through phenyl rings in A7 and through pyridine rings in A21. The distances between centroids are: dc1–c2 = 3.754 Å [c1 (C1I–C2I–C3I–C4I–C20I–C21I), c2 (C1E–C2E–C3E–C4E–C20E–C21E) and repeated for other centroids] in A7, and dc3–c4 = 3.591 Å [c3 (C3A–C4A–C5A–C22A–C23A–N5A), c4 (C3G–C4G–C5G–C22G–C23G–N5G) and repeated for other centroids]. Compounds A8 and A12 do not show these π–π interactions.
 |
| | Fig. 1 ORTEP plot for the compounds (A7, A8, A12, A21). All the non-hydrogen atoms are presented by their 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. | |
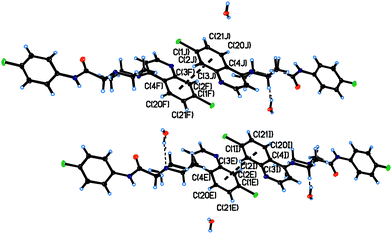 |
| | Fig. 2 Intermolecular π–π interactions in A7. Dashed lines link the centres of the π clouds involving in each interaction. | |
 |
| | Fig. 3 Intermolecular π–π interactions in A21. Dashed lines link the centres of the π clouds involving in each interaction. | |
2.3. Biology
Antiamoebic activity. The antiamoebic effect was compared with the most widely used antiamoebic medication, namely metronidazole (Mtz) which had a 50% inhibitory concentration (IC50) of 1.80 μM (Table 2). Three different types of amines viz substituted anilines (A1–A15), substituted piperazine (A16–A22) and aliphatic amines (A23–A27) were biologically evaluated against E. histolytica and the amoebicidal activity results for all the compounds were found to be very promising. The structure–activity relationship (SAR) showed that compounds (A1–A27) which contained piperazine in the acetamide group, showed IC50 values in the range of 0.52–3.24 μM; whereas for the aromatic and aliphatic amines, the IC50 values were in the range of 0.41–17.14 μM and 0.86–18.99 μM, respectively. Compound A20 possessed an IC50 value in same order as the one noticed for the reference drug Mtz. Compounds A11 and A15 showed IC50 values less than Mtz whereas A16, A21, A23 were found to be nearly as active as the standard drug. The aromatic amines showed varied results. None of the ortho substituted compounds (A2, A6, A12) were found to be active. The para substituted compound A7 (IC50 0.41 ± 0.02 μM) was the most potent of all the compounds screened and shows four folds more activity and the compound A9 (IC50 0.72 ± 0.01 μM) shows greater than two fold activity than the standard drug Mtz. The presence of the bulky group in aromatic amines showed reduced activity. The bulky nature might have imposed steric hindrance thereby, drastically reducing its efficacy. Precipitous decrease in activity was observed for bulky compound (A13; IC50 17.14 ± 0.02 μM) showing ten folds higher value than Mtz. Compounds containing acetoxy group (A2; IC50 7.90 ± 0.02 μM), bromo (A11; IC50 7.20 ± 0.02 μM) and chloro-fluoro disubstituted (A15; IC50 7.30 ± 0.01 μM) showed four folds whereas chloro substituted (A10; IC50 5.90 ± 0.02 μM) showed three folds higher IC50 values than Mtz and can be considered of less interest. It can be concluded that the piperazine containing substituents (A16–A21) in the acetamide group showed the best results in the series with the IC50 ranging from 0.52–3.24 μM.
Table 1 Crystal data and structure refinement for 2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)-N-(4-fluorophenyl)acetamide⋅H2O(A7), for 2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)-N-p-tolylacetamide (A8), for 2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)-N-o-tolylacetamide (A12) and 2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)-1-(4-(2,3-dichlorophenylpipezin-1-yl)-ethanone (A21)
| |
A7 |
A8 |
A12 |
A21 |
| R1 = Σ∣∣Fo∣ − ∣Fc∣∣/Σ∣Fo∣. wR2 = {Σ[w(∣∣Fo∣2 − ∣Fc∣2∣)2]∣/Σ[w(Fo4)]}1/2. |
| Formula |
C21H22ClFN4O2 |
C22H23ClN4O |
C22H23ClN4O |
C25H26Cl3N5O |
| Formula weight |
416.88 |
394.89 |
394.89 |
518.86 |
| T, K |
100(2) |
100(2) |
100(2) |
100(2) |
| Wavelength, Å |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Orthorhombic |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca |
P |
P21/n |
| a/Å |
6.7737(3) |
18.2066(10) |
9.0751(4) |
9.6408(3) |
| b/Å |
9.6448(3) |
11.0845(6) |
9.2785(4) |
12.9637(3) |
| c/Å |
15.8855(6) |
19.4301(10) |
14.1227(8) |
19.8442(5) |
| α/° |
97.023(2) |
90 |
105.382(3) |
90 |
| β/° |
97.739(2) |
90 |
92.801(3) |
98.1580(10) |
| γ/° |
107.511(2) |
90 |
119.213(2) |
90 |
| V Å−3 |
966.05(6) |
3921.2(4) |
978.02(8) |
2455.04(11) |
| Z |
2 |
8 |
2 |
4 |
| F000 |
436 |
1664 |
416 |
1080 |
| Dcalc/g cm−3 |
1.433 |
1.338 |
1.341 |
1.404 |
| μ/mm−1 |
0.234 |
0.215 |
0.216 |
0.402 |
| θ/(°) |
1.31 to 34.60 |
2.38 to 27.51 |
1.53 to 29.38 |
2.24 to 30.64 |
| Rint |
0.0467 |
0.0485 |
0.0456 |
0.0252 |
| Crystal size/mm3 |
0.50 × 0.47 × 0.38 |
0.50 × 0.45 × 0.35 |
0.43 × 0.30 × 0.25 |
0.4 × 0.5 × 0.5 |
| Goodness-of-fit on F2 |
1.082 |
1.025 |
1.064 |
1.039 |
| R1[I > 2σ(I)]a |
0.0474 |
0.0350 |
0.0375 |
0.0342 |
| wR2 (all data)b |
0.1267 |
0.0942 |
0.1047 |
0.0947 |
| Largest differences peak and hole (e Å−3) |
0.492 and −0.315 |
0.301 and −0.319 |
0.352 and −0.304 |
0.495 and −0.710 |
Table 2 Antiparasitic activity and toxic effects of 2-[4-(7-chloroquinolin-4-yl)piperazine-1-yl]acetamide derivatives A1–A27
| Compound |
R |
Antiamoebic Activitya (IC50 ± S.D.) units |
Antimalarial activityb (IC50 ± s day: μM) |
Cytotoxic activity (% cell death at 50 μM) |
Haemolytic activity (ratio of lysis relative to chloroquine at 50 μM) |
Inhibition of β-haematin formationc (IC50 value: μM) |
| Strains used “1HM1:IMSS of E. histolytica. 3D7 of P.falciparum” and. HEK-293 kidney epithelial cells. |
| A1 |
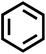 |
2.25 ± 0.03 |
3.95 ± 1.21 |
44.60 ± 10.57 |
1.04 |
54.41 ± 1.07 |
| A2 |
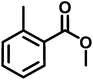 |
7.90 ± 0.02 |
29.29 ± 7.01 |
81.86 ± 3.32 |
0.98 |
34.57 ± 1.78 |
| A3 |
 |
1.38 ± 0.02 |
13.24 ± 3.24 |
28.84 ± 5.35 |
0.52 |
110.32 ± 18.93 |
| A4 |
 |
1.17 ± 0.02 |
17.96 ± 3.93 |
54.40 ± 3.48 |
0.61 |
55.62 ± 5.76 |
| A5 |
 |
6.40 ± 0.02 |
14.60 ± 2.35 |
67.33 ± 0.64 |
0.53 |
43.98 ± 3.77 |
| A6 |
 |
3.20 ± 0.01 |
19.12 ± 1.63 |
37.17 ± 5.87 |
0.66 |
44.86 ± 0.60 |
| A7 |
 |
0.41 ± 0.02 |
6.72 ± 1.45 |
48.63 ± 4.15 |
1.03 |
75.51 ± 1.71 |
| A8 |
 |
1.60 ± 0.02 |
2.84 ± 0.50 |
50.24 ± 6.10 |
0.51 |
40.36 ± 3.68 |
| A9 |
 |
0.72 ± 0.01 |
33.26 ± 7.74 |
41.22 ± 6.45 |
0.72 |
46.53 ± 5.89 |
| A10 |
 |
5.90 ± 0.02 |
14.66 ± 2.72 |
27.53 ± 10.61 |
0.67 |
68.79 ± 5.95 |
| A11 |
 |
7.20 ± 0.02 |
14.98 ± 2.35 |
71.72 ± 1.34 |
0.55 |
71.43 ± 7.91 |
| A12 |
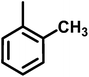 |
5.90 ± 0.01 |
33.52 ± 3.85 |
54.76 ± 3.33 |
0.68 |
95.98 ± 13.81 |
| A13 |
 |
17.14 ± 0.02 |
6.26 ± 1.53 |
28.98 ± 8.49 |
0.45 |
49.30 ± 7.72 |
| A14 |
 |
1.49 ± 0.01 |
16.42 ± 1.41 |
65.08 ± 2.12 |
0.48 |
34.28 ± 2.69 |
| A15 |
 |
7.30 ± 0.01 |
16.92 ± 1.33 |
43.10 ± 2.98 |
0.57 |
62.81 ± 3.21 |
| A16 |
 |
2.15 ± 0.01 |
11.72 ± 2.71 |
54.39 ± 3.43 |
0.88 |
17.27 ± 0.77 |
| A17 |
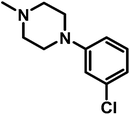 |
1.13 ± 0.02 |
23.20 ± 5.17 |
77.42 ± 3.20 |
0.52 |
42.06 ± 2.02 |
| A18 |
 |
0.52 ± 0.02 |
10.61 ± 1.41 |
70.31 ± 2.57 |
1.50 |
33.32 ± 2.10 |
| A19 |
 |
3.24 ± 0.01 |
3.91 ± 0.82 |
61.06 ± 7.75 |
1.01 |
38516 ± 7.75 |
| A20 |
 |
1.80 ± 0.01 |
15.55 ± 0.58 |
36.28 ± 3.83 |
0.74 |
>191.73 |
| A21 |
 |
2.70 ± 0.01 |
8.35 ± 1.62 |
63.57 ± 2.63 |
1.96 |
64.07 ± 5.44 |
| A22 |
 |
18.99 ± 0.02 |
0.35 ± 0.03 |
70.78 ± 4.39 |
1.65 |
35.24 ± 3.16 |
| A23 |
 |
0.86 ± 0.03 |
16.63 ± 5.26 |
63.97 ± 3.69 |
0.71 |
38.06 ± 3.36 |
| A24 |
 |
1.43 ± 0.01 |
17.88 ± 0.62 |
37.76 ± 1.67 |
1.25 |
53.29 ± 3.09 |
| A25 |
 |
2.40 ± 0.035 |
0.30 ± 0.03 |
40.07 ± 2.91 |
0.78 |
32.24 ± 3.63 |
| A26 |
 |
4.40 ± 0.02 |
9.23 ± 3.24 |
26.17 ± 4.30 |
0.45 |
106.65 ± 19.13 |
| A27 |
 |
3.55 ± 0.02 |
13.64 ± 1.79 |
54.55 ± 2.83 |
1.19 |
35.44 ± 4.48 |
| Metronidazole |
1.80 ± 0.01 |
N.a. |
38.97 ± 4.08 |
N.a. |
N.a. |
| Chloroquine |
N.a. |
0.0013 ± 0.0003 |
1.05 ± 1.27 |
1.00 |
15.32 ± 1.77 |
| Camptothecin |
N.a. |
N.a. |
74.65 ± 1.50 |
N.a. |
N.a. |
Antimalarial and cytotoxic activity. All compounds (A1–A27) were active against 3D7 strain of P. falciparum (IC50 range: 0.30–33.52 μM), with the most active compounds A22 (IC50: 0.35 μM) and A25 (IC50: 0.30 μM) almost nine fold more active than the next most active compound A8 (IC50: 2.84 μM) (Table 2). In comparison to chloroquine (IC50: 0.0013 μM), compounds A22 and A25 were on average 250 times less active in inhibiting parasite growth. The closure of the triangular side chain of the isopropyl compound A24 resulted in a 60 fold increase in antimalarial activity for the cyclopropyl compound A25. Whilst a methyl group in the para position in the aromatic amine (A8) increased the antimalarial activity 33 fold when compared to the meta (A9) and ortho (A12) substitutions. In contrast, the positioning of the side groups in compounds A4/A5 and A6/A7 did not greatly alter the inhibitory activity. As with the antiamoebic activity, the introduction of bulker groups did not improve the antimalarial activity. Similarly, the addition of halogens (Cl−, F− or Br−) on the aromatic amines did not decrease the IC50 values nor did they aid in binding to the haemin molecules to prevent β-haematin crystal formation (Table 2).All but one (A20) of the compounds inhibited β-haematin formation to varying degrees, being 1.13–12.51 fold less active than chloroquine (Table 2) – thus indicating that the 4-aminochloroquinoline group retained some ability to bind to the heme units as proposed. There was no correlation between the inhibitory effects of the compounds on the whole parasite and β-haematin formation (r2 = 0.019). The most active compounds (A22 and A25) did not primarily inhibit parasite growth by preventing β-haematin or hemozoin formation, and were found two fold less active than chloroquine (IC50: 15.32 μM). As such, the antimalarial activity could be due to the effect of the acetamide group on folic acid synthesis and needs to be investigated. The ability of the most potent compound A16 (IC50: 17.27 μM) to inhibit β-haematin formation was greatly diminished when compared to the other halogen substituted compounds (A6, A7, A10, A11, A15; IC50 range: 44.86–75.51 μM). The equipotent activity of A16 to that of chloroquine was also lost when the compound was tested against the whole parasite where the IC50 value (11.72 μM) increased greatly compared to chloroquine (9020 fold).
Of the 27 derivatives, 17 were more toxic than the dihydrofolate reductase inhibitor, methotrexate and all were more toxic than chloroquine when tested against the human kidney epithelial cells (Table 2). These 27 derivatives were also more toxic than cycloguanil and pyrimethamine which inhibited approximately 20% cell growth at 100 μg ml−1 (data not shown). The most toxic derivative was A2 (34% more toxic than methotrexate), whilst the least toxic was A26; with both of these derivatives not affecting red blood cell membrane stability (lysis ratio of 0.98 and 0.45, respectively). In contrast the derivatives which induced the most red blood cell lysis was A21 (lysis ratio 1.96) and least lytic A13 and A2 with a lysis ratio of 0.28 for both derivatives. Of the halogen substituted derivatives, overall the fluoro-substitutions were 1.6 fold less toxic to the human kidney epithelial cells compared to chloro- and bromo- substituted derivatives (Table 2). Of the two most active compounds, compounds A25 displayed the better safety profile when the inhibitory effect on the human kidney epithelial cells and haemolytic effects (40.07% inhibition; 0.78 lysis ratio) are compared to those of chloroquine (1.05% inhibition; 1.0 lysis ratio) and A22 (70.78% inhibition; 1.65 lysis ratio) (Table 2). The latter indicated that compound A25 did not alter the host red blood cell membrane integrity, and that the antimalarial activity was directed towards the intra-erythrocytic parasite. Both A22 and A25 had a higher selectivity for the malaria parasite compared to the human cell. But overall the safety profile for A25 indicates that it has potential for further structural modifications to improve both antimalarial and cytotoxic activity.
2.4. Molecular docking
The hybrid molecules capable of exhibiting dual activity i.e. β-haematin and DHFR inhibition have been designed and a homology model of PfDHFR was also generated so as to analyze the interactions of the quinoline based acetamides with the enzyme. The binding mode of four active analogues (A7, A8, A18 and A21) using the crystal structure has been predicted to understand the protein-ligand contacts and their interaction strength docking of these compounds along with WR99210, the co-crystallised compound in the active site of wild PfDHFR. The root-mean-square deviation (RMSD) between the re-scored docked ligand and original X-ray crystallised ligand WR99210 was observed to be 1.19 Å validating the docking protocol. The three compounds A7, A8 and A18 exhibited similar pose as that of co-crystallised ligand WR99210 in the binding region (Fig. 4). The Autodock binding energy of A7, A8, A18 and A21 is −9.34 kcal mol−1, −9.90 kcal mol−1, −9.67 kcal mol−1 and −9.57 kcal mol−1 respectively, in comparison to binding energy of −10.86 kcal mol−1 for WR99210 ligand bound to crystal of PfDHFR. The interaction plots generated using Ligplot (Fig. 4) indicated that the three ligands (A7, A8 and A18) exhibit H-bonding with Ile164 similar to that observed in WR99210. Also, all the four compounds showed highly conserved hydrophobic interactions with the same active site residues Ile14, Cys15, Val45, Leu46, Lys49, Asp54, Met55, Phe58, Ser111 and Ile112, expect a few exceptions in case of A21, which is due to difference in the orientation of compound within the active site of the enzyme. The molecular docking data thus provides us insight into the binding modes of ligands in the active site of PfDHFR.
 |
| | Fig. 4 Ligplot showing hydrogen bonding interaction and hydrophobic contacts (with green dashed lines and red arcs with radiating lines respectively) for the ligand (A) A7 (B) A8 (C) A18 and (D) A21. | |
3. Experimental
Most the chemicals were purchased from Aldrich Co (USA). Precoated aluminum sheets (silica gel 60 F254, Merck, Germany) were used for thin-layer chromatography (TLC) and spots were visualized under UV light. The melting points were recorded on Buchi melting point apparatus Model no. 650 and were uncorrected. Elemental analyses were performed in an Elementar Vario analyzer and the results lay within ± 0.3% of the theoretical values. IR spectra were acquired at Bruker FT-IR spectrophotometer. 1H and 13C NMR were recorded on a Bruker Spectrospin DPX 300 MHz and Bruker Spectrospin DPX 75 MHz spectrometer respectively, using CDCl3 as a solvent and trimethylsilane (TMS) as the internal standard. Splitting patterns are designated as follows; s, singlet; d, doublet; t, triplet; m, multiplet, bs, broad singlet, ss, sharp singlet. Electrospray (ES) mass spectra were carried out on Microtof-Q II 10262. Melting points were recorded on a Veego melting point apparatus (model REC-2203882) and were uncorrected.
3.1. Preparation of 7-chloro-4-(piperazin-1-yl)quinoline (2)
7-Chloro-4-(piperazin-1-yl)quinoline was prepared by the reported method.17
3.2. General procedure for the preparation of different substituted chloro acetamides
The aromatic and aliphatic acetamides were prepared by two different methods as given below:
For substituted aromatic acetamides. To a stirring solution of anilines in glacial acetic acid. 50% aqueous sodium acetate, chloro acetyl chloride was added dropwise. The reaction mixture was stirred at room temperature for 4 h. The product was precipitated by adding reaction mixture to crushed ice and was filtered on a Buckner funnel. The product formed was recrystallized in ethanol (1–15). These compounds have already been reported in the following literature.24–32
For aliphatic (2-chloro-acetamides). To a stirring solution of amine in 10 ml dichloromethane, triethylamine was added. The reaction mixture was cooled to 0 °C (under ice bath) and chloroacetyl chloride was added. The reaction mixture was then stirred for 3–4 h at room temperature and partitioned between water and dichloromethane. Organic extract was separated and dried on sodium sulphate and concentrated to yield acetylated products (16–27). All the compounds are already reported in the literature33–40 except compound 19 and 21 whose spectral data is given in the ESI.†
3.3 Synthesis of the 2-[4-(7-chloroquinolin-4-yl)piperazine-1-yl]acetamide derivatives: general synthesis
A mixture of 7-chloro-4-(piperazin-1-yl)quinoline(2) (1.0 eq.), chloroacetamides (1–27) (1.1 eq.), K2CO3 (2.5 eq.) and catalytic amount of potassium iodide in dimethyl formamide was refluxed at 90 °C for 8–10 h with continuous stirring. After reaction completion (monitored by TLC) the mixture was poured on crushed ice. The product precipitated was filtered over Buckner funnel and was recrystallized from appropriate solvent.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-phenylacetamide (A1). Yield 64% colourless solid m.p. 157–160 °C; H1 NMR (CDCl3) δppm: 9.05 (s, 1H), 8.76 (d, 1H, J = 7.8 Hz), 8.06 (s, 1H), 7.96 (d, 1H, J = 9.0 Hz), 7.60 (d, 2H, J = 7.8 Hz), 7.47–7.43 (m, 1H), 7.38 (t, 2H, J = 7.8 Hz), 7.16 (t, 1H, J = 7.5 Hz), 6.89 (d, 1H, J = 5.1 Hz), 3.31 (bs, 4H), 3.30 (s, 2H), 2.96–2.88 (m, 4H); 13C NMR (CDCl3) δppm: 167.83, 156.59, 151.84, 150.08, 137.45, 135.10, 129.90, 128.88, 126.43, 125.01, 124.41, 121.83, 119.52, 109.12, 62.05, 53.40, 52.29; IR (νmax) cm−1: 3255, 1658, 1542; ESI-MS: m/z 381.13 (M + 1); anal. calc. for C21H21N4OCl; C 66.22, H 5.56, N 14.71, found C 66.52, H 5.60, N 14.63%.
Methyl 2-(2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamido)benzoate(A2). Yield 73% yellow solid m.p. 128–131° C; H1 NMR (CDCl3) δppm: 11.79 (s, 1H), 8.72–8.64 (m, 2H), 8.06–7.97 (m, 3H), 7.64–7.53 (m, 2H), 7.201 (t, 1H, J = 7.5 Hz), 7.05 (d, 1H, J = 4.8 Hz), 3.88 (s, 3H), 3.36 (bs, 4H), 3.31 (s, 2H), 2.89 (bs, 4H); 13C NMR (CDCl3) δppm: 169.64, 167.80, 156.99, 151.91, 150.21, 140.70, 134.93, 134.39, 130.87, 128.90, 126.18, 125.20, 122.77, 121.93, 120.60, 115.91, 109.00, 62.68, 53.44, 52.15, 51.92; IR (νmax) cm−1: 3315, 1686, 1572; ESI-MS: m/z 439.15 (M + 1); anal. calc. for C23H23N4O3Cl, C 62.94, H 5.28, N 12.76, found C 62.16, H 5.28, N 12.55%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-(2,5-difluorophenyl)acetamide (A3). Yield 78% colourless solid m.p. 183–187 °C; H1 NMR (CDCl3) δppm: 9.59 (bs, 1H), 8.76 (d, 1H, J = 4.8 Hz), 8.27–8.20 (m, 1H), 8.06 (s, 1H), 7.95 (d, 1H, J = 9.0 Hz), 7.46 (d, 1H, J = 8.1 Hz), 7.09–7.01 (m, 1H), 6.90 (d, 1H, J = 4.8 Hz), 6.77–6.72 (m, 1H), 3.34 (bs, 4H), 3.19 (s, 2H), 2.96 (bs, 4H); 13C NMR (CDCl3) δppm: 168.86, 156.73, 151.95, 150.18, 138.57, 134.10, 133.94, 130.89, 129.58, 127.58, 121.76, 120.75, 115.47, 111.01, 110.91, 108.76, 61.98, 54.73, 54.27, 53.78, 52.90; IR (νmax) cm−1: 1688, 1573; ESI-MS: m/z 417.06 (M + 1); anal. calc. for C21H19N4OClF; C 60.51, H 4.59, N 13.44, found C 60.50, H 4.45, N 13.40%.
N-(3-Acetylphenyl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamide(A4). Yield 65% yellow solid m.p. 152–156 °C; H1 NMR (CDCl3) δppm: 9.29 (s, 1H), 8.85–8.77 (m, 1H), 8.07–7.93 (m, 4H), 7.73 (d, 1H, J = 6.9 Hz), 7.49 (t, 2H, J = 7.2 Hz), 6.96–6.91 (m, 1H), 3.33 (bs, 4H), 3.31 (s, 2H), 2.96 (bs, 4H), 2.63 (s, 3H); 13C NMR (CDCl3) δppm: 198.14, 169.07, 156.77, 152.62, 150.11, 139.48, 137.79, 134.04, 129.54, 128.50, 126.52, 124.52, 123.92, 121.84, 119.27, 109.87, 61.99, 52.97, 52.08, 27.19; IR (νmax) cm−1: 3295, 1726, 1671, 1568; ESI-MS: m/z 423.16 (M + 1); anal. calc. for C23H23N4O2Cl; C 65.32, H 5.48, N 13.25, found C 65.52, H 5.630, N 13.33%.
N-(4-Acetylphenyl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamide(A5). Yield 54% yellow solid m.p. 130–133 °C; H1 NMR (CDCl3) δppm: 9.28 (s, 1H), 8.77 (s, 1H), 8.070–7.92 (m, 4H), 7.71 (d, 2H, J = 7.8 Hz), 7.47–7.44 (m, 1H), 6.90 (d, 1H, J = 5.1 Hz), 3.33 (bs, 4H), 3.31 (s, 2H), 2.97 (bs, 4H), 2.59 (s, 3H); 13C NMR (CDCl3) δppm: 196.83, 168.25, 156.50, 151.84, 150.07, 141.68, 135.10, 133.04, 129.78, 128.89, 126.46, 124.96, 121.80, 118.74, 109.15, 62.06, 53.44, 52.25, 26.43; IR (νmax) cm−1: 3238, 1783, 1676, 1585; ESI-MS: m/z 423.06 (M + 1); anal. calc. for C23H23N4O2Cl; C 65.32, H 5.48, N 13.25, found C 65.40, H 5.47, N 13.70%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-(2-fluorophenyl)acetamide (A6). Yield 68% solid m.p. 133–136 °C; H1 NMR (CDCl3) δppm: 9.511 (s, 1H), 8.76 (d, 1H, J = 4.8 Hz), 8.41 (t, 1H, J = 7.8 Hz), 8.06 (s, 1H), 7.96 (d, 1H, J = 9.0 Hz), 7.46 (d, 1H, J = 9.3 Hz), 7.18–7.08 (m, 3H), 6.90 (d, 1H, J = 5.1 Hz), 3.33 (bs, 4H), 3.31 (s, 2H), 2.97 (bs, 4H); 13C NMR (CDCl3) δppm: 167.95, 156.55, 154.11, 151.91, 150.89, 135.03, 128.97, 126.38, 125.96, 124.98, 124.65, 124.45, 121.85, 121.32, 114.95, 109.20, 61.98, 53.35, 52.34; IR (νmax) cm−1: 3296, 1690, 1572; ESI-MS: m/z 399.52 (M + 1); anal. calc. for C21H20N4OClF; C 63.24, H 5.05, N 14.05, found C 63.04, H5.15, N14.25%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-(4-fluorophenyl)acetamide (A7). Yield 84% yellow solid m.p. 125–128 °C; H1 NMR (CDCl3) δppm: 9.08 (s, 1H), 8.76 (d, 1H, J = 4.8 Hz), 8.06 (s, 1H), 7.95 (d, 1H, J = 9 Hz), 7.57–7.53 (m, 2H), 7.47–7.44 (m, 1H), 7.07 (d, 2H, J = 9 Hz), 6.88 (d, 1H, J = 5.1 Hz), 3.23 (bs, 4H), 3.30 (s, 2H), 3.04 (bs, 4H); 13C NMR (CDCl3) δppm: 167.80, 161.01, 157.78, 156.56, 151.85, 150.10, 135.12, 133.48, 128.90, 126.46, 124.92, 121.84, 121.31, 115.86, 115.56, 109.12, 61.93, 53.44, 52.27; IR (νmax) cm−1: 3255, 1686, 1502; ESI-MS: m/z 399.52 (M + 1); anal. calc. for C21H20N4OClF; C 63.24, H 5.05, N 14.05, found C 63.14, H5.09, N 14.25%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-p-tolylacetamide (A8). Yield 62% colourless solid m.p. 157–160 °C; H1 NMR (CDCl3) δppm: 9.71 (s, 1H), 8.72 (d, 1H, J = 4.8 Hz), 8.04–7.99 (m, 2H), 7.56 (d, 3H, J = 7.5 Hz), 7.13 (d, 2H, J = 7.8 Hz), 7.03 (d, 1H, J = 5.1 Hz), 3.26 (bs, 4H), 3.23 (s, 2H), 2.83 (bs, 4H), 2.25 (s, 3H); 13C NMR (CDCl3) δppm: 167.64, 156.57, 151.91, 150.17, 135.08, 134.90, 134.05, 129.57, 128.97, 126.44, 124.99, 121.86, 119.54, 109.13, 62.05, 53.43, 52.31, 20.88; IR (νmax) cm−1: 3296, 1676, 1572; ESI-MS: m/z 395.11 (M + 1); anal. calc. for C22H23N4OCl; C 66.91, H 5.87, N 14.19, found C 66.89, H 5.97, N 14.09%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-m-tolylacetamide (A9). Yield 65% yellow solid m.p. 100–103 °C; H1 NMR (CDCl3) δppm: 9.00 (s, 1H), 8.76 (d, 1H, J = 5.1 Hz), 8.06 (d, 1H, J = 1.8 Hz), 7.96 (d, 1H, J = 9 Hz), 7.47–7.41 (m, 3H), 7.24 (d, 1H, J = 7.8 Hz), 6.96 (d, 1H, J = 7.8 Hz), 6.89 (d, 1H, J = 5.1 Hz), 3.32 (bs, 4H), 3.29 (s, 2H), 2.95 (bs, 4H), 2.36 (s, 3H); 13C NMR (CDCl3) δppm: 167.84, 156.82, 151.35, 150.27, 135.48, 134.63, 130.39, 128.47, 126.49, 124.84, 124.45, 121.75, 119.55, 117.48, 109.40, 61.89, 53.45, 52.30, 19.79; IR (νmax) cm−1: 3275, 1665, 1570; ESI-MS: m/z 395.19 (M + 1); anal. calc. for C22H23N4OCl; C 66.91, H 5.87, N 14.19, found C 66.71, H 5.78, N 14.09%.
N-(3-Chlorophenyl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamide(A10). Yield 65% white solid m.p. 131–135 °C; H1 NMR (CDCl3) δppm: 9.09 (s, 1H), 8.76 (d, 1H, J = 4.8 Hz), 8.06 (s, 1H), 7.95 (d, 1H, J = 9 Hz), 7.66 (s, 1H), 7.46 (d, 1H, J = 7.2 Hz), 7.29 (d, 2H, J = 8.4 Hz), 7.12 (d, 1H, J = 7.2 Hz), 6.89 (d, 1H, J = 4.5 Hz), 3.30 (bs, 4H0, 3.20 (s, 2H), 2.94 (bs, 4H); 13C NMR (CDCl3) δppm: 167.94, 156.49, 151.90, 150.17, 138.60, 135.09, 134.73, 130.10, 128.97, 126.46, 124.94, 124.41, 121.85, 119.54, 117.46, 109.16, 61.98, 53.44, 52.27; IR (νmax)cm−1: 3087, 1687, 1580; ESI-MS: m/z 416.03 (M + 1); anal. calc. for C21H20N4OCl2; C 60.73, H 4.85, N 13.49, found C 60.79, H 4.97, N 13.29%.
N-(4-Bromophenyl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamide (A11). Yield 68% colourless solid m.p. 188–191 °C; H1 NMR (CDCl3) δppm: 9.76 (bs, 1H), 8.72 (d, 1H, J = 4.8 Hz), 8.05–7.92 (m, 4H), 7.55 (d, 1H, J = 8.4 Hz), 7.31–7.28 (m, 1H), 7.03–6.97 (m, 2H), 3.36 (bs, 4H), 3.26 (s, 2H), 2.88 (bs, 4H); 13C NMR (CDCl3) δppm: 168.88, 156.75, 152.60, 150.10, 138.46, 134.03, 131.90, 128.50, 126.49, 126.20, 121.94, 121.36, 115.49, 109.85, 61.97, 52.95, 52.07; IR (νmax) cm−1: 1663, 1576; ESI-MS: m/z 460.28 (M + 1); anal. calc. for C21H20N4OBrCl; C 54.86, H 4.38 N 12.19, found C 54.98, H 4.60, N 12.19%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-o-tolylacetamide (A12). Yield 78% colorless solid m.p. 178–181 °C; H1 NMR (CDCl3) δppm: 9.20 (bs, 1H), 8.79 (d, 1H, J = 5.1 Hz), 8.15 (d, 1H, J = 8.1 Hz), 8.06 (s, 1H), 7.96 (d, 1H, J = 9.0 Hz), 7.47–7.43 (m, 1H), 7.22–7.18 (m, 2H), 7.13–7.04 (m, 1H), 6.89 (d, 1H, J = 5.1 Hz), 3.34 (bs, 4H), 3.32 (s, 2H), 3.00 (bs, 4H), 2.31 (ss, 3H); 13C NMR (CDCl3) δppm: 167.57, 156.47, 151.89, 150.16, 135.65, 135.08, 130.41, 128.96, 127.03, 126.98, 126.44, 124.99, 121.82, 121.15, 109.12, 62.17, 53.47, 52.46, 17.83l; IR (νmax) cm−1: 1684, 1576; ESI-MS: m/z 395.19 (M + 1); anal. calc. for C22H23N4OCl; C 66.91, H 5.87, N 14.91 found C 66.89, H 5.97, N 14.30%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-(naphthalen-1-yl)acetamide (A13). Yield 80% colourless solid m.p. 180–183 °C; H1 NMR (CDCl3) δppm: 9.76 (s, 1H), 8.78 (d, 1H, J = 4.8 Hz), 8.27 (d, 1H, J = 7.5 Hz), 8.07 (s, 1H), 7.99 (d, 1H, J = 7.4 Hz), 7.89–7.79 (m, 2H), 7.70 (d, 1H, J = 7.5 Hz), 7.56–7.48 (m, 4H), 6.93 (d, 1H, J = 5.1 Hz), 3.46 (bs, 4H), 3.31 (s, 2H), 3.08 (bs, 4H), 13C NMR (CDCl3) δppm: 168.01, 156.48, 151.93, 150.20, 135.10, 134.11, 132.08, 129.07, 129.01, 126.37, 126.02, 125.97, 125.08, 124.99, 121.84, 119.67, 118.64, 109.15, 62.35, 53.57, 52.48; IR (νmax) cm−1: 3280, 1690, 1571; ESI-MS: m/z 431.24 (M + 1); anal. calc. for C25H23N4OCl; C 69.68, H 5.38, N 13.00, found C 69.80, H 5.27, N 13.00%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-(2,6-dimethylphenyl)acetamide (A14). Yield 76% colorless solid m.p. 168–171 °C; H1 NMR (CDCl3) δppm: 8.75 (d, 1H, J = 4.8 Hz), 8.60 (bs, 1H), 8.06 (s, 1H), 7.97 (d, 1H, J = 8.7 Hz), 7.47–7.43 (m, 1H), 7.12–7.11 (m, 3H), 6.87 (d, 1H, J = 5.1 Hz), 3.37 (s, 2H), 3.32 (bs, 4H), 3.03 (bs, 4H), 2.25 (ss, 6H); 13C NMR (CDCl3) δppm: 168.38, 156.76, 152.64, 150.13, 135.64, 135.48, 134.03, 128.51, 128.10, 126.86, 126.55, 126.19, 121.85, 109.85, 61.59, 53.29, 52.07, 18.72; IR (νmax) cm−1: 1661, 1573; ESI-MS: m/z 409.33 (M + 1); anal. calc. for C23H25N4OCl; C 67.56, H 6.16, N 13.70, found C 67.65, H 6.56, N 13.61%.
N-(3-Chloro-4-fluorophenyl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamide (A15). Yield 76% colorless solid m.p. 177–180 °C; H1 NMR (CDCl3) δppm: 9.07 (bs, 1H), 8.76 (d, 1H, J = 5.1 Hz), 8.06 (s, 1H), 7.95 (d, 1H, J = 9.0 Hz), 7.75–7.72 (m, 1H), 7.47–7.43 (m, 2H), 7.14 (m, 1H), 6.89 (d, 1H, J = 4.8 Hz), 3.30 (bs, 4H), 3.19 (s, 2H), 2.95 (bs, 4H); 13C NMR (CDCl3) δppm: 169.01, 167.37, 156.76, 152.58, 150.11, 134.04, 132.20, 131.95, 129.11, 128.50, 126.48, 121.84, 120.79, 119.65, 117.37, 109.83, 61.90, 52.95, 52.04; IR (νmax) cm−1: 3292, 1693, 1573; ESI-MS: m/z 434.36 (M + 1); anal. calc. for C21H19N4OCl2F; C 58.21, H 4.42, N 12.93, found C 58.01, H 4.22, N 12.73%.
1-(4-Benzylpipezin-1-yl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)ethanone (A16). Yield 44% white solid m.p. 135–138 °C; H1 NMR (CDCl3) δppm: 8.72 (d, 1H, J = 5.1 Hz), 8.04 (s, 1H), 7.95 (d, 1H, J = 9 Hz), 7.44–7.40 (m, 2H), 7.33–7.31 (m, 4H), 6.84 (d, 1H, J = 5.1 Hz), 3.64 (d, 4H, J = 3.6 Hz), 3.52 (s, 2H), 3.35 (bs, 4H), 3.32 (s, 2H), 2.28 (d, 4H, J = 4.2 Hz), 2.46 (d 4H, J = 4.5 Hz); 13C NMR (CDCl3) δppm: 167.58, 156.85, 151.89, 150.17, 137.61, 134.94, 129.08, 128.89, 128.34, 127.29, 126.20, 125.16, 121.90, 108.99, 62.92, 61.02, 53.35, 52.94, 52.11, 45.65, 41.87; IR (νmax) cm−1: 1635, 1378, 1275; ESI-MS: m/z 465.01 (M + 1); anal. calc. for C26H30N5OCl; C 67.302, H 6.52, N 15.09, found C 67.16, H 6.75, N 15.18%.
1-(4-(3-Chlorophenylpipezin-1-yl)-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)ethanone (A17). Yield 34% colorless solid m.p. 116–120 °C; H1 NMR (CDCl3) δppm: 8.73 (d, 1H, J = 4.8 Hz), 8.04 (s, 1H), 7.95 (d, 1H, J = 9.0 Hz), 7.45–7.41 (m, 1H), 7.19 (d, 1H, J = 7.8 Hz), 6.89–6.79 (m, 4H), 3.79 (bs, 4H), 3.37 (s, 2H), 3.26–3.18 (m, 8H), 2.85 (bs, 4H); 13C NMR (CDCl3) δppm: 167.80, 156.48, 151.68, 150.58, 150.23, 134.92, 134.23, 128.89, 127.74, 127.57, 126.22, 125.43, 125.23, 121.78, 119.02, 109.02, 61.17, 52.98, 52.23, 52.01, 51.55, 45.75, 42.32; IR (νmax) cm−1: 1634, 1374; ESI-MS: m/z 485.43 (M + 1); anal. calc. for C25H27N5OCl2; C 61.99, H 5.62, N 14.46, found C 61.79, H 5.89, N 14.03%.
tert-Butyl 4-(2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetyl)piperzine-1-carboxylate (A18). Yield 40% white solid m.p. 104–107 °C; H1 NMR (CDCl3) δppm: 8.73 (d, 1H, J = 4.8 Hz), 8.04 (bs, 1H), 7.95 (1H, J = 6.0 Hz), 7.44 (d, 1H, J = 9.0 Hz), 6.84 (d, 1H, J = 5.1 Hz), 3.61 (bs, 4H), 3.48–3.44 (m, 4H), 3.34 (s, 2H), 1.59 (ss, 9H), 1.47 (s, 8H); 13C NMR (CDCl3) δppm: 168.02, 156.71, 154.28, 152.81, 150.01, 134.00, 128.14, 127.34, 126.80, 125.70, 121.78, 79.61, 60.88, 52.69, 52.22, 45.50, 28.97, 28.48, 27.95; IR (νmax) cm−1: 1691, 1300; ESI-MS: m/z 474.22 (M+); anal. calc. for C23H23N4O2Cl; C 60.81, H 6.80, N 14.78, found C 60.94, H 6.74, N 14.90%.
1-(4-(Benzo[1,3]dioxol-5-yl)methyl)piperzin-1-yl-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)ethanone (A19). Yield 35% white solid m.p. 192–195 °C; H1 NMR (CDCl3) δppm: 8.72 (d, 1H, J = 5.1 Hz), 8.04 (s, 1H), 7.93 (d, 1H, J = 9.0 Hz), 7.73–7.70 (m, 1H), 7.54–7.51 (m, 1H), 7.43–7.40 (m, 1H), 6.93 (d, 1H, J = 6.6 Hz), 6.85 (d, 1H, J = 4.8 Hz), 4.19 (s, 2H), 3.26 (bs, 4H), 3.12 (s, 2H), 2.83 (bs, 4H), 1.25 (s, 2H), 1.20 (bs, 4H), 1.09 (bs, 4H); 13C NMR (CDCl3) δppm: 168.62, 156.63, 151.84, 150.11, 134.94, 132.35, 130.88, 128.87, 126.27, 125.05, 121.82, 109.03, 71.74, 61.61, 53.33, 52.17, 40.78, 27.68, 22.82, 19.13; IR (νmax) cm−1: 1672, 1544; ESI-MS: m/z 508.21 (M + 1); anal. calc. for C27H30N5O3Cl; C 63.84, H 5.95, N 13.79, found C 63.47, H 5.55, N 13.75%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-1-(4-phenylpipezin-1-yl)-ethanone (A20). Yield 39% colorless solid m.p. 135–138 °C; H1 NMR (CDCl3) δppm: 8.72 (d, 1H, J = 5.1 Hz), 8.04 (s, 1H), 7.95 (d, 1H, J = 9.0 Hz), 7.44–7.40 (m, 1H), 7.39–7.23 (m, 2H), 6.95–6.85 (m, 3H), 6.83 (d, 1H, J = 5.1 Hz), 3.82–3.78 (m, 4H), 3.38 (s, 2H), 3.29–3.17 (m, 8H), 2.85 (bs, 4H); 13C NMR (CDCl3) δppm: 167.66, 156.78, 151.88, 150.94, 150.15, 134.93, 129.27, 128.89, 126.21, 125.15, 121.88, 120.59, 116.62, 109.09, 61.16, 52.98, 52.13, 50.10, 49.47, 45.61, 41.78; IR (νmax) cm−1: 1652, 1564; ESI-MS: m/z 450.09 (M + 1); anal. calc. for C25H28N5OCl; C 66.73, H 6.27, N 15.56, found C 66.70, H 6.68, N 15.44%.
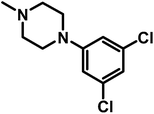
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-1-(4-(2,3-dichlorophenylpipezin-1-yl)-ethanone (A21). Yield 48% white solid m.p. 143–146 °C; H1 NMR (CDCl3) δppm: 8.72 (d, 1H, J = 4.8 Hz), 8.04 (s, 1H), 7.95 (d, 1H, J = 8.7 Hz), 7.44–7.41 (m, 1H), 7.22–7.13 (m, 2H), 6.94–6.91 (m, 1H), 6.84 (d, 1H, J = 5.1 Hz), 3.82 (bs, 4H), 3.38 (s, 2H), 3.26 (bs, 4H), 3.06–3.02 (m, 4H), 2.85 (bs, 4H); 13C NMR (CDCl3) δppm: 167.81, 156.78, 151.90, 150.58, 150.15, 134.92, 134.23, 128.89, 127.74, 127.57, 126.22, 125.23, 125.13, 121.88, 118.72, 109.02, 61.17, 52.98, 52.13, 52.01, 51.25, 45.95, 42.02; IR (νmax) cm−1: 1645, 1569; ESI-MS: m/z 518.94 (M + 1); anal. calc. for C25H26N5OCl3; C 57.85, H 5.05, N 13.50, found C 57.67, H 5.02, N 13.46%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl) 1-(4-hydroxypiperidin-1-yl)- ethanone (A22). Yield 30% green solid m.p. 102–105 °C; H1 NMR (CDCl3) δppm: 8.69 (d, 1H, J = 4.5 Hz), 8.03 (s, 1H), 7.93 (d, 1H, J = 9.0 Hz), 7.42 (d, 1H, J = 9.0 Hz), 6.82 (d, 1H, J = 4.8 Hz), 4.13 (s, 1H), 3.96 (s, 2H), 3.48–3.24 (m, 8H), 2.91–2.81 (m, 4H), 2.63 (bs, 1H), 2.14–2.01 (m, 2H), 1.72–1.50 (m, 2H); 13C NMR (CDCl3) δppm: 167.57, 156.92, 151.75, 149.97, 134.98, 128.67, 126.22, 125.19, 121.84, 108.98, 66.93, 61.08, 52.96, 52.09, 43.01, 39.33, 34.84, 34.17; IR (νmax) cm−1: 1720, 1570; ESI-MS: m/z 389.09 (M + 1); anal. calc. for C20H25N4O2Cl; C 61.77, H 6.48, N 14.41, found C 61.48, H 6.47, N 14.30%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-cyclohexylacetamide (A23). Yield 44% white solid m.p. 105–108 °C; H1 NMR (CDCl3) δppm: 8.74 (d, 1H, J = 5.1 Hz), 8.053 (s, 1H), 7.94 (d, 1H, J = 9 Hz), 7.45–7.42 (m, 1H), 7.02 (d, 1H, J = 6.9 Hz), 6.86 (d, 1H, J = 5.1 Hz), 3.85 (m, 1H), 3.26 (bs, 4H), 3.13 (s, 2H), 2.85 (bs, 4H), 1.72–1.67 (m, 4H), 1.47–1.35 (m, 2H), 1.26–1.15 (m, 4H); 13C NMR (CDCl3) δppm: 168.49, 156.63, 151.86, 150.17, 134.99, 128.93, 126.31, 125.03, 121.85, 109.04, 61.63, 53.35, 52.24, 47.39, 33.12, 25.52, 24.69; IR (νmax) cm−1: 3232, 1649, 1573; ESI-MS: m/z 387.03 (M + 1); anal. calc. for C21H27N4OCl; C 65.19, H 7.03, N 14.48, found C 65.29, H 7.34, N 14.48%.
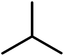
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-isopropylacetamide (A24). Yield 42% colorless solid m.p. 111–114 °C; H1 NMR (CDCl3) δppm: 8.72 (d, 1H, J = 5.1 Hz), 8.04 (s, 1H), 7.93 (d, 1H, J = 9.0 Hz), 7.43–7.40 (d, 1H, J = 7.2 Hz), 6.93 (bs, 1H), 6.85 (d, 1H, J = 4.8 Hz), 4.19–4.07 (m, 1H), 3.26 (bs, 4H), 3.12 (ss, 2H), 2.83–2.82 (m, 4H), 1.25–1.18 (m, 6H); 13C NMR (CDCl3) δppm: 167.58, 151.92, 150.18, 134.00, 131.52, 128.14, 126.80, 125.24, 121.98, 109.34, 62.61, 61.02, 52.94, 52.25, 52.11, 51.40, 45.67, 41.88; IR (νmax) cm−1: 1642, 1574. Calc. for C18H23N4OCl; 62.33, H 6.68, N 16.15.70, found C 62.30, H 6.48, N 16.35%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-cyclopropylacetamide (A25). Yield 50% colorless solid m.p. 101–104 °C; H1 NMR (CDCl3) δppm: 8.73 (d, 1H, J = 5.4 Hz), 8.15 (bs, 1H), 8.06 (d, 1H, J = 9.0 Hz), 7.95 (d, 1H, J = 8.4 Hz), 7.40 (d, 1H, J = 9.0 Hz), 6.84 (s, 1H), 3.95 (s, 2H), 3.68–3.66 (m, 1H), 3.21–3.08 (m, 8H), 2.18–2.02 (m, 4H); 13C NMR (CDCl3) δppm: 167.67, 157.23, 151.88, 150.08, 134.92, 129.04, 128.78, 126.18, 121.88, 71.78, 53.11, 45.78, 40.00, 19.13; IR (νmax) cm−1: 1662, 1568; ESI-MS: m/z 345.75 (M + 1); anal. calc. for C18H21N4OCl; C 65.32, H 5.48, N 13.25, found C 65.40, H 5.47, N 13.70%.
2-(4-(7-Chloroquinolin-4-yl)piperazine-1-yl)-N-cyclopentylacetamide (A26). Yield 45% white solid m.p. 108–111 °C; H1 NMR (CDCl3) δppm: 8.71 (d, 1H, J = 5.1 Hz), 8.02–7.97 (m, 2H), 7.72–7.65 (m, 2H), 7.56 (d, 1H, J = 7.2 Hz), 3.21 (bs, 4H), 3.02 (s, 2H), 2.72 (bs, 4H), 1.81–1.77 (m, 1H), 1.63–1.61 (m, 2H), 1.56–1.50 (m, 2H), 1.43–1.39 (m, 4H); 13C NMR (CDCl3) δppm: 168.58, 155.01, 151.46, 150.28, 133.25, 127.62, 124.93, 123.65, 121.95, 110.54, 62.03, 53.95, 52.44, 48.52, 31.96, 28.94; IR (νmax) cm−1: 1648, 1574; ESI-MS: m/z 373.51 (M + 1); anal. calc. for C20H25N4OCl; C 64.42, H 6.76, N 15.02, found C 64.41, H 6.67, N 15.00%.
N-Butyl-2-(4-(7-chloroquinolin-4-yl)piperazine-1-yl)acetamide (A27). Yield 48% colorless solid m.p. 128–131 °C; H1 NMR (CDCl3) δppm: 8.71 (d, 1H, J = 4.8 Hz), 8.03–7.97 (m, 2H), 7.77 (bs, 1H), 7.57 (d, 1H, J = 9.0 Hz), 7.02 (d, 1H, J = 4.8 Hz), 3.23 (bs, 4H), 3.14–3.07 (m, 2H), 3.03 (s, 2H), 2.72 (bs, 4H), 1.44–1.39 (m, 2H), 1.31–1.23 (m, 2H), 0.089–0.085 (m, 3H); 13C NMR (CDCl3) δppm: 169.49, 156.61, 151.85, 150.11, 134.94, 128.86, 126.28, 125.05, 121.82, 109.03, 61.59, 53.41, 52.20, 38.67, 31.76, 20.12, 13.74; IR (νmax) cm−1: 1662, 1571; ESI-MS: m/z 361.19 (M + 1); anal. calc. for C19H25N4OCl; C 63.24, H 6.98, N 15.52, found C 63.04, H 6.89, N 15.32%.
4. X-ray crystal structure determination
Three-dimensional X-ray data were collected on a Bruker Kappa Apex CCD diffractometer at low temperature for A7, A8, A12 and A21 by the ϕ–ω scan method. Reflections were measured from a hemisphere of data collected from frames each of them covering 0.3° in ω. Of the 31230 for A7, 45033 for A8, 58473 for A12 and 36222 for A21 reflections measured, all were corrected for Lorentz and polarization effects and for absorption by multi-scan methods based on symmetry-equivalent and repeated reflections, 5880, 3547, 4420 and 6536, respectively, independent reflections exceeded the significance level (∣F∣/σ∣F∣) > 4.0. Complex scattering factors were taken from the program package SHELXTL.41 The structures were solved by direct methods and refined by full matrix least-squares on F2. In A7, hydrogen atoms were included in calculation positions and refined in the riding mode, except for O (1W), which were located in difference Fourier map and fixed to oxygen atom. In A8, A12 and A21, hydrogen atoms were located in difference Fourier map and left to refine freely. Refinements were done with allowance for thermal anisotropy of all non-hydrogen atoms. Further details of the crystal structure determination are given in Table 1. A final difference Fourier map showed no residual density outside: 0.492 and −0.315 e Å−3 for A7 and 0.301 and −0.319 e Å−3 for A8, 0.352 and −0.304 e Å−3 for A12 and 0.495 and −0.710 e Å−3 for A21. A weighting scheme w = 1/[σ2(Fo2) + (0.059000P)2 + 0.058800P] for A7, w = 1/[σ2(Fo2) + (0.043600P)2 + 1.793400P] for A8, w = 1/[σ2(Fo2) + (0.050300P)2 + 0.339900P] for A12 and w = 1/[σ2(Fo2) + (0.051800P)2 + 0.625000P] for A21, where P = (|Fo|2 + 2|Fc|2)/3, were used in the latter stages of refinement. CCDC 1007368-1007370 and 1036519 contains the ESI† crystallographic data for the structure reported in this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223 336 033; or e-mail: E-mail: deposit@ccdc.cam.ac.uk.
5. Pharmacological evaluation
5.1. Antiamoebic activity
All the compounds were screened in vitro for antiamoebic activity against HM1:IMSS strain of E. histolytica by micro dilution method. E. histolytica trophozoites were cultured in wells of 96-well microtiter plate by using Diamond TYIS-33 growth medium.42 The test compounds (1 mg) were dissolved in ethanol (50 mL, level at which no inhibition of amoeba occurs).13,43 The stock solutions of the compounds were prepared freshly before use at a concentration of 1 mg mL−1. Each plate included metronidazole as standard amoebicidal drug, negative control (culture medium plus amoeba) and a blank (culture medium only). All the compounds were used in triplicate concentrations. The parasite suspension was adjusted to 105 cells per mL by adding fresh medium and then distributed to a 96-wells microtiter plate (corning). Compounds were added to the respective wells and the plate was sealed, gassed for 10 min with nitrogen and then incubated at 37 °C for 72 h. After incubation, the amoeba growth was checked under microscope. The culture medium was removed by inverting the plate and shaking gently. Plate was then immediately washed with sodium chloride (0.9%) at 37 °C, dried at room temperature and the cells fixed with methanol and once dried, stained with aqueous eosin (0.5%) for 15 min. The optical density of the resulting solution in each well was determined at 490 nm in a micro plate reader. The percentage inhibition of parasite growth was calculated from the optical densities of the control and test compounds. A non-linear regression analysis was used to determine the best fitting line from which the IC50 values were estimated. Each compound was tested at least in two independent experiments.
5.2. In vitro antimalarial activity
The chloroquine-sensitive strain of P. falciparum (3D7) was maintained continuously in vitro in supplemented RPMI-1640 culture media at 37 °C and gassed with a mixture of 5% CO2, 3% O2 and 92% N2.44,45 The culture was synchronised in the ring stage with 5% D-sorbitol.46 To determine the antimalarial activity of the various derivatives, the synchronised ring-stage parasites were adjusted to a final parasitaemia of 0.5% and 1% haematocrit, to which serial (1:2) dilutions of each derivative and positive control, chloroquine was added. Negative controls included uninfected erythrocytes and drug-free parasitized erythrocytes.47 Following a 24 h incubation period, radiolabeled [3H]-hypoxanthine isotope (Amersham) at a concentration of 0.5 μCi per well was added to each well. The microtiter plate was then incubated for a further 24 h before the parasitic [3H]-DNA was harvested onto glass fibre filter mats and the β-radioactivity counted. The counts per minute were expressed as the parasite survival rate taking the uninfected erythrocyte and untreated parasitized controls into account. Chloroquine was used as the reference agent.
5.3. Inhibition of β-haematin formation
In order to determine if the preliminary mechanism of action of the chloroquinolin-4-yl)piperazine-1-yl] acetamide derivatives was similar to chloroquine, the ability of the compounds to inhibit the synthetic equivalent of haemozoin, β-haematin was determined.48 Derivatives were incubated along with the test/control compound, 1 mg ml−1 haemin solubilised in DMSO, and a 0.5 M acetate buffer. The latter was utilized to simulate the acidic conditions (pH 4.7) of the parasitic food vacuole. The plates were then incubated for 24 h at 37 °C before the β-haematin crystals were washed with DMSO to remove the unreacted haemin. The β-haematin crystals were then dissolved in 0.2 M NaOH solution and the absorbance read at 405 nm. Percentage inhibition of β-haematin crystal formation was determined using the appropriate negative controls. Chloroquine was used as the positive control.
5.4. Toxicity assays
Cell viability (MTT) assay. Human embryonic kidney epithelial (HEK-293) cells were maintained at 37 °C in a humidified environment with 5% CO2 as a monolayer in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% of fetal bovine serum, 100 IU ml−1 penicillin and 100 μg ml−1 streptomycin).49 A cell suspension (36000 cells per well) was incubated at 37 °C for 46 h with 50 μM compounds/positive control. A final concentration of less than 1% DMSO had no effect on the viability of the cells. Thereafter, 20 μl of MTT (5 mg ml−1 in phosphate buffered saline (pH 7.3)) was added to each well and incubated for a further 2 h. DMSO was used to dissolve the formazan crystals, and then quantified by reading the absorbance at 540 nm with a reference wavelength of 690 nm (Labsystems Multiskan RC). Percent cellular viability was determined using the appropriate controls, with methotrexate and camptothecin used as the positive controls.
Haemolysis assay. A suspension of fresh red blood cells was adjusted to a 1% haematocrit in supplemented RPMI-1640 culture media and incubated for 48 h along with 25 μl test compound/control (50 μM).17 The absorbance of the supernatant was read at 412 nm. The % haemolysis was calculated using a 0.2% Triton X-100 solution as the 100% haemolytic control. Chloroquine was used as the reference agent.
5.5. Statistical analysis
Each experiment was repeated, at least, in triplicate to calculate the average and standard deviation. The concentration required to elicit a 50% effect/inhibition was determined using GraphPad Prism® (version 5) using the log sigmoid dose–response curve (variable slope) equation.
6. Molecular docking
Docking has been used extensively to determine the interacting residues within the binding pocket of enzyme with the ligand.50–52 For undertaking docking analysis, the crystal structure of wild-type Plasmodium falciparum dihydrofolate reductase–thymidylate synthase (PDB id: 1J3I) co-crystallized with ligand (WR99210) was taken.53 To identify the residues that show binding with the crystallized analogues (A7, A8, A18 and A21) with PfDHFR Autodock 4.2 as in our previous study has been used.17a Docking simulations were performed using Lamarckian Genetic Algorithm (LGA) and the grid maps representing the ligand were calculated with Autogrid. The dimensions of the grid were 60 × 60 × 60 grid points with a spacing of 0.375 Å between the grid points and centered on the ligand (27.71, 6.508 and 58.3 coordinates). Docking was performed by creating an initial population of 150 individuals, maximum number of evaluation 2500000, maximum number of generations 27000, rate of gene mutation 0.02, cross-over rate 0.8 and the remaining parameters were set as default. 10 docking conformations (poses) were generated and the best docked conformation was selected based on the Autodock binding energy. Further, the hydrogen and hydrophobic interactions between ligand–receptor complexes were identified using Ligplot.54
7. Conclusions
The present study indicates that the compounds consisting of 4-aminochloroquinoline and acetamide linked via piperazine is a viable combination as it acts against both HM1:IMSS strain of E. histolytica and 3D7 strain of P. falciparum. Antiamoebic activity indicated that out of twenty seven compounds, eleven exhibited more potent activity than the standard drug metronidazole (IC50 = 1.8 μM). Although all compounds inhibited P. falciparum growth, compounds A25 and A22 showed the most promising results. But the safety profile of A25 indicated that it should be further investigated as a lead compound for further derivatisation to improve antimalarial activity and retain safety profile.
Acknowledgements
One of the authors AI is thankful to University Grant Commission, India for providing BSR meritorious fellowship. The Faculty of Health Sciences Research Office of the University of the Wit waterstrand for financial support. This work is based on the research supported in part by the National Research Foundation of South Africa for the grant no. 87669. Any opinion, finding and conclusion or recommendation expressed in this material is that of the author(s) and the NRF does not accept any liability in this regard.
Notes and references
- J. Thwing, J. Skarbinski, R. D. Newman, A. M. Barber, S. Mali, J. M. Roberts, L. Slutsker and P. M. Arguin, Surveil–Summ., 2007, vol. 56, p. 23 Search PubMed.
- WHO, World Malaria Report, 2013 Search PubMed.
- WHO Expert Committee on Malaria, WHO Technical Report Series 20th Report, 2000 Search PubMed.
- A. Kumar, S. B. Katiyar, A. Agarwal and P. M. S. Chauhan, Curr. Med. Chem., 2003, 10, 1137 CrossRef CAS.
- R. N. Price and F. Nosten, Drug Resist. Updates, 2001, 4, 187 CrossRef CAS PubMed.
- WHO, Guidelines for the Treatment of Malaria, 2010 Search PubMed.
- C. Smith Gueye, G. Newby, J. Hwang, A. A. Phillips, M. Whittaker, J. R. MacArthur, R. D. Gosling and R. G. A. Feachem, Malar. J., 2014, 13, 286 CrossRef PubMed.
- S. M. Taylor and J. J. Juliano, J. Infect. Dis., 2014, 210, 335 CrossRef PubMed.
- P. Garner and P. M. Graves, PLoS Med., 2005, 2, 105 CrossRef PubMed.
- N. C. Araújo, V. Barton, M. Jones, P. A. Stocks, S. A. Ward, J. Davies, P. G. Bray, A. E. Shone, M. L. Cristiano and P. M. O'Neill, Bioorg. Med. Chem. Lett., 2009, 19, 2038 CrossRef PubMed.
- O. Dechy-Cabaret, F. Benoit-Vical, A. Robert and B. Meunier, ChemBioChem, 2000, 1, 281 CrossRef CAS.
- M. L. M. Gonzales, L. F. Dans and E. G. Martinez, Cochrane Database Sys. Rev., 2009, 15, CD006085 Search PubMed.
- A. T. Keene, A. Harris, J. D. Phillipson and D. C. Warhurst, Planta Med., 1986, 52, 278 CrossRef PubMed.
- F. Mansour-Ghanaei, N. Dehbashi, K. Yazdanparast and A. Shafaghi, World J. Gastroenterol., 2003, 9, 1832 Search PubMed.
-
(a) D. S. Barnett and R. Kiplin Guy, Chem. Rev., 2014, 114, 11221 CrossRef CAS PubMed;
(b) M. Njoroge, N. M. Njuguna, P. Mutai, D. S. B. Ongarora, P. W. Smith and K. Chibale, Chem. Rev., 2014, 114, 11138 CrossRef CAS PubMed.
-
(a) F. Hayat, A. Salahuddin, J. Zargan and A. Azam, Eur. J. Med. Chem., 2010, 45, 6127 CrossRef CAS PubMed;
(b) F. Hayat, E. Moseley, A. Salahuddin, R. L. Van Zyl and A. Azam, Eur. J. Med. Chem., 2011, 46, 1897 CrossRef CAS PubMed.
-
(a) A. Salahuddin, A. Inam, R. L. Van Zyl, D. C. Heslop, C. T. Chen, F. Avecilla, S. M. Agarwal and A. Azam Bioorg, Med. Chem., 2013, 21, 3080 CrossRef CAS PubMed;
(b) A. Inam, S. M. Siddiqui, T. S. Macedo, D. R. M. Moriera, A. C. Lima Liete, M. B. P. Soares and A. Azam, Eur. J. Med. Chem., 2014, 75, 67–76 CrossRef CAS PubMed;
(c) C. Nava-Zuazo, S. E. Soto, J. G. Alvarez, I. L. Rivera, G. M. M. Salinas, S. S. Fernandez, M. J. Chan-Bacab, R. C. Rivera, R. Moo-Puc, G. M. Lopez and G. N. Vazquez, Bioorg. Med. Chem., 2010, 18, 6398–6403 CrossRef CAS PubMed.
-
(a) S. Andrews, S. J. Burgess, D. Skaalrud, J. X. Kelly and D.
H. Peyton, J. Med. Chem., 2010, 53, 916 CrossRef CAS PubMed;
(b) V. K. Agrawal and S. Sharma, Indian J. Chem., 1987, 26b, 550 CAS.
- M. Garcia-Viloca, D. G. Truhlar and J. Gao, Biochemistry, 2003, 42, 13558 CrossRef CAS PubMed.
- A. Gangjee, H. D. Jain and S. Kurup, Med. Chem., 2007, 7, 524 CAS.
-
(a) G. Rastelli, S. Pacchioni, W. Sirawaraporn, R. Sirawaraporn, M. D. Parenti and A. M. Ferrari, J. Med. Chem., 2003, 46, 2834 CrossRef CAS PubMed;
(b) S. Madapa, Z. Tusi, D. Sridhar, A. Kumar, M. I. Siddiqi, K. Srivastava, A. Rizvi, R. Tripathi, S. K. Puri, G. B. Shiva Keshava, P. K. Shukla and S. Batra, Bioorg. Med. Chem., 2009, 17, 203 CrossRef CAS PubMed.
-
(a) S. Cho, Y. Park, J. Kim, S. Lee, C. Ma, S. Song, W. Joo, J. R. Falck, M. Shiro, D. Dong-Soo Shin and Y. A. Yoon, J. Org. Chem., 2003, 68, 7918 CrossRef CAS PubMed;
(b) S. Ganguly and K. Somakala, J. Inst. Chem., 2007, 79, 33 Search PubMed.
- M. S. Balakrishna, P. P. George and J. T. Mague, J. Chem. Res., 2003, 576 CrossRef CAS.
- Z. Xiaohe, Q. Yu, Y. Hong, S. Xiuqing and Z. Rugang, Chem. Biol. Drug Des., 2010, 76, 330–339 Search PubMed.
- J. Qinggang, D. Yang, X. Wang, C. Chen, Q. Deng, G. Zhiqiang, L. Yuan, X. Yang and F. Liao, Bioorg. Med. Chem., 2014, 22, 3405–3413 CrossRef PubMed.
- M. E. Gonzalez-Rosende, T. Olivar, E. Castillo and J. Sepulveda-Arques, Arch. Pharm., 2008, 341, 690–695 CrossRef CAS PubMed.
- H. Behbehani and H. M. Ibrahim, Molecules, 2012, 17, 6362–6385 CrossRef CAS PubMed.
- R. Kumar, M. Kaur, M. S. Bahia and O. Silakari, Eur. J. Med. Chem., 2014, 80, 83–91 CrossRef CAS PubMed.
- T. R. K. Reddy, C. Li, X. Guo, P. M. Fischer and L. V. Dekker, Bioorg. Med. Chem., 2014, 22, 5378–5391 CrossRef CAS PubMed.
- N. P. Jain, C. D. Upasani, R. S. Kalkotwar and U. N. Jain, Res. J. Pharm., Biol. Chem. Sci., 2013, 3, 1470–1480 Search PubMed.
- C. Hong, W. Luo, D. Yao, Y.-B. Su, X. Zhang, R.-G. Tian and C.-J. Wang, Bioorg. Med. Chem., 2014, 22, 3213–3219 CrossRef CAS PubMed.
- M. Zhang, W. Zhu and Y. Li, Eur. J. Med. Chem., 2013, 62, 301–310 CrossRef CAS PubMed.
- C. Zhang, C. Tan, X. Zu, X. Zhai, F. Liu, B. Chu, X. Ma, Y. Chen, P. Gong and Y. Jiang, Eur. J. Med. Chem., 2011, 46, 1404–1414 CrossRef CAS PubMed.
- N. A. Colabufo, F. Berardi, R. Perrone, S. Rapposelli, M. Digiacomo, M. Vanni and A. Balsamo, J. Med. Chem., 2008, 51, 7602–7613 CrossRef CAS PubMed.
- S. J. Shuttleworth, D. Nasturica, C. Gervais, M. Arshad Siddiqui, R. F. Rando and N. Lee, Bioorg. Med. Chem. Lett., 2000, 10, 2501–2504 CrossRef CAS.
- D. A. Brown, P. S. Kharkar, I. Parrington, M. E. A. Reith and A. K. Dutta, J. Med. Chem., 2008, 51, 7806–7819 CrossRef CAS PubMed.
- P. Wessig and K. Möllnitz, J. Org. Chem., 2008, 73, 4452–4457 CrossRef CAS PubMed.
- C.-F. Wu, X. Zhao, W.-X. Lan, C. Cao, J.-T. Liu, Xi-K. Jiang and Z.-T. Li, J. Org. Chem., 2012, 77, 4261–4270 CrossRef CAS PubMed.
- D. L. Musso, F. R. Cochran, J. L. Kelley, E. W. McLean, J. L. Selph, G. C. Rigdon, G. F. Orr, R. G. Davis, B. R. Cooper, V. L. Styles, J. B. Thompson and W. R. Hall, J. Med. Chem., 2003, 46, 3–6 Search PubMed.
- C. Jöst, C. Nitsche, T. Scholz, L. Roux and C. D. Kle, J. Med. Chem., 2014, 57, 7590–7599 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-97: An Integrated System for Solving and Refining Crystal Structures from Diffraction Data (Revision 5.1), University of Göttingen, Germany, 1997 Search PubMed.
- L. S. Diamond, D. R. Harlow and C. Cunnick, Trans. R. Soc. Trop. Med. Hyg., 1978, 72, 431 CrossRef CAS.
- F. D. Gillin, D. S. Reiner and M. Suffness, Antimicrob. Agents Chemother., 1982, 22, 342 CrossRef CAS.
- J. B. Jensen and W. Trager, Science, 1976, 193, 673 Search PubMed.
- R. L. Van Zyl, S. T. Seatlholo and A. M. Viljoen, S. Afr. J. Bot., 2010, 76, 667 Search PubMed.
- C. Lambros and J. P. Vanderberg, J. Parasitol., 1979, 65, 418 CrossRef CAS.
- R. E. Desjardins, C. J. Canfield, J. D. Haynes and J. D. Chulay, Antimicrob. Agents Chemother., 1979, 16, 710 CrossRef CAS.
- S. M. Chemaly, C.-T. Chen and R. L. van Zyl, J. Inorg. Biochem., 2007, 101, 764 CrossRef CAS PubMed.
- T. J. Mosmann, Immunol. Methods, 1983, 65, 55 CrossRef CAS.
- I. S. Yadav, P. P. Nandekar, S. Srivastavaa, A. Sangamwar, A. Chaudhury and S. M. Agarwal, Gene, 2014, 539, 82 CrossRef CAS PubMed.
- A. Salahuddin, S. M. Agarwal, F. Avecillac and A. Azam, Bioorg. Med. Chem. Lett., 2012, 22, 5694 CrossRef CAS PubMed.
- S. M. Agarwal, R. Jain, A. Bhattacharya and A. Azam, Int. J. Parasitol., 2008, 38, 137 CrossRef CAS PubMed.
- J. Yuvaniyama, P. Chitnumsub, S. Kamchonwongpaisan, J. Vanichtanankul, W. Sirawaraporn, P. Taylor, M. D. Walkinshaw and Y. Yuthavong, Nat. Struct. Biol., 2003, 10, 357 CrossRef CAS PubMed.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., 1995, 8, 127 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.