DOI:
10.1039/C5RA02750K
(Paper)
RSC Adv., 2015,
5, 48983-48991
Synthesis of g-C3N4/Bi2O3/TiO2 composite nanotubes: enhanced activity under visible light irradiation and improved photoelectrochemical activity†
Received
12th February 2015
, Accepted 26th May 2015
First published on 26th May 2015
Abstract
g-C3N4 and Bi2O3 were successfully incorporated into TiO2 nanotubes (TiO2-NT) to produce a composite nanotube material designated as g-C3N4/Bi2O3/TiO2. The photoelectrochemical (PEC) activity under visible light irradiation with respect to bleaching of methylene blue (MB) and degradation of phenol were determined. The UV-vis absorption spectrum of the composite material, g-C3N4/Bi2O3/TiO2-NT, was red-shifted toward a narrower band gap energy (Eg). The valence band (VB) of g-C3N4/Bi2O3/TiO2-NT was shifted to a more positive potential resulting in an increased driving force for water oxidation. The photocurrent generated by g-C3N4/Bi2O3/TiO2-NTs was approximately 15 times higher and the incident photon-to-current efficiency (IPCE at λ = 400 nm) was higher than that of the naked TiO2-NTs. This response is attributed to increasing the lifetimes of photo-generated electron–hole pairs. A significantly higher PEC response in terms of the bleaching of MB was observed on g-C3N4/Bi2O3/TiO2-NT. The roles of improved charge separation and subsequent higher electron-transfer efficiencies for in g-C3N4/Bi2O3/TiO2-NT electrodes were examined.
1. Introduction
Semiconductor photocatalysis has been extensively explored for a variety of environmental control applications1–3 and for solar energy conversion in terms of photocatalytic water-splitting4,5 and artificial photosynthesis including CO2 reduction to hydrocarbon fuels.6 Photoelectrochemical (PEC) activity under an applied potential bias was applied for splitting water or degradation of environmental pollutants. Since 43% of incoming solar radiation is within the visible region, improving solar conversion efficiency over this region is important.7 For large-scale applications, sustained and in-depth attention has been paid to TiO2 because the cost is low, stability is excellent and band edge potential is suitable.8,9 However, since UV light (4% of the solar spectrum) at λ ≤ 385 nm is required to activate anatase TiO2, the potential practical applications are limited.10 The modified TiO2 has been investigated to improve the photocatalytic activity induced by visible light. For instance, doping TiO2 with metal ions11,12 or nonmetal ions13,14 have been employed. Moreover, some non-oxide photocatalysts (e.g., metal sulfides, (oxy) nitrides and oxysulfides)15,16 and various metal complexes (e.g., Pt, Au or Ag)17 are quite active under visible light irradiation. However, rapid electron–hole recombination, reduced stability, and high costs of production of the doped metal oxides prevent their larger-scale practical applications.
Recently, compositing with two or more different substances together on TiO2 has drawn increasing attention to researchers and been confirmed to be an effective approach. Polypyrrole-decorated Ag–TiO2 nanofibers,18 Bi2O3/Bi4Ti3O12/TiO2 nanobelts,19 Bi2O3/TiO2/graphene composite systems20 have shown promise for achieving higher visible light photocatalytic activity. In addition, electrochemistry improving the photocatalysis system also has been developed. N and S co-doped into TiO2 nanotube array films21 and B and P co-doped TiO2 nanotube arrays22 exhibited excellent PEC properties and photocatalytic activities than undoped TiO2.
Recently, graphitic carbon nitride (g-C3N4) was studied having capable of oxidation of pollutants under visible light irradiation.23,24 Moreover, g-C3N4 has high chemical and thermal stability with narrower band gap energy (Eg) of 2.7 eV (ref. 23) than TiO2. Other researchers have synthesized TiO2 modified with g-C3N4 by chemical vapor deposition25 or electrodeposition methods26 to obtain materials that were photoelectrocatalytically active under visible light illumination. Bi2O3 is also being explored as a potentially useful photocatalyst due to its lower bandgap energy (2.8 eV) and beneficial electrical properties.27 Bi2O3 has also been combined with TiO2 for improved visible light activity.28,29 A multi-material composite incorporating g-C3N4 and Bi2O3 into titanium dioxide nanotubes was prepared and tested in our paper attempting to improve both the photocatalytic and photoelectrochemical activities under visible light irradiation.
Modification of TiO2 nanotubes (TiO2-NT) via incorporation of g-C3N4 and Bi2O3 is expected to improve the overall visible light activity when compared to naked TiO2. Compared to the VB edge of TiO2 (2.7 V vs. NHE (pH = 7)),30 the band edge of g-C3N4 is more negative (1.4 V vs. NHE (pH = 7)),30 while the band edge of Bi2O3 is more positive (3.13 V vs. NHE (pH = 7)).31 This combination should improve overall electron–hole separation and improve electron transfer characteristics.
In order to prepare the composite catalyst, we employed a thermal poly-condensation and dip-coating method to modify TiO2-NTs with g-C3N4 and Bi2O3. The degradation of methylene blue (MB) and phenol by PEC performance was investigated using a composite catalyst as anode. The PEC performances of the composite electrodes were examined and the photocatalytic (PC) and electrocatalytic (EC) activities under identical reaction conditions were also compared. A mechanism to account for enhanced PEC activity was finally proposed.
2. Experimental methods
2.1. Chemical reagents
Oxalic acid dehydrate ((COOH)2·2H2O) (≥99.5% purity), ammonium fluoride (NH4F) (≥96.0% purity), melamine (C3H6N6) (≥98.5% purity), bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) (≥99.0% purity), methyl alcohol (CH3OH) (≥99.9% purity, HPLC, TEDIA company), ethylene glycol (C2H6O2) (≥99.5% purity), sodium sulfate (Na2SO4) (≥99.0% purity), sodium sulfite (Na2SO3) (≥97.0% purity) were all purchased from Hangzhou Huipu Chemical Reagent Co., Ltd.
2.2. Synthesis of g-C3N4/Bi2O3/TiO2 nanotubes
Electrochemical anodization method was used to prepare TiO2-NTs electrodes. The Ti sheet (0.5 mm thick, 99.5% purity) was washed by ultrasonic irradiation for 15 min. The electrochemical reaction was operated using Ti sheet as anode and Ni sheet as cathode with the voltage of 20 V for 120 min. The electrolyte was a mixed solution of 1/12 M (COOH)2·2H2O and 0.5 wt% NH4F.32,33 Metal-free g-C3N4 powders were prepared by calcination of melamine to 520 °C for 4 h.34
The g-C3N4/Bi2O3/TiO2-NTs composites were prepared by a dip-coating method in which 0.2 g g-C3N4 and 0.243 g Bi(NO3)3·5H2O were dispersed in 20 mL of ethylene glycol solution; the TiO2-NTs that were still attached to the Ti sheet were slowly dipped up and down from the above solution for 30 min. The dipped coated sheets were then annealed at 400 °C for 2 h.33 When g-C3N4/TiO2-NTs or Bi2O3/TiO2-NTs were prepared, the precursor solution was changed to 0.2 g g-C3N4 or 0.243 g Bi(NO3)3·5H2O in 20 mL of ethylene glycol solution.
2.3. Characterization of the synthesized g-C3N4/Bi2O3/TiO2 nanotubes
The synthesized materials (TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs) were characterized by X-ray diffractometer (XRD), X-ray photoelectron spectroscope (XPS), emission scanning electron microscope (FE-SEM, Hitachi S-4700 II) with an energy-dispersive X-ray spectrometer (EDX), transmission electron microscope (TEM) and UV-vis diffuse reflectance spectra (DRS).
2.4. PEC experimental techniques
The PEC properties of each sample were determined using an electrochemical workstation (CHI 660D). Modified TiO2-NTs electrodes were used as working electrodes with saturated Ag/AgCl electrode as a reference electrode and Pt sheet as a counter electrode, respectively. Visible light at an intensity of 100 mW cm−2 was obtained by using a Xe-lamp light source with UV filter (λ > 420 nm). Photocurrents and incident-photon-to-current efficiencies (IPCE) were tested in mixed 0.1 M Na2SO4 and Na2SO3 solutions at pH of 10.5. IPCE measurements were taken at the visible wavelengths of 400, 430, 450, 475, 500, 550, 600 and 633 nm with a light intensity meter (model FZ-A). Mott–Schottky (M–S) plots and Nyquist plots were taken in 0.1 M Na2SO4 solutions at pH = 7.6. M–S plots were obtained at frequencies of 500, 1000 and 3000 Hz at an AC amplitude of 5 mV. Nyquist plots were obtained both under dark and visible light conditions. The experiments including Nyquist plots, LSVs and IPCE plots were repeated for 3 times.
The activity of the prepared catalyst was assessed in three different modes of operation that include (1) photocatalytic (PC), (2) electrocatalytic (EC) and photoelectrocatalytic (PEC). The applied potential in EC and PEC process was 3.0 V. In each case, an electrolyte solution consisting of 0.1 M Na2SO4 was irradiated with visible light that had an average light intensity of 200 mW cm−2. MB (100 mL, 1 × 10−5 M, without regulating pH) and phenol (100 mL, 10 mg L−1, pH = 3) fitted with an active electrode with a 4.5 cm2 effective surface area of g-C3N4/Bi2O3/TiO2-NTs. Before initiating the reactions under illumination, the solution reacted with catalyst electrode for 30 min without light. At given time intervals, 3 mL MB and 1 mL phenol aliquot samples were collected. The variation in the MB concentrations was recorded using an UV-vis spectrophotometer. The concentrations of phenol were analyzed by HPLC with a Diamonsil C18 column and ultraviolet detection at 278 nm with ratio of the deionized water and methanol of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v) and a flow rate of 1.0 mL min−1. The overall MB bleaching process and phenol degradation was fitted to pseudo first-order kinetics.
:4 (v/v) and a flow rate of 1.0 mL min−1. The overall MB bleaching process and phenol degradation was fitted to pseudo first-order kinetics.
3. Results and discussion
3.1. SEM and TEM analyses
Fig. 1 shows the morphologies of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs, g-C3N4/Bi2O3/TiO2-NTs, and pure g-C3N4. The TiO2-NTs electrode has almost uniform holes and a highly ordered tubular structure. The average of the inner diameter of TiO2-NTs is close to 80 nm, and their average of outer diameter is near 110 nm. After modification, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs, g-C3N4/Bi2O3/TiO2-NTs have very similar tubular structures. The g-C3N4 powders are likely sheet structures, as shown in Fig. 1(e), accumulating on the surface of g-C3N4/TiO2-NTs. The cross section of the TiO2 nanotubes was depicted in Fig. 1(f) showing that the mean length of the TiO2 nanotubes was about 850 nm. Moreover, the EDX analysis (Fig. S1 in ESI†) confirms that Ti, O, C, N, Bi exist in composite material and shows that g-C3N4 and Bi2O3 are deposited on TiO2 tubular substrates.
 |
| | Fig. 1 SEM images of TiO2-NTs (a), g-C3N4/TiO2-NTs (b), Bi2O3/TiO2-NTs (c), g-C3N4/Bi2O3/TiO2-NTs (d), g-C3N4 powders (e) and cross section of the TiO2 nanotubes (f). | |
Fig. 2 shows the TEM analysis for g-C3N4/Bi2O3/TiO2 composite. The particles also detected in g-C3N4/TiO2 and Bi2O3/TiO2 composites, so g-C3N4 and Bi2O3 could be found in the nanotubes. And combined with SEM in Fig. 1, the g-C3N4 powders also accumulate on the surface of g-C3N4/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs composites, but little Bi2O3 was accumulated on the surface of Bi2O3/TiO2-NTs. It was probably due to that Bi2O3 was prepared by anneal of Bi(NO3)3·5H2O, which may permeate into the tube of TiO2 and Bi2O3 was formed. But g-C3N4 was prepared at first and was like the sheet structures, so some small sheets were deposited into the tubes of TiO2 and some were accumulated on the surfaces of the tubes.
 |
| | Fig. 2 TEM analysis for g-C3N4/TiO2 (a), Bi2O3/TiO2 (b) and different magnifications of g-C3N4/Bi2O3/TiO2 (c and d) composites. | |
3.2. XRD and XPS analyses
The XRD analysis was applied to detect the formation of TiO2, g-C3N4 and Bi2O3 in the composite electrode. However, due to the very low level of incorporated Bi2O3, the characteristic peak of Bi2O3 in g-C3N4/Bi2O3/TiO2-NTs was not seen. In another synthesis the amount of Bi(NO3)3·5H2O was increased to 2.910 g in 20 mL of ethylene glycol, which was then dipped coated and annealed to get the fully loaded g-C3N4/Bi2O3/TiO2-NTs. Fig. 3 shows the XRD patterns for the TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs electrodes that were annealed at 400 °C. The peak of 2θ = 25.2° for anatase TiO2 can be seen in both of the XRD patterns. The characteristic peaks corresponding to g-C3N4, Bi2O3 are also evident in Fig. 3, which confirms that g-C3N4, Bi2O3 had been successfully synthesized.
 |
| | Fig. 3 XRD patterns of TiO2-NTs (a) and g-C3N4/Bi2O3/TiO2-NTs (b). | |
In addition, the XPS analysis of g-C3N4/Bi2O3/TiO2-NTs electrode is also depicted in Fig. 4, which showed the existence of Ti 2p, O 1s, C 1s, N 1s and Bi 4f elements. In the inset figure of Fig. 3, two peaks centered at 164.3 and 158.9 eV belong to Bi 4f5/2 and Bi 4f7/2 region, which should be determined as Bi2O3 species.35 While, near the main peaks due to Bi3+, there are other two small peaks centered at 157.2 eV and 162.6 eV, which probably belong to reduced Bi oxidation phase or Bi0.36,37 Based on several studies about the reduced Bi phase, it could interact with the structure of Bi oxidation and titania to form the electronic effect between these two oxides.36,38
 |
| | Fig. 4 XPS spectra of g-C3N4/Bi2O3/TiO2-NTs. | |
3.3. UV-vis DRS analyses
UV-vis DRS of the composite TiO2-NTs electrodes modified by g-C3N4 and Bi2O3 are shown in Fig. 5. The spectrum of g-C3N4/TiO2-NTs is similar to that of unmodified TiO2-NTs from 220 to 600 nm. While from 220 to 370 nm, the spectrum shows Bi2O3/TiO2-NTs has a lower absorbance when compared to the unmodified TiO2-NTs, while g-C3N4/Bi2O3/TiO2-NTs showed an increase in absorbance compared to naked TiO2. When wavelength is larger than 370 nm, however, the absorbance of Bi2O3/TiO2-NTs, g-C3N4/Bi2O3/TiO2-NTs are higher than that of the naked TiO2-NTs. In the case of g-C3N4/Bi2O3/TiO2-NTs electrode, the absorbance is significantly higher than for the other electrodes in UV-visible region. All of the samples have some degree of visible light absorbance (λ > 400 nm), however this may be due, in part, to the roughness of the electrode surfaces with pores.39–41
 |
| | Fig. 5 UV-vis DRS of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs. | |
From Fig. 5, the absorption edges of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs are located approximately at 382 nm, 388 nm, 400 nm, and 410 nm, indicating that addition of g-C3N4 and Bi2O3 to the nanotube array extends the absorption to visible range. The corresponding band gap energies were determined using the standard equation as shown belong in eqn (1):42
where
α,
h,
ν,
Eg and
A are the absorption coefficient, Planck's constant, light frequency, band gap energy, and a constant, respectively. For TiO
2,
n is 4 for the indirect transition.
24 Thus, plots of (
αhν)
1/2 versus photon energy (
hν) are obtained. As shown in Fig. S2,
† the
Eg of TiO
2-NTs is 3.25 eV, which is similar to anatase TiO
2,
43 while the
Eg values for g-C
3N
4/TiO
2-NTs, Bi
2O
3/TiO
2-NTs and g-C
3N
4/Bi
2O
3/TiO
2-NTs are 3.20 eV, 3.10 eV and 3.02 eV, respectively. The UV-vis DRS analysis was detected three times and the
Eg was changed a little. It was probably that there was a small amount of Bi
2O
3 and C
3N
4 were incorporated with TiO
2 using a dip-coating method, which leads the red shift not significant.
44,45
3.4. Nyquist and M–S plots
In order to investigate the capacitance and resistance of the electrodes, Nyquist plots of the TiO2-NTs electrodes as modified by g-C3N4 and Bi2O3 are shown in Fig. 6. Only one arc was observed for each electrode while the arc radius of g-C3N4/Bi2O3/TiO2-NTs was the smallest in dark and visible light irradiation. It implies that interfacial charge transfer is enhanced by a higher efficiency of charge separation.
 |
| | Fig. 6 Nyquist plots of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTsin 0.1 M Na2SO4 aqueous solution (pH 7.6) in dark (a) and visible light (b) conditions with a frequency range of 10−2 to 105 Hz and a scan rate of 5 mV s−1. Light intensity: 100 mW cm−2. | |
The M–S plots, as shown in Fig. 7, reveal the flat band potential (Efb) and interfacial charge transfer of the electrode, in which Csc2 means the space charge capacitance of the electrode.46,47 The Efb shifts positively from ca. −0.75 V for TiO2-NTs to −0.40 V for g-C3N4/Bi2O3/TiO2-NTs. Similarly, the onset potential of anodic photocurrent shifts positively from ca. −0.54 V for TiO2-NTs to −0.35 V for g-C3N4/Bi2O3/TiO2-NTs as shown in Fig. S3.† In both of samples, photocurrent onset potentials are more positive than the Efb. It is probably due to the recombination of electron–hole more quickly for water oxidation, so more positive potentials are achieved for transmission of charge.48
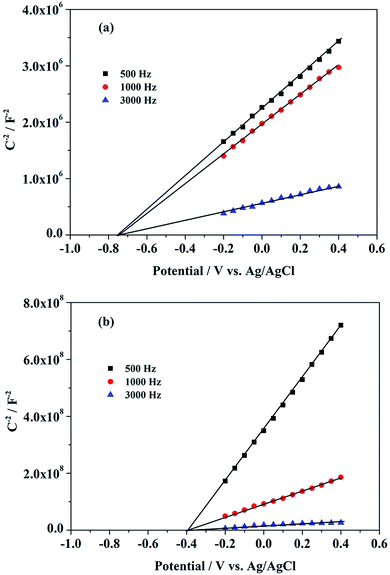 |
| | Fig. 7 M–S plots of the TiO2-NTs (a) and g-C3N4/Bi2O3/TiO2-NTs (b) electrodes in 0.1 M Na2SO4 aqueous solution (pH 7.6) under dark condition with an AC amplitude of 5 mV at each potential. | |
The g-C3N4/Bi2O3/TiO2-NTs electrode has the highest absorbance and narrowest Eg, as shown in Fig. 5. Combined with Efb and Eg,32 the VB edge of TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs electrode is calculated at 2.50 V and 2.62 V, respectively. As a result, the improvement of water oxidation ability of g-C3N4/Bi2O3/TiO2-NTs was achieved for the enhancing of PEC properties.49
3.5. PEC properties
Linear sweep voltammetry (LSV) method was used to analyze the PEC activities of the composite TiO2-NTs electrodes. In comparison, LSVs without irradiation were also tested, which shows the surface catalytic property of electrodes for O2 evolution.50 From Fig. 8, most of the composite TiO2-NTs electrodes show a current response until the bias potential is at least 1.0 V without irradiation. The g-C3N4/Bi2O3/TiO2-NTs electrode shows a positive shift of the onset potential (Fig. S3†) implying that the surface became easier for O2 evolution than the un-modified TiO2-NTs electrodes surface. Moreover, it indicates that the composite electrodes reduced the charge transfer resistance,50 which is consistent with result of EIS Nyquist analysis in Fig. 6.
 |
| | Fig. 8 LSVs of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs in 0.1 M Na2SO4 and Na2SO3 mixed aqueous solution (pH 10.5) under dark condition. Scan rate: 10 mV s−1. | |
Fig. 9 shows LSVs of the composite TiO2-NTs electrodes under visible light irradiation in 0.1 M Na2SO4 and Na2SO3 mixture solution. The photocurrent of g-C3N4/Bi2O3/TiO2-NTs electrode is ca. 15 times larger than that of TiO2-NTs. Bi2O3/TiO2-NTs electrode has a lightly lower photocurrent compared to g-C3N4/Bi2O3/TiO2-NTs, but its photocurrent is still over 8 times larger than that of TiO2-NTs. The recombination of photogenerated electron–hole decrease with the bias potential increase, and the driving force for photo-generated electrons transferring to the external circuit enhanced. The enhancement of photo-response of g-C3N4/Bi2O3/TiO2-NTs electrode under visible light irradiation is probably due to: (1) the higher absorbance and the wider absorption edge; (2) the improved driving force for O2 production; (3) the improved separation of photo-generated electron–hole pairs.
 |
| | Fig. 9 LSVs of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs in 0.1 M Na2SO4 and Na2SO3 mixed aqueous solution (pH 10.5) under chopped visible light (λ > 420 nm) irradiation. Scan rate: 10 mV s−1. Light intensity: 100 mW cm−2. | |
3.6. IPCE
Fig. 10 shows the IPCE plots as function of wavelength for the composite TiO2-NTs electrodes calculated by eqn (2):51,52| | |
IPCE (%) = 1240 × (iph/λPin) × 100
| (2) |
where iph is the photocurrent at 0.40 V (mA), λ is the wavelength (nm) of light, and Pin is the light power intensity at λ (mW). The IPCE values decreased with increasing wavelength from 400 to 633 nm. The g-C3N4/Bi2O3/TiO2-NTs electrodes show the highest IPCE at 7.59% at 400 nm, which amounts to an enhancement of ca. 55% when compared to the unmodified TiO2-NTs (4.9%).
 |
| | Fig. 10 IPCE plots of TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs and g-C3N4/Bi2O3/TiO2-NTs calculated from the photocurrent in 0.1 M Na2SO4 and Na2SO3 mixed aqueous solution (pH 10.5) at an applied potential of 0.40 V vs. Ag/AgCl. Light intensity: 100 mW cm−2. | |
Zhou's and co-workers22 have developed boron and phosphor co-doped TiO2 nanotube arrays (BP-TNTs) by an anodization process on a Ti sheet, achieving the highest IPCE value 3.8% at 400 nm. The same team also prepared a CN/TNT composite heterojunction photocatalyst, which had a maximum IPCE of 7.3% at 400 nm.26 Jia et al.53 have synthesized polypyrrole (PPy) onto self-organized TiO2 nanotube arrays (TiO2-NTs), and the PPy/TiO2-NTs electrode showed the maximum IPCE of 5% at 410 nm. In contrast, the g-C3N4/Bi2O3/TiO2-NTs electrode has higher IPCE in the visible.
3.7. Pollutants degradation
MB and phenol are degraded on the g-C3N4/Bi2O3/TiO2-NTs electrode in photolysis, EC, PC and PEC processes, respectively. Fig. 11 shows the removal efficiency of MB at 6.4%, 8.8%, 50.2% and 77.9% after 3 h of reaction in photolysis, EC, PC and PEC process. The corresponding apparent rate constants k = 0.029, 0.24 and 0.50 h−1 in EC, PC and PEC processes, respectively. The value of k for MB degradation in PEC process is roughly 1.9 times as high as that in (EC + PC) process, indicating that synergistic effect occurred during PEC process. Fig. S4† shows the absorbance spectra of MB degradation at g-C3N4/Bi2O3/TiO2-NTs electrode in PEC process. There are three absorption peaks at 292, 246 and 665 nm, in which peak at 665 nm belongs to the auxochrome group of MB and the peaks at 292 nm and 246 nm belong to the substituted benzenes ring structures.54,55 It can be see that all of the peaks decreased during the reaction, which implied that the MB was bleached and the benzenes ring structures were also decomposed. In addition, before 235 nm, the absorbance spectra of MB increased with time increasing, which possibly shows that some substituted benzene derivatives or other intermediate products may be formed. The analysis of the intermediates needs further investigation.
 |
| | Fig. 11 Variation of [MB] concentration vs. time at the g-C3N4/Bi2O3/TiO2-NTs electrode in EC, PC and PEC processes. Applied potential: 3.0 V. Electrolyte: 0.1 M Na2SO4, pH = 7.6. | |
In addition, the colorless organic phenol also was degraded efficiently in PEC process by using g-C3N4/Bi2O3/TiO2-NTs electrode as shown in Fig. S5.† Fig. S6† shows HPLC of phenol degradation at the g-C3N4/Bi2O3/TiO2-NTs electrode in PEC process. Compared with the standard compounds, hydroquinone and benzoquinone were produced after degradation as depicted in Fig. S6.† It probably indicated that phenol was degraded to form hydroquinone and then transformed to benzoquinone, and then the benzene rings were opened to form small molecular acids and finally could be mineralized.
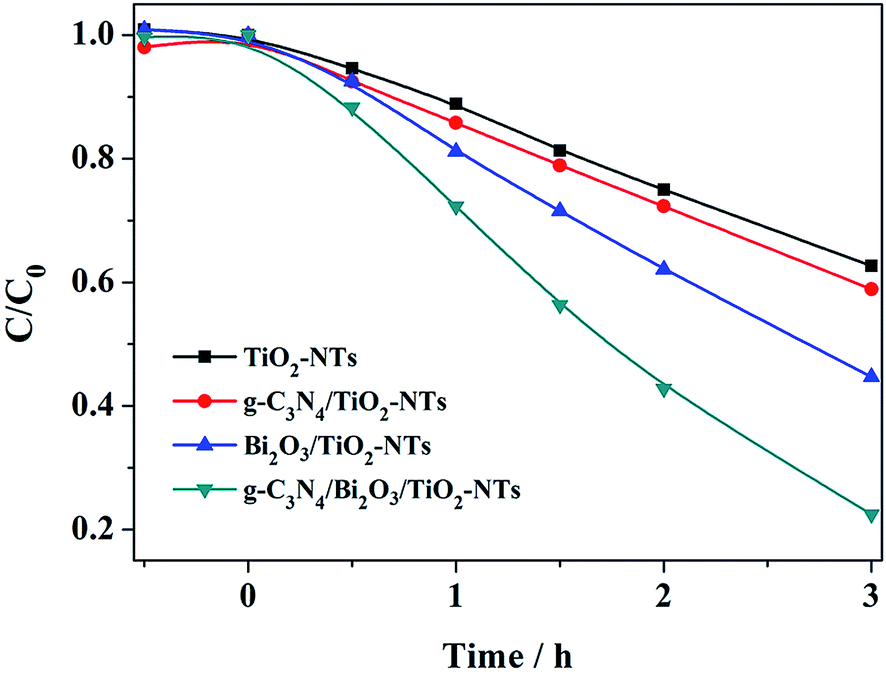
Fig. 12 shows the concentration change of MB with different modified TiO2-NTs electrodes in PEC process. Correspondingly, the removal efficiency of MB is 37.3%, 41.1%, 55.3% and 77.5% for TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs, and g-C3N4/Bi2O3/TiO2-NTs after 3 h of reaction. Hence, the g-C3N4/Bi2O3/TiO2-NTs electrode could contribute to the promotion of the PEC performance compared with TiO2-NTs electrode.
 |
| | Fig. 12 The variation of [MB] vs. time at the TiO2-NTs, g-C3N4/TiO2-NTs, Bi2O3/TiO2-NTs, g-C3N4/Bi2O3/TiO2-NTs electrodes in PEC process with visible light irradiation. Applied potential: 3.0 V. Electrolyte: 0.1 M Na2SO4, pH = 7.6. | |
To check the stability of g-C3N4/Bi2O3/TiO2-NTs electrode, MB degradation using the PEC process was repeated for four times. As shown in Fig. 13, the degradation reaction is repeatable through four PEC reaction cycles. This indicates that g-C3N4/Bi2O3/TiO2-NTs electrode is reasonably stable during PEC processing.
 |
| | Fig. 13 Stability of the g-C3N4/Bi2O3/TiO2-NTs electrode for PEC degradation of MB experiments with visible light irradiation at an applied potential of 3.0 V. Electrolyte: 0.1 M Na2SO4, pH = 7.6. | |
3.8. Possible considerations
Based on the above results, suggested mechanism for charge separation and electron transfer in g-C3N4/Bi2O3/TiO2-NTs electrode is depicted schematically in Fig. 14. The bottoms of the conduction band (CB) of TiO2, Bi2O3 and g-C3N4 are located at about −0.5 V, +0.33 V, −1.3 V vs. NHE (pH = 7), respectively.30,31 The Eg of Bi2O3 (2.8 eV) and g-C3N4 (2.7 eV) are smaller than that for TiO2 (3.2 eV). Thus, only Bi2O3 and g-C3N4 can absorb photons and excite photoelectrons under visible light irradiation. One of possible mechanisms is that TiO2 is placed between Bi2O3 and C3N4, which has advantage to electron–holes transfer. As shown in Fig. 14(a), with the accumulation of valence holes of Bi2O3, the photo-induced holes of Bi2O3 flow into the VB of the TiO2 layer and then into the VB of the g-C3N4. Holes on the g-C3N4 surface can also degrade MB, giving rise to enhance photocatalytic activity.56,57 Meanwhile, the electron injection is transferred from the CB of g-C3N4 into that of TiO2 and subsequently could be shuttled freely along g-C3N4/Bi2O3/TiO2-NTs matrix of the electrodes to the external circuit, enhancing the separation of electron–hole pairs. Moreover, photo-generated electrons of Bi2O3 can react with oxygen molecules to generate superoxide O2˙−, which leads to H2O2 production and eventual bleaching of MB. Even though the TiO2 interlayer isn't induced by visible light, it is used as a support and connector between Bi2O3 layers and g-C3N4 particles to improve the separation of electron–hole pairs. Key steps of the sequence of PEC reactions can be summarized as follows:| | |
Bi2O3 + hν → Bi2O3 (h+ + e−)
| (3) |
| | |
g-C3N4 + hν → g-C3N4 (h+ + e−)
| (4) |
| | |
Bi2O3 (h+) + TiO2 → TiO2 (h+) + Bi2O3
| (5) |
| | |
TiO2 (h+) + g-C3N4 → g-C3N4 (h+) + TiO2
| (6) |
| | |
g-C3N4 (e−) + TiO2 → TiO2 (e−) + g-C3N4
| (7) |
| | |
TiO2 (e−) + bias voltage → TiO2 + external circuit
| (8) |
| | |
Bi2O3 (e−) + O2 → Bi2O3 + O2˙−
| (9) |
 |
| | Fig. 14 One of suggested mechanism of charge separation and electron transfer in g-C3N4/Bi2O3/TiO2-NTs electrodes under visible light irradiation. | |
Of course, two of these three substances (TiO2, Bi2O3 and C3N4) composited together, the electron–holes also could be transferred from high energy band to low energy band. So some other phenomenon, such as electrons transferred from C3N4 to TiO2 or from TiO2 to C3N4, was described in Fig. 14(b).
4. Conclusion
g-C3N4 and Bi2O3 with TiO2-NTs have been coupled into a composite photocatalytic and electrocatalytic material through a sequential dip-coating procedure followed by high-temperature annealing. After adding g-C3N4 and Bi2O3, in to the host matrix, the absorption spectrum of g-C3N4/Bi2O3/TiO2-NTs electrode red-shifted in to visible region of electromagnetic spectrum, resulting in an increase in light absorbance. Compared to naked TiO2-NTs, photocurrent response of g-C3N4/Bi2O3/TiO2-NTs was enhanced by 15 times and PEC activity for pollutants degradation was also improved. The enhancement in the PEC and IPCE activities was most likely due to the narrowing of the effective Eg coupled with a positive shift of Efb (ca. 0.35 V). These results indicated that the g-C3N4/Bi2O3/TiO2-NTs composite electrodes have the potential for wastewater treatment during PEC process under visible light irradiation.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21276235, 21477114). In addition support was provided by the Educational Commission of Zhejiang Province (Y201432049).
References
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- H. Bao, F. F. Li, L. C. Lei, B. Yang and Z. J. Li, RSC Adv., 2014, 4, 27277–27280 RSC.
- A. J. Bard and M. A. Fox, Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. L. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 CAS.
- Q. Liu, D. Wu, Y. Zhou, H. B. Su, R. Wang, C. F. Zhang, S. C. Yan, M. Xiao and Z. G. Zou, ACS Appl. Mater. Interfaces, 2014, 6, 2356–2361 CAS.
- Q. Li, B. D. Guo, J. G. Yu, J. R. Ran, B. H. Zhang, H. J. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- G. Liu, L. Z. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- H. Yamashita, M. Harada, J. Misaka, M. Takeuchi, K. Ikeue and M. Anpo, J. Photochem. Photobiol. A: Chem., 2002, 148, 257–261 CrossRef CAS.
- J. C. Yu, G. S. Li, X. C. Wang, X. L. Hu, C. W. Leung and Z. D. Zhang, Chem. Commun., 2006, 25, 2717–2719 RSC.
- C. W. H. Dunnill, Z. A. Aiken, J. Pratten, M. Wilson, D. J. Morgan and I. P. Parkin, J. Photochem. Photobiol. A: Chem., 2009, 207, 244–253 CrossRef CAS PubMed.
- Y. X. Zhao, X. F. Qiu and C. Burda, Chem. Mater., 2008, 20, 2629–2636 CrossRef CAS.
- Q. Kang, S. H. Liu, L. X. Yang, Q. Y. Cai and C. A. Grimes, ACS Appl. Mater. Interfaces, 2001, 3, 746–749 CrossRef PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- K. Mogyorósi, Á. Kmetykó, N. Czirbus, G. Veréb, P. Sipos and A. Dombi, React. Kinet. Catal. Lett., 2009, 98, 215–225 CrossRef.
- Y. C. Yang, J. W. Wen, J. H. Wei, R. Xiong, J. Shi and C. X. Pan, ACS Appl. Mater. Interfaces, 2013, 5, 6201–6207 CAS.
- Z. H. Zhao, J. Tian, D. Z. Wang, X. L. Kang, Y. H. Sang, H. Liu, J. Y. Wang, S. W. Chen, R. I. Boughton and H. D. Jiang, J. Mater. Chem., 2012, 22, 23395–23403 RSC.
- J. G. Hou, C. Yang, Z. Wang, S. Q. Jiao and H. M. Zhu, Appl. Catal., B, 2013, 129, 333–341 CrossRef CAS PubMed.
- G. T. Yan, M. Zhang, J. Hou and J. J. Yang, Mater. Chem. Phys., 2011, 129, 553–557 CrossRef CAS PubMed.
- X. S. Zhou, B. Jin, S. S. Zhang, H. J. Wang, H. Yu and F. Peng, Electrochem. Commun., 2012, 19, 127–130 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–82 CrossRef CAS PubMed.
- J. Y. Zhang, Y. H. Wang, J. Jin, J. Zhang, Z. Lin, F. Huang and J. G. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 10317–10324 CAS.
- J. Wang and W. D. Zhang, Electrochim. Acta, 2012, 71, 10–16 CrossRef CAS PubMed.
- X. S. Zhou, B. Jin, L. D. Li, F. Peng, H. J. Wang, H. Yu and Y. P. Fang, J. Mater. Chem., 2012, 22, 17900–17905 RSC.
- S. Shamaila, A. K. L. Sajjad, F. Chen and J. L. Zhang, Appl. Catal., B, 2010, 94, 272–280 CrossRef CAS PubMed.
- Y. Bessekhouad, D. Robert and J.-V. Weber, Catal. Today, 2005, 101, 315–321 CrossRef CAS PubMed.
- Z. F. Bian, J. Zhu, S. H. Wang, Y. Cao, X. F. Qian and H. X. Li, J. Phys. Chem. C, 2008, 112, 6258–6262 CAS.
- Y. J. Cui, Z. X. Ding, P. Liu, M. Antonietti, X. Z. Fu and X. C. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC.
- J. L. Hu, H. M. Li, C. J. Huang, M. Liu and X. Q. Qiu, Appl. Catal., B, 2013, 142–143, 598–603 CrossRef CAS PubMed.
- A. Ghicov, H. Tsuchiya, J. M. Macak and P. Schmuki, Electrochem. Commun., 2005, 7, 505–509 CrossRef CAS PubMed.
- Y. L. Su, S. Han, X. W. Zhang, X. Q. Chen and L. C. Lei, Mater. Chem. Phys., 2008, 110, 239–246 CrossRef CAS PubMed.
- S. S. Zhao, S. Chen, H. T. Yu and X. Quan, Sep. Purif. Technol., 2012, 99, 50–54 CrossRef CAS PubMed.
- K. H. Reddy, S. Martha and K. M. Parida, RSC Adv., 2012, 2, 9423–9436 RSC.
- M. N. Gómez-Cerezo, M. J. Mssũnoz-Batista, D. Tudelab, M. Fernández-García and A. Kubacka, Appl. Catal., B, 2014, 156–157, 307–313 CrossRef PubMed.
- K. Uchida and A. Ayame, Surf. Sci., 1996, 357, 170–178 CrossRef.
- A. Hameed, T. Montini, V. Gombac and P. Fornasiero, J. Am. Chem. Soc., 2008, 130, 9658–9659 CrossRef CAS PubMed.
- H. M. Zhu, B. F. Yang, J. Xu, Z. P. Fu, M. W. Wen, T. Guo, S. Q. Fu, J. Zuo and S. Y. Zhang, Appl. Catal., B, 2009, 90, 463–469 CrossRef CAS PubMed.
- Y. Zhang, J. N. Lu, X. P. Wang, Q. Xin, Y. Q. Cong, Q. Wang and C. J. Li, J. Colloid Interface Sci., 2013, 409, 104–111 CrossRef CAS PubMed.
- G. P. Dai, J. G. Yu and G. Liu, J. Phys. Chem. C, 2011, 115, 7339–7346 CAS.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS PubMed.
- S. Kumar, A. G. Fedorov and J. L. Gole, Appl. Catal., B, 2005, 57, 93–107 CrossRef CAS PubMed.
- Z. W. Tong, D. Yang, T. X. Xiao, Y. Tian and Z. Y Jiang, Chem. Eng. J., 2015, 260, 117–125 CrossRef CAS PubMed.
- M. N. Gómez-Cerezoa, M. J. Mũnoz-Batista, D. Tudela, M. Fernández-García and A. Kubacka, Appl. Catal., B, 2014, 156–157, 307–313 CrossRef PubMed.
- W. D. Zhang, L. C. Jiang and J. S. Ye, J. Phys. Chem. C, 2009, 113, 16247–16253 CAS.
- Y. Liu, Y. X. Yu and W. D. Zhang, Electrochim. Acta, 2012, 59, 121–127 CrossRef PubMed.
- S. D. Tilley, M. Cornuz, K. Sivula and M. Gratzel, Angew. Chem., Int. Ed., 2010, 49, 6405–6408 CrossRef CAS PubMed.
- Y. Q. Cong, M. M. Chen, T. Xu, Y. Zhang and Q. Wang, Appl. Catal., B, 2014, 147, 733–740 CrossRef CAS PubMed.
- R. L. Spray, K. J. McDonald and K.-S. Choi, J. Phys. Chem. C, 2011, 115, 3497–3506 CAS.
- Y. Q. Cong, H. S. Park, H. X. Dang, F.-R. F. Fan, A. J. Bard and C. B. Mullins, Chem. Mater., 2012, 24, 579–586 CrossRef CAS.
- H. Ye, J. Lee, J. S. Jang and A. J. Bard, J. Phys. Chem. C, 2010, 114, 13322–13328 CAS.
- Y. C. Jia, P. Xiao, H. C. He, J. Y. Yao, F. L. Liu, Z. F. Wang and Y. H. Li, Appl. Surf. Sci., 2012, 258, 6627–6631 CrossRef CAS PubMed.
- X. J. Yu, L. Z. Huang, Y. C. Wei, J. Zhang, Z. Z. Zhao, W. Q. Dai and B. H. Yao, Mater. Res. Bull., 2015, 64, 410–417 CrossRef CAS PubMed.
- M. A. Rauf, M. A. Meetani, A. Khaleel and A. Ahmed, Chem. Eng. J., 2010, 157, 373–378 CrossRef CAS PubMed.
- Z. W. Tong, D. Yang, T. X. Xiao, Y. Tian and Z. Y. Jiang, Chem. Eng. J., 2015, 260, 117–125 CrossRef CAS PubMed.
- Y. F. Chen, W. X. Huang, D. L. He, Y. Situ and H. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 14405–14414 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02750k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.