DOI:
10.1039/C5RA02295A
(Paper)
RSC Adv., 2015,
5, 43669-43686
Identification of unusual C–Cl⋯π contacts in 2-(alkylamino)-3-chloro-1,4-naphthoquinones: effect of N-substituents on crystal packing, fluorescence, redox and anti-microbial properties†
Received
5th February 2015
, Accepted 24th April 2015
First published on 27th April 2015
Abstract
The chemo-selective reaction of 2,3-dichloro-1,4-naphthoquinone with different primary amines affords access to a series of derivatives, such as 2-(alkylamino)-3-chloro-1,4-naphthoquinone (1–6) and 2-(benzylamino)-1,4-naphthoquinone (7), in good yields. All the compounds 1–7 were characterized thoroughly by microanalysis, standard spectroscopy and thermogravimetric methods. The supramolecular structures of 1–4 and 7 were studied by means of single-crystal X-ray diffraction to gauge the influence of substituents that are present on the amine functionality on the association of molecules in the solid state. The study showed that the introduction of various amine N-substituents induces conformational changes that apparently modify the nature and number of donor–acceptor sites for noncovalent interactions, leading to diverse crystal packing patterns. Interestingly, the introduction of 2-(benzylamino)- and 2-(2-pyridylmethylamino)- substituents in 2 and 4 successfully switched on the C–Cl⋯π synthon, which is scarcely seen in the crystal packing of organic molecules. Compounds 1, 2, 4 and 5 fluoresced in the range of 350–620 nm with concomitant Stokes shifts of 81, 131, 141 and 131 nm, respectively, and their cyclic voltammograms evidenced two quasi-reversible single-electron waves. All the compounds (except 5) exhibited their first endothermic peak on the DTA curves without any mass loss due to the phase change, attributable to the melting points of the respective compounds. Remarkably, compound 5 exhibited an enhanced antibacterial activity against S. aureus and proved to be a more potent antibacterial agent than the well-known drug “ciprofloxacin”.
1. Introduction
The intermolecular forces that hold the molecules in the solid state are inadequately understood and hard to predict, particularly for organic solids.1 Because the properties of a material are often governed by the way in which its constituent molecules are arranged,2 any step taken into predicting molecular packing is a practical movement towards the ultimate goal of structural chemistry to design new solids with desired properties. Hydrogen bonds and stacking interactions, including weak hydrogen bonds such as C–H⋯O,3 N–H⋯O,4 C–H⋯π5 and π⋯π6 interactions, are considered as the main directing tools in the organization of molecules in both chemistry as well as in biology. These have been implicated successfully in both the crystal engineering and supramolecular assemblies. However, the ability of halogen atoms to function as reliable sites for directing intermolecular recognition processes was largely overlooked until the 1990s. Halogen bonds have been recognized recently as another type of non-covalent interaction that can be used as a new tool for the organization of supramolecular systems and molecular crystals.7 They have been shown to be accountable for the formation of a variety of stable supramolecular assemblies in crystals,7c,8 biological systems,9 solutions,10 and even in the gas phase.11 A competition between hydrogen bonding and halogen bonding has been observed by Professor Aakeroy's group and others12 during the supramolecular assembly of organic molecules in the solid state. More interestingly, rational modifications of the hierarchy of intermolecular interactions in molecular crystal structures using tuneable halogen bonds have been developed.13 In particular, halogen bonds of the type C–H⋯Cl7b,14 have been investigated significantly; however, literature shows only a few reports8,15 on the existence of C–Cl⋯π interactions. A survey of the protein data bank (PDB) suggested only a limited number of C–Cl⋯π interactions have been retrieved from crystal structures of protein–ligand complexes.15 Thus, a considerably larger amount of data on C–Cl⋯π contacts should help us better evaluate the characteristics of such interactions in biomolecules and in structural chemistry.
Apart from this, considerable attention has been paid to the functionalization of naphthoquinone derivatives and evaluation of their biological properties16 mainly due to their involvement in intermolecular interactions during multiple biological oxidative processes,17 and thus they have a great impact on the biological systems. A large part of the biological activity of quinonoid systems is shown to be related to its capacity to generate free radicals via redox reactions, which involve electrogenerated radical anion species (semiquinone) with long half-life periods and the ability to transfer the electron to another species in vivo.18 Reportedly, naphthoquinone derivatives possess diverse biological properties that include antibacterial and antifungal,19 antiviral,20 antimalarial21 and anticancer22 activity, which have stimulated the study of these bioactive compounds in the field of medicinal chemistry.
This study outlines a facile synthesis method and the spectral, optical and thermal characterization of 2-(alkylamino)-3-chloro-1,4-naphthoquinones 1–6 and 2-(benzylamino)-1,4-naphthoquinone 7 (Chart 1). The single crystal X-ray diffraction technique has been used to facilitate the understanding of structures and to review the influence of various N-substituents on the crystal packing patterns in the solid state. We have determined the crystal structure for five 2-substituted 3-chloro-1,4-naphthoquinone derivatives and have analyzed the propensity of the formation of C–Cl⋯π, C–H⋯Cl and C–H⋯π contacts in the presence of dominating C–H⋯O contacts and the directional preference in terms of crystal packing. These derivatives form an interesting class of compounds with synthetic versatility and effective anti-microbial activities. Due to the increasing importance of halogen bonding in biological systems,16 the identification of C–Cl⋯π, C–H⋯Cl intermolecular interactions in 2 and 4 would add merit and assist the evaluation of the characteristics of such interactions in biomolecules and in structural chemistry.
 |
| | Chart 1 Compounds 1–7 under investigation. | |
2. Experimental section
2.1 Material and physical measurements
All the chemicals and solvents used in this work were of laboratory grade and were made available from various commercial sources and used without further purification. Melting points were recorded in open capillaries and are uncorrected. Thin layer chromatography was performed on Merck 60 F254 aluminium coated plates. Elemental analyses (C, H, N) were performed on a Perkin-Elmer 2400 analyzer. Mass spectra were obtained on a Thermo-Fisher DSQ II GCMS instrument. FT-IR (KBr pellets) spectra were recorded in the 4000–400 cm−1 range using a Perkin-Elmer FT-IR spectrometer. The NMR spectra were obtained on a Bruker AV-III 400 MHz spectrometer in CDCl3 solvent. UV-visible spectra were recorded on a Perkin Elmer Lambda 35 UV-visible spectrophotometer and the optical characterization of solid samples was performed using the UV-visible transmittance measurements. Fluorescence was recorded on a spectrofluorometer model FP-6300 made by JASCO. TGA/DTA plots were obtained using SII TG/DTA 6300 in flowing N2 with a heating rate of 10 °C min−1. Electrochemical measurements were performed on a CH Instruments 600C potentiostat using a Pt disk as the working electrode, Ag/AgCl as the reference electrode and a Pt wire as the counter electrode. Voltammograms were recorded using anhydrous solutions of the amines (1–7) in CH2Cl2 solutions (1.0 mM) containing tetra-n-butylammonium hexafluorophosphate (0.1 M) as the supporting electrolyte. The synthesized compounds were screened for their in vitro antibacterial activity against S. aureus, B. subtilis, E. coli, P. aeruginosa and antifungal activity against C. albicans and A. niger at the Division of Central Kashiba Advance Diagnostic Laboratory, Surat, Gujarat, India.
2.2 Synthesis of compounds 1–7
2.2.1 General procedure for the synthesis of 2-(alkylamino)-3-chloro-1,4-naphthoquinones (1–6). To yield a series of 2-(alkylamino)-3-chloro-1,4-naphthoquinone derivatives, the corresponding amine (cyclohexyl amine (0.5 mL, 4.404 mmol), benzyl amine (0.481 mL, 4.404 mmol), furfuryl amine (0.427 mL, 4.404 mmol), 2-picolyl amine (0.476 mL, 4.404 mmol), 3-picolyl amine (0.475 mL, 4.404 mmol) or n-butyl amine (0.460 mL, 4.404 mmol)) was added in 30 mL of absolute ethanol containing 1 equivalent of 2,3-dichloro-1,4-napthaquinone (1 g, 4.404 mmol). The reaction mixture was stirred at room temperature for 6 hours. During these hours, a change in the colour of the reaction mixture was observed from yellow to red. The progress of the reaction was monitored by TLC. The reaction mixture was dried under vacuum and the residue was washed several times with saturated Na2CO3 solution followed by 3 × 10 mL of distilled water. The red-coloured product was finally dried under vacuum and preserved in desiccator for analysis. The elemental analysis, FT-IR and NMR data for compounds 1–6 are given as follows:
1. Yield 1260.25 mg, 99%. M.p. 116.7 °C. Elemental analysis: calcd for C16H16ClNO2 (289.09): C, 66.32; H, 5.57; N, 4.83. Found: C, 66.80; H, 5.68; N, 4.79. ES-MS: 290.61 (M + H), (50%); 287.76 (M − 2H), (100%). IR (KBr disc, cm−1): 3303s, 2931m, 2852m, 1675s, 1634m, 1596s, 1564s, 1515s, 1451m, 1331s, 1291s, 1251m, 1234m, 1154m, 1132m, 1079w, 963w, 847w, 822w, 783w, 722s, 679m, 647w, 619m, 560w, 544w, 467w. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.17 (dd, 1H, –Ph), 8.06 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.63 (td, 1H, –Ph), 6.12 (s, 1H; NH), 4.44 (m, 1H, CH), 2.09 (dd, 2H, CH2), 1.80 (m, 2H, CH2), 1.38 (m, 6H, CH2). 13C NMR: δ (ppm) 180.6 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 177 (CO), 134.9 (C–N), 132.8, 132.4, 129.7, 126.8 (all correspond to the carbons of Ph), 52.5, 34.6, 25.3, 24.5 (all correspond to the carbons of cyclohexyl moiety).
2. Yield: 1242.61 mg, 95%. M.p. 112.4 °C. Elemental analysis: calcd for C17H12ClNO2 (297.06): C, 68.58; H, 4.06; N, 4.70. Found: C, 68.35; H, 4.22; N, 4.65. ES-MS: 297.78 (M + H); (40%). IR (KBr disc, cm−1): 3279s, 2935w, 1683s, 1638m, 1598s, 1565s, 1516s, 1443m, 1332s, 1299s, 1254s, 1136m, 1066m, 1029m, 934w, 913w, 867w, 823m, 753m, 725s, 699s, 680m, 607m, 595m, 545m, 494m, 445m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18 (dd, 1H, –Ph), 8.058 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.65 (td, 1H, –Ph), 6.26 (s, 1H, NH), 7.3 (m, 5H, –Ph), 5.07 (s, 2H, CH2). 13C NMR: δ (ppm) 180.4 (CO), 176.9 (CO), 144 (C–N), 137.8, 134.9, 132.6, 129.7, 129, 128, 127.7, 126.9 (all correspond to the carbons of Ph), 48.9 (benzylic CH2).
3. Yield: 1175.32 mg, 93%. M.p. 147.3 °C. Elemental analysis: calcd for C15H10ClNO3 (287.03): C, 62.62; H, 3.50; N, 4.87. Found: C, 62.55; H, 3.49; N, 4.95. ES-MS: 286.27 (M − H), (100%); 287.20 (M), (56%); 288.19 (M + H), (37%). IR (KBr disc, cm−1): 3324s, 3147w, 3125w, 1680s, 1639m, 1599s, 1569s, 1519s, 1431m, 1357m, 1334m, 1300s, 1250s, 1205m, 1139s, 1064s, 1008m, 933m, 901w, 864w, 823w, 756s, 721s, 693m, 679m, 605m, 550m, 478m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.16 (dd, 1H, –Ph), 8.05 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.65 (td, 1H, –Ph), 6.23 (s, 1H; NH), 7.42 (d, 1H, CH of furanyl moiety), 6.35–6.38 (m, 2H, CH of furanyl moiety), 5.08 (s, 2H, CH2). 13C NMR: δ (ppm) 180.3 (CO), 176.9 (CO), 150.6 (C–N), 143.8, 142.8, 134.9, 132.6, 132.5, 129.8, 126.9, 110.6, 108.4 (all correspond to the carbons of Ph/furanyl moiety), 41.8 (benzylic CH2).
4. Yield: 1194.17 mg, 91%. M.p. 160.4 °C. Elemental analysis: calcd for C16H11ClN2O2 (298.05): C, 64.33; H, 3.71; N, 9.38. Found: C, 64.55; H, 3.79; N, 9.35. ES-MS: 298.55 (M + H), (5%), 262.79 (100%). IR (KBr disc, cm−1): 3252s, 3067w, 1683s, 1605s, 1575s, 1566s, 1496s, 1480m, 1444m, 1432m, 1330s, 1295s, 1288s, 1251m, 1208m, 1139m, 1016m, 839m, 760m, 719s, 679m, 639w, 547m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.68 (d, 1H, –Py), 8.19 (dd, 1H, –Ph), 8.10 (dd 1H, –Ph), 7.75 (td, 1H, –Ph), 7.67 (td, 1H, –Ph), 7.34 (m, 3H, Py), 7.82 (s, 1H, NH), 5.22 (s, 2H, CH2). 13C NMR: δ (ppm) 180.6 (CO), 176.8 (CO), 155.1, 148.7, 144.3, 137.3, 134.8, 132.6, 132.5, 129.9, 126.8, 126.7, 122.8, 121.9 (all correspond to the carbons of Ph/pyridyl moiety), 48.4 (benzylic CH2).
5. Yield: 1181.28 mg, 90%. M.p. 151.8 °C. Elemental analysis: calcd for C16H11ClN2O2 (298.05): C, 64.33; H, 3.71; N, 9.38. Found: C, 64.37; H, 3.68; N, 9.31. ES-MS: 297.65 (M − H), (5%). IR (KBr disc, cm−1): 3171m, 3007w, 1683s, 1644m, 1601s, 1572s, 1521s, 1444m, 1424m, 1328s, 1292s, 1255s, 1184w, 1136s, 1063m, 1035w, 1028w, 820w, 795w, 723m, 708m, 680w, 635w, 612m, 547m, 503w, 451w. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.66 (d, 1H, –Py), 8.61 (dd, 1H, –Py), 8.18 (dd 1H, –Ph), 8.07 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.67 (td, 1H, –Ph), 7.40 (m, 1H, –Py), 7.75 (m, 1H, –Py), 6.31 (s, 1H, NH), 5.11 (s, 2H, CH2). 13C NMR: δ (ppm) 180.3 (CO), 176.9 (CO), 149.4, 149.0, 143.7, 135.2, 135, 135, 133.7, 132.7, 129.7, 126.9, 123.8 (all correspond to the carbons of Ph/pyridyl moiety), 46.2 (benzylic CH2).
6. Yield: 1077.61 mg, 93%. M.p. 109.8 °C. Elemental analysis: calcd for C14H14ClNO2 (263.07): C, 63.76; H, 5.35; N, 5.31. Found: C, 63.95; H, 5.29; N, 5.35. ES-MS: 262.04 (M − H), (26%). IR (KBr disc, cm−1): 3279s, 3064m, 1651s, 1559s, 1507w, 1499m, 1456m, 1427s, 1365m, 1237s, 1172w, 1164w, 1063s, 1031m, 1003w, 924w, 785w, 744m, 726s, 698s, 591m, 556s, 505s, 418w. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18 (dd, 1H, –Ph), 8.059 (dd, 1H, –Ph), 7.74 (td, 1H, –Ph), 7.64 (td, 1H, –Ph), 6.10 (s, 1H, NH), 3.88 (t, 2H, CH2), 1.70 (m, 2H, CH2), 1.48 (m, 2H, CH2), 1.43 (t, 3H, CH3). 13C NMR: δ (ppm) 180.6 (CO), 176.9 (CO), 134.9, 132.8, 132.4, 129.7, 126.8 (all correspond to the carbons of Ph), 44.7, 33, 19.8 (all correspond to CH2), 13.7 (CH3).
O), 177 (CO), 134.9 (C–N), 132.8, 132.4, 129.7, 126.8 (all correspond to the carbons of Ph), 52.5, 34.6, 25.3, 24.5 (all correspond to the carbons of cyclohexyl moiety).
2. Yield: 1242.61 mg, 95%. M.p. 112.4 °C. Elemental analysis: calcd for C17H12ClNO2 (297.06): C, 68.58; H, 4.06; N, 4.70. Found: C, 68.35; H, 4.22; N, 4.65. ES-MS: 297.78 (M + H); (40%). IR (KBr disc, cm−1): 3279s, 2935w, 1683s, 1638m, 1598s, 1565s, 1516s, 1443m, 1332s, 1299s, 1254s, 1136m, 1066m, 1029m, 934w, 913w, 867w, 823m, 753m, 725s, 699s, 680m, 607m, 595m, 545m, 494m, 445m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18 (dd, 1H, –Ph), 8.058 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.65 (td, 1H, –Ph), 6.26 (s, 1H, NH), 7.3 (m, 5H, –Ph), 5.07 (s, 2H, CH2). 13C NMR: δ (ppm) 180.4 (CO), 176.9 (CO), 144 (C–N), 137.8, 134.9, 132.6, 129.7, 129, 128, 127.7, 126.9 (all correspond to the carbons of Ph), 48.9 (benzylic CH2).
3. Yield: 1175.32 mg, 93%. M.p. 147.3 °C. Elemental analysis: calcd for C15H10ClNO3 (287.03): C, 62.62; H, 3.50; N, 4.87. Found: C, 62.55; H, 3.49; N, 4.95. ES-MS: 286.27 (M − H), (100%); 287.20 (M), (56%); 288.19 (M + H), (37%). IR (KBr disc, cm−1): 3324s, 3147w, 3125w, 1680s, 1639m, 1599s, 1569s, 1519s, 1431m, 1357m, 1334m, 1300s, 1250s, 1205m, 1139s, 1064s, 1008m, 933m, 901w, 864w, 823w, 756s, 721s, 693m, 679m, 605m, 550m, 478m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.16 (dd, 1H, –Ph), 8.05 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.65 (td, 1H, –Ph), 6.23 (s, 1H; NH), 7.42 (d, 1H, CH of furanyl moiety), 6.35–6.38 (m, 2H, CH of furanyl moiety), 5.08 (s, 2H, CH2). 13C NMR: δ (ppm) 180.3 (CO), 176.9 (CO), 150.6 (C–N), 143.8, 142.8, 134.9, 132.6, 132.5, 129.8, 126.9, 110.6, 108.4 (all correspond to the carbons of Ph/furanyl moiety), 41.8 (benzylic CH2).
4. Yield: 1194.17 mg, 91%. M.p. 160.4 °C. Elemental analysis: calcd for C16H11ClN2O2 (298.05): C, 64.33; H, 3.71; N, 9.38. Found: C, 64.55; H, 3.79; N, 9.35. ES-MS: 298.55 (M + H), (5%), 262.79 (100%). IR (KBr disc, cm−1): 3252s, 3067w, 1683s, 1605s, 1575s, 1566s, 1496s, 1480m, 1444m, 1432m, 1330s, 1295s, 1288s, 1251m, 1208m, 1139m, 1016m, 839m, 760m, 719s, 679m, 639w, 547m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.68 (d, 1H, –Py), 8.19 (dd, 1H, –Ph), 8.10 (dd 1H, –Ph), 7.75 (td, 1H, –Ph), 7.67 (td, 1H, –Ph), 7.34 (m, 3H, Py), 7.82 (s, 1H, NH), 5.22 (s, 2H, CH2). 13C NMR: δ (ppm) 180.6 (CO), 176.8 (CO), 155.1, 148.7, 144.3, 137.3, 134.8, 132.6, 132.5, 129.9, 126.8, 126.7, 122.8, 121.9 (all correspond to the carbons of Ph/pyridyl moiety), 48.4 (benzylic CH2).
5. Yield: 1181.28 mg, 90%. M.p. 151.8 °C. Elemental analysis: calcd for C16H11ClN2O2 (298.05): C, 64.33; H, 3.71; N, 9.38. Found: C, 64.37; H, 3.68; N, 9.31. ES-MS: 297.65 (M − H), (5%). IR (KBr disc, cm−1): 3171m, 3007w, 1683s, 1644m, 1601s, 1572s, 1521s, 1444m, 1424m, 1328s, 1292s, 1255s, 1184w, 1136s, 1063m, 1035w, 1028w, 820w, 795w, 723m, 708m, 680w, 635w, 612m, 547m, 503w, 451w. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.66 (d, 1H, –Py), 8.61 (dd, 1H, –Py), 8.18 (dd 1H, –Ph), 8.07 (dd, 1H, –Ph), 7.75 (td, 1H, –Ph), 7.67 (td, 1H, –Ph), 7.40 (m, 1H, –Py), 7.75 (m, 1H, –Py), 6.31 (s, 1H, NH), 5.11 (s, 2H, CH2). 13C NMR: δ (ppm) 180.3 (CO), 176.9 (CO), 149.4, 149.0, 143.7, 135.2, 135, 135, 133.7, 132.7, 129.7, 126.9, 123.8 (all correspond to the carbons of Ph/pyridyl moiety), 46.2 (benzylic CH2).
6. Yield: 1077.61 mg, 93%. M.p. 109.8 °C. Elemental analysis: calcd for C14H14ClNO2 (263.07): C, 63.76; H, 5.35; N, 5.31. Found: C, 63.95; H, 5.29; N, 5.35. ES-MS: 262.04 (M − H), (26%). IR (KBr disc, cm−1): 3279s, 3064m, 1651s, 1559s, 1507w, 1499m, 1456m, 1427s, 1365m, 1237s, 1172w, 1164w, 1063s, 1031m, 1003w, 924w, 785w, 744m, 726s, 698s, 591m, 556s, 505s, 418w. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18 (dd, 1H, –Ph), 8.059 (dd, 1H, –Ph), 7.74 (td, 1H, –Ph), 7.64 (td, 1H, –Ph), 6.10 (s, 1H, NH), 3.88 (t, 2H, CH2), 1.70 (m, 2H, CH2), 1.48 (m, 2H, CH2), 1.43 (t, 3H, CH3). 13C NMR: δ (ppm) 180.6 (CO), 176.9 (CO), 134.9, 132.8, 132.4, 129.7, 126.8 (all correspond to the carbons of Ph), 44.7, 33, 19.8 (all correspond to CH2), 13.7 (CH3).
2.2.2 General procedure for the synthesis of 2-(benzylamino)-1,4-naphthoquinone (7). 2,3-Dichloro-1,4-napthaquinone (300 mg, 1.321 mmol) and benzyl amine (0.288 mL, 2.642 mmol) were added in 10 mL of DMF and the reaction mixture was allowed to reflux for 4 hours. Reaction progress was monitored by TLC. The reaction mixture was cooled to room temperature. The reaction mixture was then quenched with ice cold water and the crude product was recovered by vacuum filtration. The desired compound was purified by column chromatography (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ethyl acetate:petroleum ether) to yield a red-coloured solid.
7. Yield: 271.24 mg, 78%. M.p. 162 °C. Elemental analysis: calcd for C17H13NO2 (263.09): C, 77.55; H, 4.98; N, 5.32. Found: C, 77.67; H, 5.05; N, 5.41. ES-MS: 262.85, 263 (M + H); (100%). IR (KBr disc, cm−1): 3333s, 3061w, 1683s, 1594s, 1563s, 1503s, 1452m, 1440m, 1360s, 1338s, 1305m, 1259s, 1216m, 1151w, 1124s, 1097w, 1066m, 1027w, 1006m, 943s, 846s, 782m, 728s, 694m, 609m, 547m, 482m, 458w, 451w, 416m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.115 (dd, 1H, –Ph), 8.082 (dd, 1H, –Ph), 7.755 (td, 1H, –Ph), 7.66 (td, 1H, –Ph), 6.25 (s, 1H, NH), 7.38 (m, 5H, Ph), 5.82 (s, 1H, –Ph), 4.07 (s, 2H, CH2). 13C NMR: δ (ppm) 183.1 (CO), 181.9 (CO), 147.7, 135.9, 134.8, 133.5, 132.1, 130.5, 129, 128.2, 127.7, 126.3, 126.2, 101.7 (all correspond to the carbons of Ph), 46.8 (CH2).
:5 ethyl acetate:petroleum ether) to yield a red-coloured solid.
7. Yield: 271.24 mg, 78%. M.p. 162 °C. Elemental analysis: calcd for C17H13NO2 (263.09): C, 77.55; H, 4.98; N, 5.32. Found: C, 77.67; H, 5.05; N, 5.41. ES-MS: 262.85, 263 (M + H); (100%). IR (KBr disc, cm−1): 3333s, 3061w, 1683s, 1594s, 1563s, 1503s, 1452m, 1440m, 1360s, 1338s, 1305m, 1259s, 1216m, 1151w, 1124s, 1097w, 1066m, 1027w, 1006m, 943s, 846s, 782m, 728s, 694m, 609m, 547m, 482m, 458w, 451w, 416m. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.115 (dd, 1H, –Ph), 8.082 (dd, 1H, –Ph), 7.755 (td, 1H, –Ph), 7.66 (td, 1H, –Ph), 6.25 (s, 1H, NH), 7.38 (m, 5H, Ph), 5.82 (s, 1H, –Ph), 4.07 (s, 2H, CH2). 13C NMR: δ (ppm) 183.1 (CO), 181.9 (CO), 147.7, 135.9, 134.8, 133.5, 132.1, 130.5, 129, 128.2, 127.7, 126.3, 126.2, 101.7 (all correspond to the carbons of Ph), 46.8 (CH2).
2.3 X-ray crystallography and data collection
Crystals of 1–4 and 7 suitable for X-ray crystallographic study were obtained from dichloromethane by slow evaporation at 4 °C. Intensity data were collected on an Oxford diffraction X Calibur diffractometer equipped with Eos CCD detector at 150 K for [C16H16ClNO2] 1, [C17H12ClNO2] 2, [C15H10ClNO3] 3, [C16H11ClN2O2] 4 and [C17H13NO2] 7. Monochromatic Mo-Kα X-ray (λ = 0.71073 Å) was used for the measurements. Data were collected and reduced using the “CrysAlispro” program.23 An empirical absorption correction using spherical harmonics was implemented in “SCALE3 ABSPACK” scaling algorithm. The crystal structures were solved by direct methods using SHELXL-9724 and the refinement was carried out against F2 using SHELXL-97 program package.25 All non-hydrogen atoms were refined anisotropically.
3. Results and discussion
3.1 Synthesis and characterization
The chemo-selective reaction of 2,3-dichloro-1,4-naphthoquinone with different primary amines efficiently yielded a series of novel 2-substituted-1,4-naphthoquinone derivatives, namely, 2-(alkylamino)-3-chloro-1,4-naphthoquinones (1–6) and 2-(benzylamino)-1,4-naphthoquinone (7), in good yields (Scheme 1). The dual role of amine as a nucleophile and as a base facilitated the formation of products in EtOH via the removal of one chloro substituent with ease. Remarkably, only the reaction of 2,3-dichloro-1,4-naphthoquinone with benzyl amine in refluxing DMF led to the formation of 3-hydro analogue 7. Compounds 2, 6 and 7 were synthesized by modified literature procedures26 and this modification was scalable to 95% for 2, 90% for 6 and 70% for 7 and the reactions took less time than the original methods.26a,b,d
 |
| | Scheme 1 Synthetic protocol for 2-(alkylamino)-3-chloro-1,4-naphthoquinones (1–6) and 2-(benzylamino)-1,4-naphthoquinone (7). | |
All the compounds 1–7 have been characterized thoroughly by microanalysis, standard spectroscopy and a thermogravimetric method. The spectroscopic analysis data, such as GC-MS, IR, NMR and UV-visible spectra of the compounds 1–7 are consistent with their chemical formulas determined by elemental analysis, and the structures of 1–4 and 7 were elucidated from single crystal X-ray diffraction study.
In the IR spectra of 1–7, the most characteristic bands observed between 3250–3350 cm−1 are diagnostic of υ(N–H) stretching vibrations. The weak intensity bands that appeared in the regions of 3147–2931 cm−1 are attributable to the aromatic υ(C–H) stretching vibrations, whereas bands that appeared in the regions of 850–820 cm−1 are assignable to the aromatic υ(C–H) out-of plane bending vibrations, which is a characteristic feature of the phenyl ring of the naphthoquinone moiety. In addition, 1–7 display strong bands (1651–1683 cm−1) and medium intensity bands (1172–1124 cm−1) due to υ(CO) and υ(C–N) vibrations.27 Unlike 7, compounds 1–6 display a sharp band in the region of 725–719 cm−1 due to υ(C–Cl) stretching vibrations. The 1H NMR spectra of 1–7 exhibit most of their characteristic signals in the range of δ = 6.10–7.82 ppm due to amine –NH moiety along with signals due to the aromatic protons of the naphthoquinone moiety and corresponding N-substituent with proper splitting patterns. Among these, –NH signal in compound 4 is significantly downfield shifted due to the high electron withdrawing nature of N-(2-pyridylmethyl) substituent. A singlet signal appeared at δ = 5.82 ppm, which is assignable to the proton present at the 3-position of 2-(benzylamino)-1,4-naphthoquinone (7). In the 13C spectra of 1–7, the most characteristic signals appeared in the range of δ = 181–176 ppm, which are attributable to the two quinonic carbonyl moieties. Compound 1 displayed signals due to methine and methylene groups at 52.5 ppm and δ = 35–24 ppm regions, respectively. In the case of compounds 2–5, signals in the range of 41–49 ppm are assigned to methylene carbon atoms, whereas signals observed in the δ = 155–108 ppm regions are due to aromatic carbons. Compound 6 shows three upfield signals in δ = 44–19 ppm regions due to –CH2, while a signal appeared at δ = 13.7 ppm, which corresponds to the –CH3 moiety of the n-butyl substituent. The IR, 1H and 13C NMR spectral data for 1–7 are consistent with similar compounds reported in the literature. All the compounds 1–7 were further characterized by mass spectroscopy. The molecular ion peak (m/z) corresponds to either (M + H) or (M − H) and gives the evidence for the formation of the desired compounds.
Block-shaped colourless crystals for 1, 2, 4 and golden-yellow crystals for compound 3, suitable for single crystal X-ray diffraction study, were grown from a dichloromethane solution by slow evaporation at 4 °C. Our efforts to obtain single crystals of 5 and 6 suitable for XRD analysis using different crystallization techniques were unsuccessful. The crystal data and structure refinement for compounds 1–4 and 7 are given in Table 1.
Table 1 Crystal data and structure refinement for compounds 1–4 and 7
| Identification code |
1 |
2 |
3 |
4 |
7 |
| Formula |
C16H16ClNO2 |
C17 H12ClNO2 |
C15H10ClNO3 |
C16 H11ClN2O2 |
C17H13NO2 |
| Formula weight |
289.75 |
297.73 |
287.69 |
298.72 |
263.30 |
| Temperature (K) |
150(2) |
293(2) |
150(2) |
150(2) |
293 |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Orthorhombic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P21/c |
P21/n |
Pna21 |
| a (Å) |
8.1167(10) |
10.2681(4) |
7.4914(2) |
5.0576(4) |
10.8597(13) |
| b (Å) |
8.3589(10) |
7.7447(3) |
20.7986(5) |
11.4615(7) |
24.115(4) |
| c (Å) |
10.5321(13) |
17.5818(9) |
7.9056(2) |
23.3465(11) |
5.0303(7) |
| α (°) |
92.178(10) |
90 |
90 |
90 |
90 |
| β (°) |
104.565(11) |
93.836(4) |
93.019(2) |
91.898(6) |
90 |
| γ (°) |
94.094(10) |
90 |
90 |
90 |
90 |
| Volume (Å3) |
688.68(15) |
1395.03(10) |
1230.07(5) |
1352.60(15) |
1317.3(3) |
| Z |
2 |
4 |
4 |
4 |
4 |
| Calculated density (mg m−3) |
1.397 |
1.418 |
1.553 |
1.467 |
1.3275 |
| Absorption coefficient (m mm−1) |
0.278 |
0.277 |
0.317 |
0.288 |
0.088 |
| F(000) |
304 |
616 |
592 |
616 |
52.3 |
| Crystal size (mm3) |
0.23 × 0.18 × 0.13 |
0.32 × 0.27 × 0.23 |
0.23 × 0.18 × 0.13 |
0.23 × 0.17 × 0.13 |
0.14 × 0.11 × 0.06 |
| θ range for data collection (°) |
3.07–25.00°. |
2.95–25.00°. |
3.24–25.00°. |
3.17–24.99°. |
6.76–55.84° |
| Index ranges |
−9 ≤ h ≤ 9 |
−11 ≤ h ≤ 12 |
−8 ≤ h ≤ 8 |
−6 ≤ h ≤ 5 |
−13 ≤ h ≤ 5 |
| −9 ≤ k ≤ 9 |
−5 ≤ k ≤ 9 |
−24 ≤ k ≤ 21 |
−13 ≤ k ≤ 13 |
−31 ≤ k ≤ 28 |
| −12 ≤ l ≤ 12 |
−20 ≤ l ≤ 20 |
−9 ≤ l ≤ 9 |
−27 ≤ l ≤ 27 |
−6 ≤ l ≤ 3 |
| Reflections collected/unique |
5112/2429 [R(int) = 0.0424] |
10267/2451 [R(int) = 0.0451] |
8482/2164 [R(int) = 0.0165] |
8228/2368 [R(int) = 0.0779] |
1366 [R(int) = 0.0306] |
| Completeness to theta = 25.00 (%) |
99.8 |
99.9 |
99.8 |
99.9 |
99.9 |
| Data/restraints/parameters |
2429/0/181 |
2451/0/194 |
2164/0/181 |
2368/0/191 |
1366/0/181 |
| Goodness-of-fit on F2 |
1.036 |
1.004 |
1.082 |
1.120 |
1.118 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0637, wR2 = 0.1655 |
R1 = 0.0441, wR2 = 0.1085 |
R1 = 0.0295, wR2 = 0.0707 |
R1 = 0.0652, wR2 = 0.1740 |
R1 = 0.0718, wR2 = 0.1410 |
| Largest diff. peak and hole/e Å−3 |
0.335 and −0.387 |
0.210 and −0.233 |
0.299 and −0.265 |
0.445 and −0.672 |
0.28 and −0.33 |
3.2 Structural descriptions
A careful analysis of single crystal X-ray diffraction data reveals that a change in the N-substituents in 1–4 and the absence of a chloro substituent in 7 changes the electronic nature as well as the stereochemical conformations of the molecules. This can be clearly visualized from the different dihedral angles between the least squares plane through the naphthoquinone rings and that of amine substituents. The 3-chloro-1,4-naphthoquinone moiety, having an extended conjugation, holds the two naphthoquinone rings in a virtually planar direction. The angle between the least squares planes through the benzene and quinone rings of naphthoquinones appeared in a range of 0.99–2.29° in 1–4 and 7. However, the angle between the least squares planes passing through the quinone ring and various N-substituent ring falls in the range of 61.88–88.04° in all the molecules (except 4), attaining a ‘gauche’ conformational mode. Notably, the absence of a chloro substituent in 7 diminishes the n–π electrostatic repulsive interactions and this molecule largely deviates from a stable ‘gauche’ conformational mode, which is revealed by the dihedral angle (88.04°) between naphthoquinone ring and benzene ring of the N-benzyl substituent. Contrarily, the presence of a N-(2-pyridylmethyl) substituent in 4 changes the dihedral angle between the planes of naphthoquinone ring and pyridine ring to 0.99°, and thus induces coplanarity in the molecular framework. These conformational changes induced by various amine N-substituents in 1–4 and 7 apparently modify the nature and number of donor–acceptor sites for noncovalent interactions, leading to diverse crystal packing patterns. This generates a scope for the control and fine tuning of the supramolecular assemblies that arise due to non-covalent interactions in these molecules. The supramolecular architectures that arise due to a number of weak noncovalent intermolecular interactions in 1–4 and 7 can be described as follows.
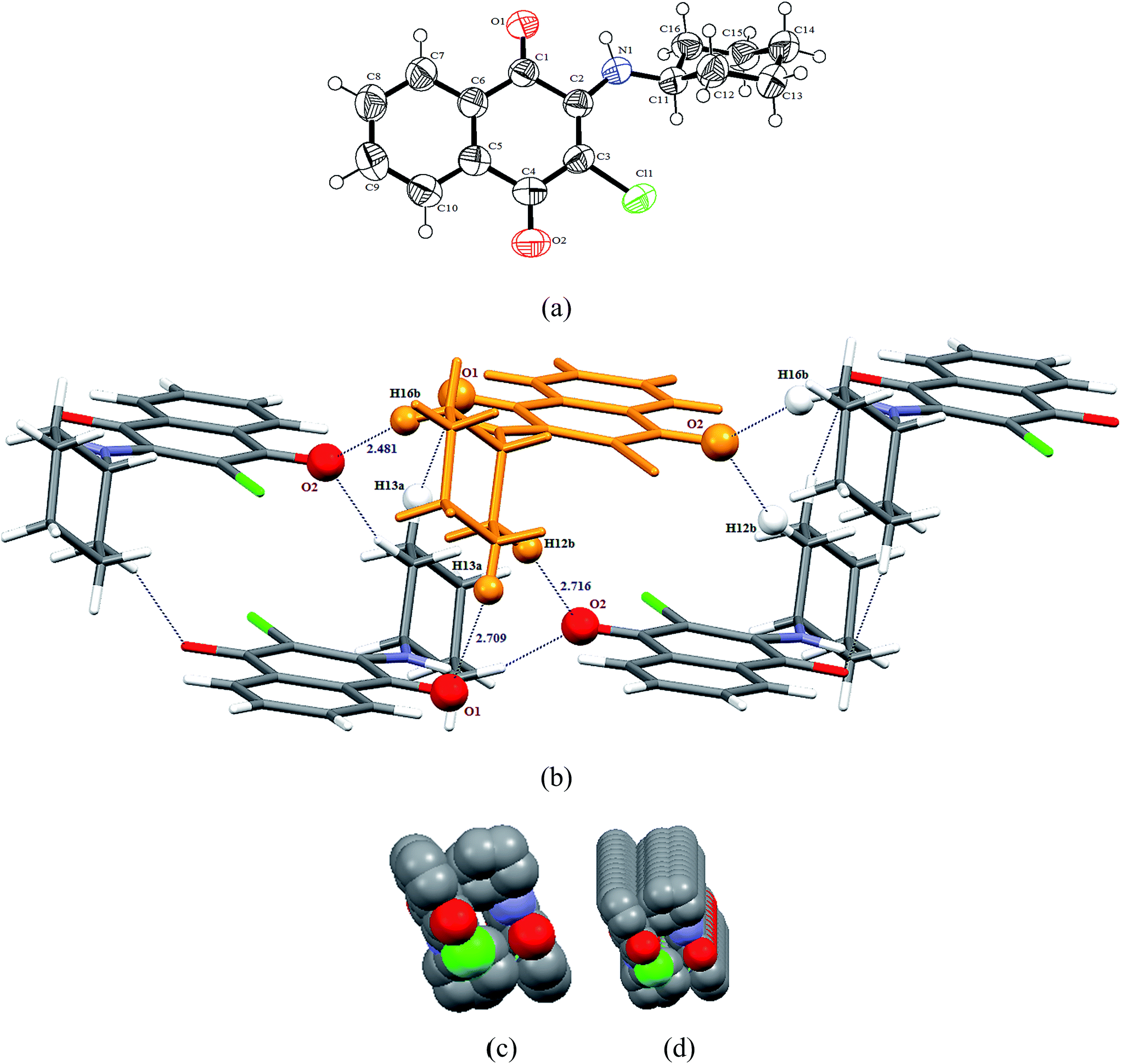
3.2.1 X-ray structure of 2-(cyclohexylamino)-3-chloro-1,4-naphthoquinone [C16H16ClNO2] (1). Compound 1 crystallizes in chiral triclinic P space group and the X-ray crystal structure shows an asymmetric unit that contains a full molecule of [C16H16ClNO2], as shown in Fig. 1(a). There are two such molecules in the unit cell. The selected bond distances (Å) and bond angles (°) for 1 are: C1–C6 1.475(4), C1–O1 1.211(3), C1–C2 1.522(3), C4–C5 1.499(4), C4–O2 1.223(3), C2–C3 1.370(4), C3–Cl1 1.738(2), C2–N1 1.337(3), N1–C11 1.463(3) Å and C6–C1–O1 122.0(2), C2–C1–O1 119.0(2), C5–C4–O2 119.5(2), C3–C4–O2 122.6(3), C2–C3–Cl1 122.7(2), C1–C2–N1 110.5(2), C2–N1–C11 131.2(2)°, respectively. The structural parameters are found to be in the normal range and have a good agreement with those observed for similar compounds.28 Interestingly, 1 containing a cyclohexyl substituent on the amine functionality adopts a ‘gauche’ conformation, which is predominantly stabilized via a number of intermolecular CH⋯O interactions. The cyclohexyl group primarily behaves as a C–H donor and the quinone group as a C–H acceptor. Molecules of 1 are interconnected along the a-axis in anti fashion through a number of CH⋯O interactions viz. C13–H13A⋯O1 (2.709 Å), C12–H12B⋯O2 (2.716 Å) and C16–H16B⋯O2 (2.481 Å) donor–acceptor interactions. In fact, one of the ketonic oxygens (O2) is involved in bifurcated hydrogen bonding, arranging a molecule parallel and another molecule anti-parallel along the a-axis. The relevant parameters for the weak interactions are tabulated in Table 2. The molecules present in the asymmetric unit form a supramolecular unit, consisting of five molecules aggregated, as shown in Fig. 1(b). Interestingly, these supramolecular units can be used to generate infinite networks along the a-axis, forming a fascinating 1D tubular architecture, as shown in Fig. 1(c) and (d).
 |
| | Fig. 1 (a) Thermal ellipsoidal plot of compound 1 with atom labeling scheme; (b) supramolecular unit consisting of five molecule aggregates involving CH⋯O interactions; (c) spacefill representation of molecular packing along a-axis forming a 1D tubular architecture; (d) view of molecular packing in spacefill model along an axis slightly tilted down from a-axis. | |
Table 2 Significant intermolecular interactions [interatomic distances (Å), and bond angles (°)] found in compounds 1–4 and 7
| Compounds |
D─H⋯A |
D─H |
H⋯A |
D⋯A |
<DHA (β) |
<α |
| 1 |
C13─H13a⋯O1 |
0.970 |
2.709 |
3.471 |
135.81 |
— |
| C12─H12b⋯O2 |
0.970 |
2.716 |
3.628 |
155.96 |
| C16─H16b⋯O2 |
0.970 |
2.481 |
3.383 |
154.61 |
| 2 |
C6─H6⋯O1 |
0.930 |
2.303 |
3.218 |
168.61 |
— |
| N1─H1N⋯O2 |
0.862 |
2.295 |
3.051 |
146.38 |
| C17─H17⋯O2 |
0.930 |
2.715 |
3.533 |
147.26 |
| C9─Cl1⋯Cg [Cg: C16 C17 C12 C13 C14 C15] |
1.732 |
3.879 |
5.545 |
160.96 |
31.10 |
| 3 |
N1─H1⋯O1 |
0.860 |
2.279 |
2.988 |
139.93 |
— |
| C11─H11⋯O2 |
0.970 |
2.570 |
3.356 |
138.19 |
| C15─H15⋯O2 |
0.930 |
2.572 |
3.331 |
139.13 |
| C14─H14⋯O2 |
0.930 |
2.601 |
3.507 |
164.89 |
| C15─H15⋯Cl1 |
0.930 |
2.916 |
3.472 |
119.69 |
| 4 |
C4─H4⋯O1 |
0.951 |
2.715 |
3.309 |
148.56 |
— |
| C6─H6B⋯O1 |
0.990 |
2.460 |
3.400 |
126.66 |
| C2─H2A⋯O2 |
0.951 |
2.593 |
3.302 |
131.58 |
| C8─Cl1⋯Cg1 [Cg1: C10C11 C12 C13 C14 C15] |
1.736 |
3.768 |
5.468 |
165.87 |
6.15 |
| C6─ H6A⋯Cg2 [Cg2: C7 C8 C9 C10 C15 C16 |
0.991 |
3.472 |
3.808 |
102.30 |
21.2 |
| C6─H6B⋯Cg3 [Cg3: C1 N1 C2 C3 C4 C5] |
0.990 |
3.039 |
3.668 |
122.53 |
3.03 |
| 7 |
C2─H2⋯O1 |
0.930 |
2.443 |
3.321 |
157.32 |
— |
| N1─H1⋯O2 |
0.860 |
2.168 |
2.901 |
143.03 |
| C13─H13⋯O2 |
0.929 |
2.709 |
3.511 |
144.98 |
| C16─ H16⋯Cg1 [Cg1: C4 C5 C6 C7 C8 C9] |
0.930 |
2.522 |
4.407 |
159.94 |
36.36 |
| C11─ H11b⋯Cg3 [Cg2: C1 C2 C3 C4 C9 C10] |
0.970 |
3.480 |
4.280 |
143.62 |
11.66 |
| Cg1⋯Cg2 [Cg3: C12 C13 C14 C15 C16 C17] |
— |
4.187 |
— |
— |
— |
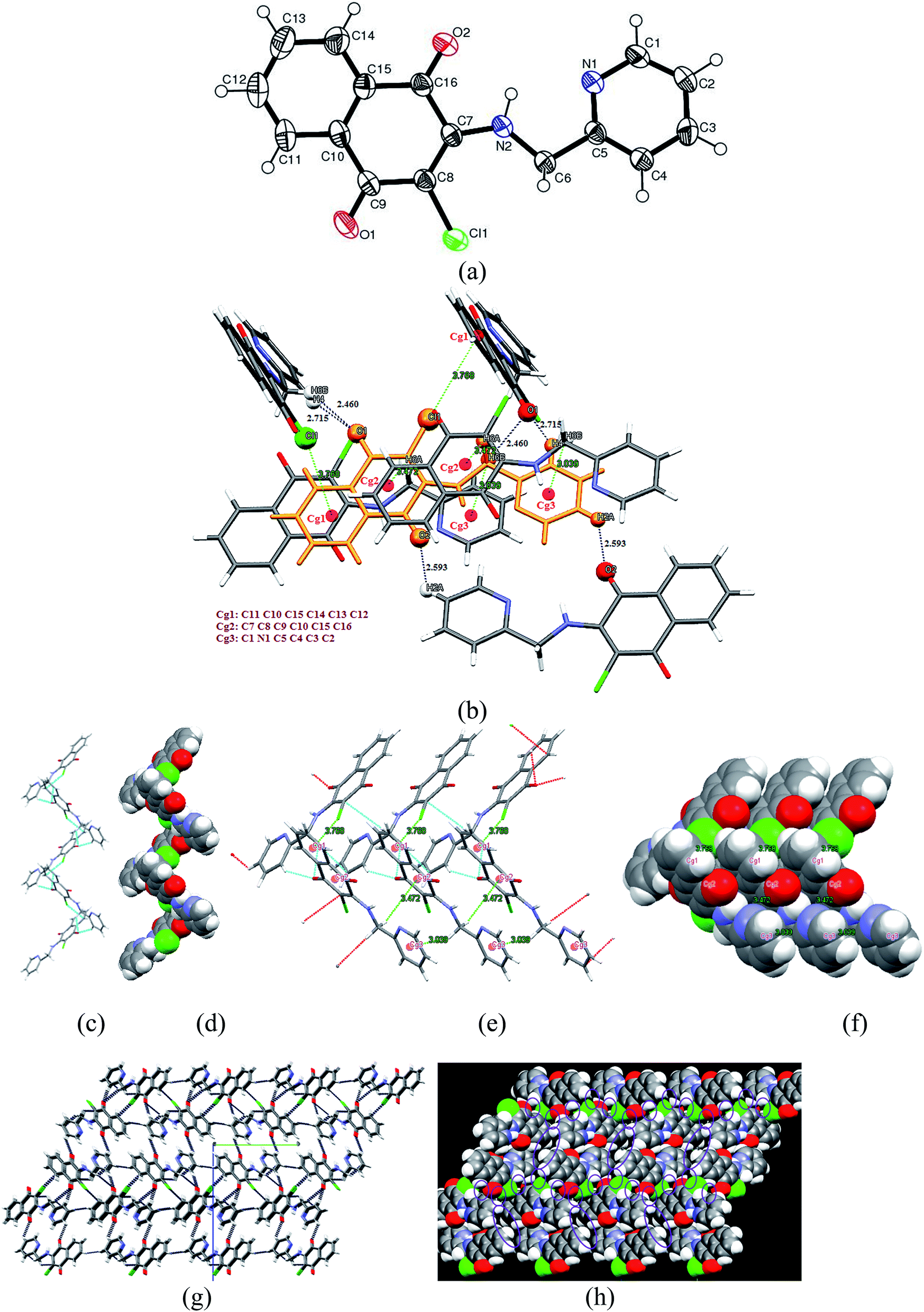
3.2.2 X-ray structure of 2-(benzylamino)-3-chloro-1,4-naphthoquinone [C17 H12ClNO2] (2). Compound 2 crystallizes in the centrosymmetric monoclinic P21/c space group and the X-ray crystal structure shows that the asymmetric unit contains a full molecule of [C17 H12ClNO2], as shown in Fig. 2(a). There are four such molecules in the unit cell. The relevant parameters are tabulated in Table 1. The selected bond distances (Å) and bond angles (°) for 2 are: C1–C2 1.471(3), C1–O1 1.210(3), C1–C10 1.519(3), C7–C8 1.501(3), C8–O2 1.227(3), C8–C9 1.444(3), C9–Cl1 1.732(2), C10–N1 1.344(3), N1–C11 1.467(3), C11–C12 1.518(3) Å and C2–C1–O1 121.5(2), C10–C1–O1 119.1(2), C2–C1–Cl0 119.38(18), C7–C8–O2 119.6(2), C9–C8–O2 122.2(2), C7–C8–C9 118.16(18), C8–C9–C11 113.22(16), C10–C9–Cl1 122.83(18), C10–N1–Cl1 131.7(2), N1–C11–C12 112.56(17)°, respectively. These values followed the normal range and were found consistent with similar types of compounds reported in the literature (vide supra). Notably, the presence of an N-benzyl substituent efficiently changes the molecular orientation, which appeared to play a great role on crystal packing pattern of 2. The molecules of 2 appeared to be involved in an unusual C–Cl⋯π donor–acceptor interactions (Fig. 2(b)) along with CH⋯O and N–H⋯O {C6–H6⋯O1 (2.303 Å), N1–H1⋯O2 (2.295 Å) and C17–H17⋯O2 (2.715 Å)} donor–acceptor interactions. It may be noted that similar to compound 1, O2 is involved in the bifurcated CH⋯O interactions. The relevant parameters for weak interactions present in compound 2 are given in Table 2. Probably, the anti-parallel orientations of phenyl and naphthoquinone rings, adopting a ‘gauche’ conformation with a dihedral angle of 72.79°, is responsible for the efficient C–Cl⋯π interactions. For such interactions to occur, it is not mandatory that the chlorine atom should be positioned directly above the phenyl ring plane with an orthogonal orientation. Statistical data retrieved from PDB suggest that in the majority cases of protein–ligand complexes,15 the frequency of distributions of α angle (angles between the vector along the Cg–Cl line and the normal to the plane of the ring) and β angle (∠C–Cl–Cg°), as shown in Fig. 3, varies in the range of 20–30° and 160–180°. Compound 2 displays a Cl⋯Cg distance (3.879 Å) and favourable α angle (31.10°) in the standard range.15,29 The α angle was calculated from α = 90° − θ, where θ is the angle of intersection of the phenyl ring plane (58.90°) and the β angle C9–Cl1⋯Cg (Cg: C16 C17 C12 C13 C14 C15) of 160.96° is adequately large to facilitate the effective C–Cl⋯π interactions. In the solid state, the molecule present in the asymmetric unit forms four donor–acceptor contacts, viz., C6–H6(naphthoquinone)⋯O1, N1–H1⋯O2, C17–H17(benzylic)⋯O2 and C11–Cl11⋯π(benzylic) interactions, which lead to a five molecule supramolecular aggregate, as shown in Fig. 2(b). Notably, C–Cl⋯π donor–acceptor interactions efficiently manage the anti orientation of packed molecules, limiting the growth of self-assembly along the c-axis. However, the dimensionality has been extended along the b-axis through C–H⋯O and N–H⋯O interactions, leading to the fascinating 1D infinite supramolecular chair-like architecture, as shown in Fig. 2(c) and (d).
 |
| | Fig. 2 (a) Thermal ellipsoidal plot of compound 2 with atom labeling scheme. (b) Supramolecular unit consists of five molecule aggregates involving CH⋯O, NH⋯O and Cl⋯π interactions. (c) Formation of 1D infinite supramolecular chair-like architecture (d) Spacefill representation of molecular packing along the b-axis. | |
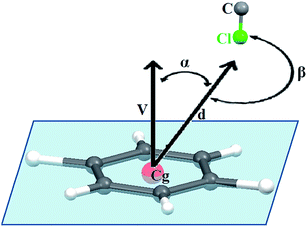 |
| | Fig. 3 Protocol for Cl⋯π interactions; d is the distance between the phenyl ring centroid (Cg) and the H atom; V is the vector normal to the plane of phenyl ring; α is the angle between the d and V vectors; and β is the C–Cl⋯π angle. | |
3.2.3 X-ray structure of 2-(furfurylamino)-3-chloro-1,4-naphthoquinone [C15H10ClNO3] (3). Compound 3 crystallizes in the centrosymmetric monoclinic P21/c space group and the X-ray crystal structure shows an asymmetric unit that contains a full molecule of [C15H10ClNO3], as shown in Fig. 4(a). Similar to the compound 2, there are four molecules in the unit cell. The relevant parameters are tabulated in Table 1. The selected bond distances (Å) and bond angles (°) for 3 are: C1–C2 1.454(2), C4–O1 1.220(18), C2–C3 1.370(2), C1–O2 1.228(18), C2–Cl1 1.734(15), C3–N1 1.341(19), N1–C11 1.469(19), C11–C12 1.487(2) Å and C2–C1–O2 121.74(14), C10–C1–O2 120.19(14), C1–C2–Cl1 114.14(11), C3–C2–Cl1 122.46(12), C5–C4–O1 121.75(13), C3–C4–O1 118.81(13), C3–N1–C11 130.63(13), N1–C11–C12 113.46(12), C2–C3–N1 130.82(14), C4–C3–N1 111.01(12)°, respectively, and the structural parameters were found consistent with similar types of compounds reported in the literature (vide supra). The amino, methylene and furfuryl groups of 3 behave as X–H donors, whereas the quinone group acts essentially as an X–H acceptor. For instance, the C14–H14 and C15–H15 groups associated with furfuryl and O2 and Cl11 groups of naphthoquinone are involved in the formation of close contacts with two different molecules via C14–H14⋯O2 and C15–H15⋯Cl11 donor–acceptor contacts, respectively, and these interactions evidently lead to attractive spiral-like arrangements along the a-axis, as shown in Fig. 4(b) and (c). It may be noted that the C15–H15 group is also involved in the interaction with the ketonic O2 group of naphthoquinone. The ketonic group of compound 3 is involved in trifurcated hydrogen bonding (unlike compounds 1 and 2), holding two molecules along the a-axis through C14–H14(furfuryl)⋯O2 and C15–H15(furfuryl)⋯O2 contacts and another molecule through C11–H11a(methylene)⋯O2 along c-axis. This tri-furcated C–H⋯O hydrogen bonding interaction arranged the molecules in the ac plane into an attractive comb-shaped assembly, as shown in Fig. 4(d). The second ketonic group O1 of the molecule present in the asymmetric unit forms an N1–H1⋯O1 donor–acceptor close contact with another molecule and arranges in an ‘anti’ fashion in the bc plane. The relevant parameters for weak interactions present in compound 3 are tabulated in Table 2. The supramolecular unit consists of eight aggregated molecules, as shown in Fig. 4(e), which on multiplication leads to a fascinating 3D supramolecular architecture (Fig. 4(f)).
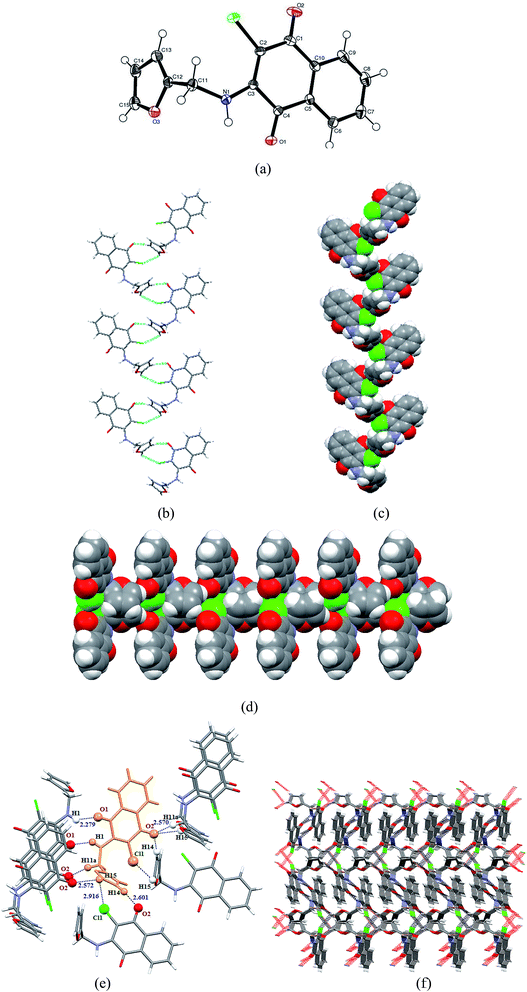 |
| | Fig. 4 (a) Thermal ellipsoidal plot of 3 with atom labelling scheme; (b) Formation of spiral shaped molecular arrangement of 3 through C15–H15⋯Cl and C14–H14⋯O2 donor–acceptor contacts along the a-axis; (c) spacefill representation of spiral-shaped assembly; (d) spacefill representation of an attractive comb-shaped assembly via trifurcated C–H⋯O hydrogen bonding in the ac plane; (e) supramolecular unit consisting of eight aggregated molecules involving NH⋯O, CH⋯O, and CH⋯Cl hydrogen bonding interactions; (f) formation of 3D infinite supramolecular architecture in capped stick model, view along c-axis. | |
3.2.4 X-ray structure of 2-(2-pyridylmethylamino)-3-chloro-1,4-naphthoquinone [C16H11ClN2O2] (4). Similar to the compounds 2 and 3, compound 4 bearing a N-(2-pyridylmethyl) substituent also crystallizes in the centrosymmetric monoclinic P21/c space group, and the X-ray crystal structure shows an asymmetric unit that contains a full molecule of [C16H11ClN2O2], as shown in Fig. 5(a). There are four such molecules orthogonally arranged in the unit cell. The relevant parameters are tabulated in Table 1. The selected bond distances (Å) and bond angles (°) for 4 are: C9–C10 1.492(5), C9–O1 1.226(4), C15–C16 1.483(5), C16–O2 1.212(4), C9–C8 1.452(5), C16–C7 1.518(5), C8–Cl1 1.736(3), C8–C7 1.371(3), C7–N2 1.338(4), N2–C6 1.449(4), C5–C6 1.513(4) Å and C10–C9–O1 120.4(3), C8–C9–O1 121.6(3), C9–C8–Cl1 114.6(2), C7–C8–Cl1 122.2(3), C15–C16–O2 122.0(3), C7–C16–O2 119.1(3), C7–N1–C6 130.2(3), N2–C7–C16 111.0(3), C5–C6–N2 109.5(3)°, respectively, and the structural parameters are found consistent with similar types of compounds reported in the literature (vide supra). More interestingly, the 2-pyridylmethyl substituent on the amino functionality of 4 drastically decreases the dihedral angle and preferentially adopts a lower energy molecular conformation, which is nearly coplanar to that of the naphthoquinone moiety (dihedral angle 11.12°), offering a large number of sites for weak interactions. The molecules present in the asymmetric unit form a supramolecular unit consisting of a six-molecule aggregate, as shown in Fig. 5(b). For instance, molecules of 4 are arranged in a zig–zag fashion, forming a chain along the b-axis through an emerging C–Cl1⋯π (Cg1: C10 C11 C12 C13 C14 C15; 3.768 Å) interaction and a cooperative bifurcated hydrogen bonding C4–H4⋯O1 (2.715 Å), C6–H6B⋯O1 (2.460 Å) donor–acceptor interaction, as shown in Fig. 5(c) and (d), which is indeed extended along the c-axis via C2–H2A⋯O2 (2.593 Å) donor–acceptor contacts. These zig–zag chains are apparently translated into orthogonally arranged layers along the c-axis through cooperative C6–H6A⋯π (Cg2: C7 C8 C9 C10 C15 C16; 3.472 Å) and C6–H6B⋯π (Cg3: C1 N1 C2 C3 C4 C5; 3.039 Å) donor–acceptor interactions. The structural parameters for these interactions are summarized in Table 2. All the three types of donor–acceptor interactions provide an opportunity to extend the dimensionality of supramolecular architecture and develop a 3D infinite network (Fig. 5(g)), which illustrates a number of voids along the a-axis in the spacefill model, as shown in Fig. 5(h).
 |
| | Fig. 5 (a) Thermal ellipsoidal plot of 4 with atom labelling scheme; (b) Supramolecular unit consisting of a six-molecule aggregate involving CH⋯O, CH⋯π and CCl⋯π interactions; (c) Capped stick and (d) spacefill representation of orthogonal molecular arrangement of 4 forming a zig–zag chain along the b-axis through cooperative C–Cl⋯Cg1 (C12 C11 C10 C15 C14 C13) and bifurcated C–H⋯O interactions; (e) Capped stick and (f) spacefill representation showing the translation of zig–zag chains into layers along the c-axis through cooperative C6H6A⋯Cg2 (quinone) and C6H6B⋯Cg3 (pyridine) donor–acceptor interactions; (g) Capped stick and (h) spacefill representation of 3D stacking of molecules of 4 along the a-axis, illustrating voids. | |
3.2.5 X-ray structure of 2-(benzylamino)-1,4-naphthoquinone [C17H13NO2] (7). Unlike other compounds, compound 7 crystallizes in the polar orthorhombic Pna21 space group, and the X-ray crystal structure shows an asymmetric unit that contains a full molecule of [C17H13NO2], as shown in Fig. 6(a). There are four such molecules arranged in parallel along the b-axis in the unit cell. The relevant parameters are tabulated in Table 1. The selected bond distances (Å) and bond angles (°) for 7 are: C9–C10 1.473(7), C10–O1 1.219(6), C10–C1 1.496(7), C1–C2 1.359(7), C3–C4 1.496(8), C3–O2 1.243(6), C2–C3 1.419(8), C1–N1 1.337(6), N1–C11 1.439(6), C11–C12 1.503(8) Å and C9–C10–O1 122.6(5), C1–C10–O1 119.6(5), C4–C3–O2 119.2(5), C2–C3–O2 121.7(6), C1–N1–C11 124.1(5), N1–C11–C12 116.2(5)°, respectively, and the structural parameters were found to be consistent with earlier structures.26b Interestingly, the presence of the N-benzyl substituent and the absence of the 3-chloro substituent from naphthoquinone greatly affected the crystal packing pattern of 7. Surprisingly, it appears that the absence of a chloro substituent did not affect the distortion of the naphthoquinone moiety, and its conformation was found almost consistent to that of compound 2. However, the absence of repulsive interactions between chloro group and aromatic substituent leads to a nearly orthogonal arrangement of the rings (naphthoquinone and phenyl), possessing a dihedral angle of 88.04°. The molecule present in the asymmetric unit exhibits a vast number of intermolecular contacts, which includes two CH⋯O, one NH⋯O, two CH⋯π and one π⋯π donor–acceptor interactions: C2–H2⋯O1 (2.443 Å), C13–H13⋯O2 (2.709 Å), N1–H1⋯O2 (2.168 Å), and C16–H16⋯π(Cg1) (3.522 Å), C11–H11b⋯π(Cg3) (3.480 Å) and π(Cg1)⋯π(Cg2) (4.187 Å) (where, Cg1: C4 C5 C6 C7 C8 C9; Cg2: C1 C2 C3 C4 C9 C10; Cg3: C12 C13 C14 C15 C16 C17), respectively. Similar to other compounds, one of the ketonic oxygens O2 is involved in bifurcated CH⋯O interactions. As a result of all of these interactions, the molecules present in the asymmetric unit forms a supramolecular unit, consisting of a nine-molecule aggregate, as shown in Fig. 6(b). The structural parameters for the weak interactions observed in compound 7 are tabulated in Table 2. Individually, C2–H2⋯O1 and C13–H13⋯O2 donor–acceptor interactions mutually connect the molecules along the a-axis, while N1–H1⋯O2 donor–acceptor interactions lead to a 1D network in the ac plane. Moreover, the π(Cg1)⋯π(Cg2) and C11–H11B⋯π(Cg3) interactions are mutually involved in the formation of a cavity ∼4.187 × 6.922 Å2, and these donor–acceptor interactions contribute to the replication of cavities along the c-axis. Fascinatingly, C16–H16⋯π(Cg1) donor–acceptor interactions predominantly contribute to a 1D stacking of molecules, leading to a right-handed helical packing along the b-axis with a helical pitch of 24.634 Å, as shown in Fig. 6(c). When these interactions are extended, it leads to the formation of layers containing helically arranged molecules. Each layer consists of parallel helices (either P or M helical) separated by ∼3.769 Å, and the lower layer contains opposite helices to that of the upper layer. These two layers form a grid-like structure with a cross-sectional angle of 23.57°, and the separation distance between the layers is ∼2.901 Å. A view along the a axis is shown in Fig. 6(d). The mutual effect of all the interactions resulted in a fascinating three dimensional architecture, which contains a number of openings along the a–axis, as shown in Fig. 6(e) and (f).
 |
| | Fig. 6 (a) Thermal ellipsoidal plot of 7 with atom labelling scheme; (b) supramolecular unit consisting of a nine-molecule aggregate involving CH⋯O, CH⋯π and π⋯π interactions; (c) helical packing of 7 along the b-axis through C16–H16⋯π interactions; (d) grid like packing of P and M helical layers in 7, view along the a-axis; (e) Capped stick view of three dimensional arrangement of molecules along the c-axis; (f) View of molecular packing in spacefill model along the c-axis illustrating voids in the packed molecules. | |
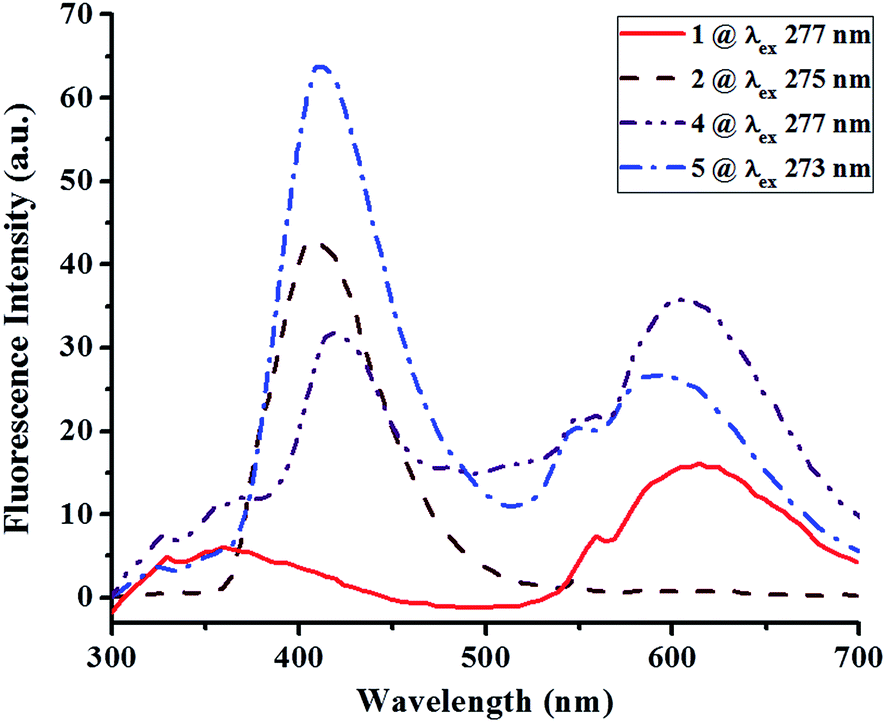
3.3 UV-visible absorption and fluorescence properties
The UV-visible absorption (Fig. 7) and fluorescence spectra (Fig. 8) of all the compounds 1–7 were obtained at room temperature from 10−5 M CH2Cl2 solution and 10−5 M DMF solution samples, respectively, and the pertinent results are summarized in Table 3. The assignments of UV-visible absorption and emission bands were performed based upon the absorption and emission spectra of closely related compounds.30–32
 |
| | Fig. 7 UV-visible absorption spectra of compounds 1–7 in 10−5 M CH2Cl2 solution. | |
 |
| | Fig. 8 Fluorescence spectra of compounds 1, 2, 4 and 5 in 10−5 M DMF solution. | |
Table 3 UV-visible absorption and fluorescence spectral data for 1–7
| Entry |
UV-visible spectral data (10−5 M CH2Cl2) |
Fluorescence spectral data (10−5 M DMF) |
| λmax nm (ε, L Mol−1 cm−1) |
Transitions |
Wavenumber (cm−1) |
λex (nm) |
λem (nm) (intensity) |
| 1 |
277 (100205) |
π → π* |
36036 |
277 |
358(5), 613(16) |
| 334 (8014) |
π → π* |
29940 |
| 473 (12985) |
n → π* |
21142 |
| 2 |
275 (68146) |
π → π* |
36364 |
275 |
406(43) |
| 331 (6685) |
π → π* |
30211 |
| 461 (8873) |
n → π* |
21678 |
| 3 |
273 (102221) |
π → π* |
36630 |
— |
Not fluorescent |
| 334 (10534) |
π → π* |
29940 |
| 457 (12476) |
n → π* |
21881 |
| 4 |
277 (77518) |
π → π* |
36140 |
277 |
418(32), 553(29), 605(36) |
| 327 (15019) |
π → π* |
30581 |
| 465 (8304) |
n → π* |
21551 |
| 5 |
273 (90855) |
π → π* |
36523 |
273 |
412(64), 546(21) |
| 327 (14341) |
π → π* |
30581 |
| 459 (10808) |
n → π* |
21786 |
| 6 |
276 (80642) |
n → π* |
36231 |
— |
Not fluorescent |
| 332 (4558) |
π → π* |
30120 |
| 470 (8474) |
n → π* |
21276 |
| 7 |
270 (76100) |
π → π* |
37037 |
— |
Not fluorescent |
| 330 (8191) |
π → π* |
30303 |
| 440 (10312) |
n → π* |
22727 |
The UV-visible absorption spectra of 1–7 showed the expected benzene and naphthoquinone bands at 270–277 nm and 327–334 nm regions, respectively, which mainly arise due to π → π* electronic transitions.26c In addition, a broad low energy band is observed in the visible region centered between 440 nm and 473 nm. This latter absorption is typical of amino-substituted benzoquinones, naphthoquinones and anthraquinones33 and is assigned to CT transitions and weak n → π* transitions of the carbonyl group in the quinone (Table 3). Notably, this broad band in the visible region for compound 1 and 6 showed a significant bathochromic shift relative to all other compounds probably due to the +I effects of N-cyclohexyl and N-n-butyl substituents, respectively.
Apart from this, fluorescence spectra indicate that the compounds 1, 2, 4 and 5 fluoresced in the range of 350–620 nm upon excitation at their respective λmax values of 277, 275, 277 and 273 nm with concomitant Stokes shifts of 81, 131, 141 and 131 nm, respectively. The spectrum of each compound is indeed composed of two broad bands and are comparable to those of closely related compounds.34 Compound 1 bearing cyclohexyl substituents exhibits a weak emission (5 a.u. at 358 nm and 16 a.u. at 613 nm) from locally excited (π* → π) states at room temperature. However, the alteration of cyclohexyl substituents to benzyl substituents in compound 2 causes a significant increase in fluorescence (43 a.u. at 406 nm). Surprisingly, the fluorescence of compounds 4 (32 a.u. at 418 nm; 29 a.u. at 553 nm) and 5 (64 a.u. at 412 nm; 21 a.u. at 546 nm) differ significantly and this suggests a great role of the position of pyridyl nitrogen on their fluorescence property. The earlier studies35,36 revealed that the fluorescence behavior of a molecule greatly depends upon the arrangement of molecular fragments, leading to polymorphism, conformational rigidity of the fluorophore (dihedral angles), and intermolecular interactions π⋯π or C–H⋯π interactions, and it also depends upon the nature of substituents.
3.4 Thermogravimetric studies for 1–7
A thermogravimetric study of 1–7 performed under N2 atmosphere from room temperature to 750 °C at a heating rate of 10 °C min−1, and thermal analysis data of all the compounds is given in Table 4.
Table 4 Thermogravimetric data for 1–7
| Comp. no. |
Mp °C |
Mass loss% (temperature range °C) |
Residual content |
DTG (°C) |
DTA (°C) |
| 1 |
116.7 |
64% (200–550) |
36% char |
250.6 |
116.7(endo), 253.3(exo) |
| 2 |
112.4 |
62.7% (180–700) |
37.3% char |
201.6, 460 |
112.4(endo), 201.9(exo) |
| 3 |
147.3 |
44.7% (150–700) |
Mass loss continues after 750 °C |
173.7, 220.1 |
147.3(endo), 177.4(exo) |
| 4 |
160.4 |
42% (150–700) |
Mass loss continues after 750 °C |
161.4 |
160.4(endo), 192.7(exo) |
| 5 |
151.8 |
35.1% (150–700) |
Mass loss continues after 750 °C |
146.3 |
151.8(endo) |
| 6 |
109.8 |
64.5% (200–500) |
35.5% char |
110.2, 126.9 |
109.8(endo), 220.6(exo) |
| 7 |
153.9 |
92.4% (205–400) |
7.6% char |
275.4 |
153.9(endo) |
Single (compound 1, 5–7) or multi (compound 2–4) stage thermal degradation patterns were observed on TG curves. All the compounds (except 5) exhibit their first endothermic peak on the DTA curves without any significant mass loss on the corresponding TG curves due to the phase change, attributable to the melting points of the respective compounds. However, other DTA peaks are attributed to the exothermic elimination of molecular fragments due to the thermal degradation, confirmed by the DTG curves. Compounds 3–5, bearing aromatic heterocyclic N-substituents appeared to be thermally unstable and the mass loss for these started at ∼150 °C; however, other compounds were thermally stable up to 200 °C (ESI†). Despite the similar type of molecular framework, the diversity in the thermal stability of these compounds could be attributed to the presence of various intermolecular interactions in the solid state, leading to diverse crystal packing patterns (vide supra).
3.5 Cyclic voltammetric study
The electrochemical investigations of 1–7 were performed in the potential range from +0.5 to −2.0 V at a scan rate of 50 mV s−1 in 1.0 mM CH2Cl2 solutions containing nBu4NPF6 (0.1 M) as the supporting electrolyte. The voltammograms for 1–7 are shown in Fig. 9, and electrochemical data is presented in Table 5. The electrochemical examination of 1–7 clearly demonstrates the quasi-reversible redox behavior of 1, 2, 4 and 5. Similar to the electrochemical behavior of methyl-halogenated naphthoquinones,37 these compounds primarily exhibit two single-electronic waves due to the electroreduction of the naphthoquinone functionality to a semiquinone radical anion and then to a dianion. Both the waves are reversible in nature. The separation between each cathodic and corresponding anodic peak, ΔEp at 50 mV s−1 scan rate was larger than 59 mV (Table 5), and the ratio of the current intensity of the cathodic and anodic peaks were different from unity, suggesting a quasi-reversible nature of both the redox couples of 1, 2, 4 and 5. Notably, compound 4, bearing a N-(2-methylpyridine) substituent, displays an additional peak in the cathodic scan (Ep#c) as well as in the corresponding anodic scan (Ep#a), which resulted in a significant cathodic shift of the electroreduction peaks associated with naphthoquinone moieties probably due to increased electron density caused by the initial electroreduction of 2-pyridine center.38 However, the voltammograms of other compounds display similar features, except for the differences in the cathodic/anodic current peak heights (Fig. 9). Unlike these compounds, the cyclic voltammograms of compounds 3, 6 and 7 display only one single-electronic wave in a cathodic scan due to the electroreduction of the naphthoquinone functionality to a semiquinone radical anion under similar conditions, as shown in Fig. 9.
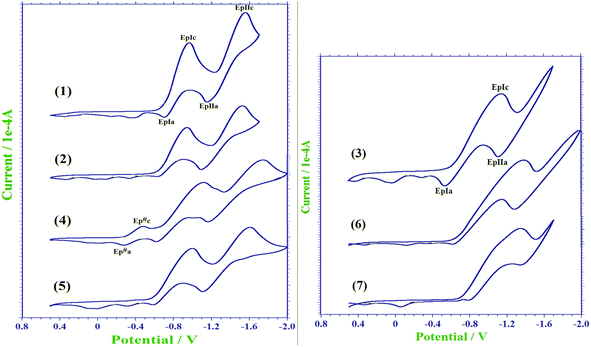 |
| | Fig. 9 Cyclic voltammograms of a 1 mM solution of compounds 1–7 in CH2Cl2 containing 0.1 M tetra-n-butylammonium hexafluorophosphate as the supporting electrolyte. | |
Table 5 Electrochemical data for the compounds 1–7
| Entry |
EpIc (V) |
EpIIc (V) |
E#pc (V) |
EpIa (V) |
EpIIa (V) |
E#pa (V) |
ΔEpI (V) |
ΔEpII (V) |
ΔE#p (V) |
| 1 |
−0.962 |
−1.555 |
— |
−0.698 |
−1.151 |
|
0.264 |
0.404 |
— |
| 2 |
−0.941 |
−1.526 |
— |
−0.620 |
−1.089 |
|
0.321 |
0.437 |
— |
| 3 |
−1.145 |
— |
— |
−0.537 |
−1.112 |
|
0.608 |
— |
— |
| 4 |
−1.118 |
−1.746 |
−0.480 |
−0.619 |
−1.167 |
−0.274 |
0.499 |
0.579 |
0.206 |
| 5 |
−0.998 |
−1.606 |
— |
−0.585 |
−1.108 |
|
0.413 |
0.498 |
— |
| 6 |
−1.373 |
— |
— |
−0.619 |
−1.284 |
|
0.754 |
— |
— |
| 7 |
−1.348 |
— |
— |
−0.785 |
−1.343 |
|
0.563 |
— |
— |
3.6 Biological evaluation
In the light of the antibacterial action of quinones,39 all the naphthoquinone derivatives 1–7 were screened for their antibacterial activity against two gram positive bacteria S. aureus and B. subtilis and two gram negative bacteria E. coli and P. aeruginosa by the Broth dilution method.40 These compounds were further evaluated for their in vitro antifungal activity against C. albicans and A. niger. The concentration of compounds was varied from 10 μg mL−1 to 600 μg mL−1. Analysis suggests that solvent alone had no antibacterial or antifungal activities against any of the tested microorganisms. For comparison purposes, two standard drugs ciprofloxacin and flucanazole were taken as reference and tested under the similar conditions. The lowest concentrations of the compounds that prevented visible growth i.e. minimum inhibitory concentration (MIC) of the naphthoquinone derivatives 1–7 against inhibited organisms is summarized in Table 6. A careful comparison of the antibacterial activity of compounds 1–7 with the well-known antibacterial drug ciprofloxacin (MIC against S. aureus: 15 μg mL−1 and MIC against B. subtilis: 5 μg mL−1) showed that compounds 4 and 5 showed the best activity against both the gram positive bacteria S. aureus and B. subtilis (Table 6). However, compounds 1, 3, 6 and 7 displayed less activity against both the gram positive bacteria, except compound 6, which exhibited moderate activity against B. subtilis. Further results suggest that all compounds exhibit moderate activity against both the gram negative bacteria, except compound 2, which showed less activity against E. coli. The presence of N-methylpyridine substituents in 4 and 5 apparently endowed them with extremely potent activity against both the gram positive bacteria. Remarkably, compound 5 exhibited an enhanced antibacterial activity against S. aureus and proved to be a more potent antibacterial agent than ciprofloxacin. All these naphthoquinone derivatives showed high MIC values (600 μg mL−1) against both the fungi, except compounds 4 and 5, which bear pyridine N-substituents, exhibited moderate (MIC 300 μg mL−1) activity against C. albicans (Fig. 10). In general, the naphthoquinone derivatives were found to exhibit better anti-microbial properties than a number of recently reported coumarin derivatives41 against the same set of microbes. Further comparison of antibacterial properties of 4 and 5 with those of closely related compounds16e,42 against S. aureus suggest that these compounds showed better activity than a number of 1,4-naphthoquinone derivative such as 2-arylamino-3-chloro-1,4-naphthoquinones, 2-amino-3-arylsulfanyl-1,4-naphthoquinones and 2-arylamino-3-arylsulfanyl-1,4-naphthoquinones.
Table 6 MIC determination of antibacterial and antifungal agent (μg mL−1)
| Entry |
MIC (μg mL−1) |
| S. aureus |
B. subtilis |
E. coli |
P. aeruginosa |
C. albicans |
A. niger |
| (G +ve bacteria) |
(G −ve bacteria) |
(Fungi) |
| 1 |
>600 |
600 |
200 |
200 |
>600 |
>600 |
| 2 |
200 |
100 |
600 |
200 |
>600 |
>600 |
| 3 |
>600 |
600 |
300 |
200 |
>600 |
>600 |
| 4 |
20 |
10 |
300 |
300 |
300 |
>600 |
| 5 |
10 |
10 |
200 |
200 |
300 |
>600 |
| 6 |
600 |
300 |
300 |
200 |
600 |
>600 |
| 7 |
>600 |
>600 |
200 |
200 |
>600 |
>600 |
| Ciprofloxacin |
15 |
5 |
15 |
10 |
— |
— |
| Flucanazole |
— |
— |
— |
— |
10 |
40 |
 |
| | Fig. 10 Zone of inhibition of compound 4 and 5 against Staphylococcus aureus. | |
4. Conclusions
This study allows us to conclude that the introduction of various amine N-substituents in 1–4 and 7 induces conformational changes, which apparently modify the nature and number of donor–acceptor sites for noncovalent interactions, leading to diverse crystal packing patterns. The N-(phenylmethyl)- and N-(2-pyridylmethyl)-substituents in 2 and 4 successfully lead to the opening of an emerging C–H⋯π synthon, scarcely seen in the crystal packing of organic molecules. This synthon can efficiently manage the anti orientation of molecules of 2 and limit the growth of self-assembly along the c-axis; however, the self-assembly can be extended along the b-axis through cooperative C–H⋯O and N–H⋯O interactions, leading to a fascinating 1D infinite supramolecular chair-like architecture. The molecules of 4 are arranged in a zig–zag fashion, forming a chain along the b-axis via cooperative C–Cl⋯π and bifurcated hydrogen bonding. The mutual effect of all interactions in 4 resulted into the formation of a fascinating 3D infinite network, illustrating a number of openings along the a-axis in the spacefill model. The fluorescence study indicates that the compounds 1, 2, 4 and 5 fluoresce in the range of 350–620 nm with the concomitant Stokes shifts of 81, 131, 141 and 131 nm, respectively, and their cyclic voltammograms evidence two quasi-reversible single-electron waves. All the compounds (except 5) exhibit their first endothermic peak on the DTA curves without any significant mass loss on the corresponding TG curves due to the phase change, attributable to the melting points of the respective compounds. A careful comparison of the antibacterial activity of compounds 1–7 with a well-known antibacterial drug ciprofloxacin (MIC against S. aureus: 15 μg mL−1 and MIC against B. subtilis: 5 μg mL−1) showed that 4 and 5 showed the best activity against both the gram positive bacteria S. aureus and B. subtilis. The presence of N-pyridylmethyl substituents in 4 and 5 apparently endowed them with extremely potent activity against S. aureus and B. subtilis. Interestingly, compound 5 exhibited an enhanced antibacterial activity against S. aureus and proved to be a more potent antibacterial agent than ciprofloxacin.
Acknowledgements
VKS acknowledges CSIR, New Delhi for financial support (Project no. 01/(2733)/13/EMR-II), SKV and RK acknowledge the UGC, New Delhi for fellowships.
References
-
(a) A. T. Hulme, S. L. Price and D. A. Tocher, J. Am. Chem. Soc., 2005, 127, 1116 CrossRef CAS PubMed
 ;
(b) M. T. Kirchner, D. Bläser and R. Boese, Chem.–Eur. J., 2010, 16, 2131 CrossRef CAS PubMed .
;
(b) M. T. Kirchner, D. Bläser and R. Boese, Chem.–Eur. J., 2010, 16, 2131 CrossRef CAS PubMed . - G. R. Desiraju, Curr. Opin. Solid State Mater. Sci., 1997, 2, 451 CrossRef CAS .
-
(a) Y. Gu, T. Kar and S. Scheiner, J. Am. Chem. Soc., 1999, 121, 9411 CrossRef CAS ;
(b) Z. S. Derewenda, L. Lee and U. Derewenda, J. Mol. Biol., 1995, 252, 248 CrossRef CAS ;
(c) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, in Structural Chemistry and Biology, Oxford University Press, New York, 2001 Search PubMed ;
(d) P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253 CrossRef CAS PubMed ;
(e) G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS PubMed ;
(f) R. Taylor and O. Kennard, J. Am. Chem. Soc., 1982, 104, 5063 CrossRef CAS .
-
(a) G. Mukherjee and K. Biradha, J. Chem. Sci., 2014, 126, 1285 CrossRef CAS ;
(b) S. Scheiner, Phys. Chem. Chem. Phys., 2011, 13, 13860 RSC .
-
(a) M. Nishio, CrystEngComm, 2004, 6, 130 RSC ;
(b) M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757 RSC ;
(c) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049 CrossRef CAS PubMed ;
(d) S. K. Nayak, R. Sathishkumar and T. N. Guru Row, CrystEngComm, 2010, 12, 3112 RSC ;
(e) O. Takahashi, Y. Kohno, S. Iwasaki, K. Saito, M. Iwaoka, S. Tomoda, Y. Umezawa, S. Tsuboyama and M. Nishio, Bull. Chem. Soc. Jpn., 2001, 74, 2421 CrossRef CAS ;
(f) A. K. Tewari and R. Dubey, Bioorg. Med. Chem., 2008, 16, 126 CrossRef CAS PubMed ;
(g) Y. Kobayashi and K. Saigo, J. Am. Chem. Soc., 2005, 127, 15054 CrossRef CAS PubMed ;
(h) K. Saigo and Y. Kobayashi, Chem. Rec., 2007, 7, 47 CrossRef CAS PubMed ;
(i) P. Sozzani, A. Comotti, S. Bracco and R. Simonutti, Chem. Commun., 2004, 768 RSC ;
(j) J. P. Hill, R. Scipioni, M. Boero, Y. Wakayama, M. Akada, T. Miyazaki and K. Ariga, Phys. Chem. Chem. Phys., 2009, 11, 6038 RSC ;
(k) E. R. T. Tiekink and J. Z. Schpector, The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering, John Wiley & Sons Inc., 2012 Search PubMed ;
(l) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC ;
(m) M. Nishio, J. Mol. Struct., 2012, 1018, 2 CrossRef CAS PubMed .
-
(a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS ;
(b) C. A. Hunter, Chem. Soc. Rev., 1994, 23, 101 RSC ;
(c) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC ;
(d) T. F. Headen, C. A. Howard, N. T. Skipper, M. A. Wilkinson, D. T. Bowron and A. K. Soper, J. Am. Chem. Soc., 2010, 132, 5735 CrossRef CAS PubMed ;
(e) R. Kadu, V. K. Singh, S. K. Verma, P. Raghavaiah and M. M. Shaikh, J. Mol. Struct., 2013, 1033, 298 CrossRef CAS PubMed .
-
(a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed ;
(b) K. Rissanen, CrystEngComm, 2008, 10, 1107 RSC ;
(c) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 CrossRef CAS PubMed ;
(d) P. Metrangolo, Y. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461 CrossRef CAS PubMed ;
(e) R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677 CrossRef CAS PubMed .
-
(a) D. Swierczynski, R. Luboradzki, G. Dolgonos, J. Lipkowski and H. J. Schneider, Eur. J. Org. Chem., 2005, 1172 CrossRef CAS PubMed ;
(b) L. Brammer, G. M. Espallagras and S. Libri, CrystEngComm, 2008, 10, 1712 RSC .
-
(a) P. Auffinger, F. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789 CrossRef CAS PubMed ;
(b) A. R. Voth, F. A.Hays and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6188 CrossRef CAS PubMed .
-
(a) R. Glaser, N. Chen, H. Wu, N. Knotts and M. Kaupp, J. Am. Chem. Soc., 2004, 126, 4412 CrossRef CAS PubMed ;
(b) O. V. Shishkin, V. N. Khrustalev, S. V. Lindeman, L. Y. Ukhin, Z. I. Orlova and T. N. Gribanova, Z. Kristallogr., 1998, 213, 296 CrossRef .
-
(a) A. C. Legon, Chem. Phys. Lett., 1999, 314, 472 CrossRef CAS ;
(b) A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686 CrossRef .
-
(a) C. B. Aakeroy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772 CrossRef PubMed ;
(b) K. Bouchmella, B. Boury, S. G. Dutremez and A. van der Lee, Chem.–Eur. J., 2007, 13, 6130 CrossRef CAS PubMed .
- G. M. Espallargas, F. Zordan, L. A. Marin, H. Adams, K. Shankland, J. van der Streek and L. Brammer, Chem.–Eur. J., 2009, 15, 7554 CrossRef PubMed .
-
(a) Y. Nie, J. Miao, H. Pritzkow, H. Wadepohl and W. Siebert, J. Organomet. Chem., 2013, 747, 174 CrossRef CAS PubMed ;
(b) V. R. Hathwar, S. M. Roopan, R. Subashini, F. N. Khan and T. N. Gururow, J. Chem. Sci., 2010, 122, 677 CrossRef CAS PubMed ;
(c) S. Yamada, N. Sako, M. Okuda and A. Hozumi, CrystEngComm, 2013, 15, 199 RSC ;
(d) C. B. Aakeröy, N. Schultheiss, A. Rajbanshi, J. Desper and C. Moore, Cryst. Growth Des., 2009, 9, 432 CrossRef PubMed .
-
(a) Y. Imai, Y. Inoue, I. Nakanishi and K. Kitaura, Protein Sci., 2008, 17, 1129 CrossRef CAS PubMed ;
(b) Y. Lu, Y. Wang and W. Zhu, Phys. Chem. Chem. Phys., 2010, 12, 4543 RSC .
-
(a) S. L. de Castro, F. S. Emery and E. N. da Silva Júnior, Eur. J. Med. Chem., 2013, 69, 678 CrossRef CAS PubMed ;
(b) V. F. Ferreira, S. B. Ferreira and F. D. C. da Silva, Org. Biomol. Chem., 2010, 8, 4793 RSC ;
(c) D. A. Lanfranchi, E. C. Rodo, B. Bertrand, H. H. Huang, L. Day, L. Johann, M. Elhabiri, K. Becker, D. L. Williams and E. D. Charvet, Org. Biomol. Chem., 2012, 10, 6375 RSC ;
(d) V. R. Campos, E. A. dos Santos, V. F. Ferreira, R. C. Montenegro, M. C. B. V. de Souza, L. V. C. Lotufo, M. O. de Moraes, A. K. P. Regufe, A. K. Jordao, A. C. Pinto, J. A. L. C. Resende and A. C. Cunha, RSC Adv., 2012, 2, 11438 RSC ;
(e) V. K. Tandon, H. K. Maurya, M. K. Verma, R. Kumar and P. K. Shukla, Eur. J. Med. Chem., 2010, 45, 2418 CrossRef CAS PubMed .
-
(a) G. Powis, Pharmacol. Ther., 1987, 35, 57 CrossRef CAS ;
(b) P. J. O'Brien, Chem.-Biol. Interact., 1991, 80, 1 CrossRef .
- C. Frontana, A. V. Mayagoitia, J. Garza, R. Vargas and I. Gonzalez, J. Phys. Chem. A, 2006, 110, 9411 CrossRef CAS PubMed .
-
(a) J. R. E. Hoover and A. R. Dag, J. Am. Chem. Soc., 1954, 76, 4148 CrossRef CAS ;
(b) S. A. Vichkanova, S. B. Izosimova, V. V. Adgina and L. D. Shipulina, Rastit. Resur., 1979, 15, 167 CAS ;
(c) N. G. Clark, Pestic. Sci., 1985, 16, 23 CrossRef CAS PubMed ;
(d) V. K. Tandon, H. K. Maurya, A. Tripathi, G. B. ShivaKesva, P. K. Shukla, A. Srivastava and D. Panda, Eur. J. Med. Chem., 2009, 44, 1086 CrossRef CAS PubMed ;
(e) V. K. Tandon, H. K. Maurya, D. B. Yadav, A. Tripathi, M. Kumar and P. K. Shukla, Bioorg. Med. Chem. Lett., 2006, 16, 5883 CrossRef CAS PubMed .
-
(a) K. W. Stagliano and A. Emadi, US20040087663A1, 2004 ;
(b) S. Ganapaty, P. S. Thomas, G. Karagianis, P. G. Waterman and R. Brun, Phytochemistry, 2006, 67, 1950 CrossRef CAS PubMed .
- C. Biot, H. Bauer, R. H. Schirmer and E. D. Charret, J. Med. Chem., 2004, 47, 5972 CrossRef CAS PubMed .
-
(a) A. B. Pardee, Y. Z. Li and C. J. Li, Cancer Drug Targets, 2002, 2, 227 CrossRef CAS ;
(b) B. Hazra, M. D. Sarma and U. Sanyal, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 812, 259 CrossRef CAS PubMed .
- Oxford Diffraction, CrysAlis PRO, Oxford Diffraction Ltd., Yarnton, England, 2009.
- G. M. Sheldrick, SHELXTL Reference Manual: Version 5.1, Bruker AXS, Madison, WI, 1997 Search PubMed .
- G. M. Sheldrick, SHELXL-97: Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed .
-
(a) J. C. Lien, L. J. Huang, J. P. Wang, C. M. Teng, K. H. Lee and S. C. Kuo, Bioorg. Med. Chem., 1997, 5, 2111 CrossRef CAS ;
(b) S. Cunha, L. Fernandes, P. Santos, Z. N. Rocha, R. Rivelino, J. Ferrari, I. Vencato and C. Lariucci, Quim. Nova, 2010, 33, 2108 CrossRef CAS PubMed ;
(c) O. Pawar, A. Patekar, A. Khan, L. Kathawate, S. Haram, G. Markad, V. Puranik and S. S. Gawali, J. Mol. Struct., 2014, 1059, 68 CrossRef CAS PubMed ;
(d) G. M. Neelgund and M. L. Budni, Spectrochim. Acta, Part A, 2005, 61, 1729 CrossRef PubMed .
- S. Bittner, S. Gorohovsky, O. Paz-Tal (Levi) and J. Y. Becker, Amino Acids, 2002, 22, 71 CrossRef CAS .
-
(a) S. Pal, M. Jadhav, T. Weyhermuller, Y. Patil, M. Nethaji, U. Kasabe, L. Kathawate, V. B. Konkimalla and S. S. Gawali, J. Mol. Struct., 2013, 1049, 355 CrossRef CAS PubMed . (in n-butyl crystal structure also);
(b) J. C. Knight, T. Tatchell and I. A. Fallis, Acta Crystallogr., 2007, 63, o1226–o1227 CAS .
- F. A. Shah, M. N. Tahir, S. Alia and M. A. Kashmiri, Acta Crystallogr., 2008, 64, o787 CAS .
- E. A. Perpete, C. Lambert, V. Wathelet, J. Preat and D. Jacquemin, Spectrochim. Acta, Part A, 2007, 68, 1326 CrossRef PubMed .
- E. Leyva, L. I. López, S. E. L. Carrillo, M. R. Kessler and A. M. Rojas, J. Fluorine Chem., 2011, 132, 94 CrossRef CAS PubMed .
- E. Leyva, S. J. S. Sobeck, S. E. L. Carrillo and D. A. M. Lara, J. Mol. Struct., 2014, 1068, 1 CrossRef CAS PubMed .
- T. Win and S. Bittner, Tetrahedron Lett., 2005, 46, 3229 CrossRef CAS PubMed .
- C. Ibis and N. G. Deniz, J. Chem. Sci., 2012, 124, 657 CrossRef CAS .
- Y. Zheng, K. Ma, H. Li, J. Li, J. He, X. Sun, R. Li and J. Ma, Catal. Lett., 2009, 128, 465 CrossRef CAS .
-
(a) J. A. O'Meara, N. Gardee, M. Jung, R. N. Ben and T. Durst, J. Org. Chem., 1998, 63, 3117 CrossRef ;
(b) F. Valot, F. Fache, R. Jacquot, M. Spagnol and M. Lemaire, Tetrahedron Lett., 1999, 40, 3689 CrossRef CAS ;
(c) A. K. Szardening, T. S. Burkoth, G. C. Look and D. A. Campbell, J. Org. Chem., 1996, 61, 6720 CrossRef ;
(d) C. Chiappe and D. Pieraccini, Green Chem., 2003, 5, 193 RSC .
- F. C. D. Abreu, A. C. O. Lopes, S. A. D. Monte, N. A. Soares and M. O. F. Goulart, J. Electroanal. Chem., 2003, 560, 79 CrossRef PubMed .
- A. Marjolin and J. A. Keith, ACS Catal., 2015, 5, 1123 CrossRef CAS .
-
(a) C. A. Colwell and M. Mccall, Science, 1945, 101, 592 CAS ;
(b) O. H. Ostenhof, Science, 1947, 105, 549 Search PubMed ;
(c) A. Bondi Jr, C. C. Dietz and E. H. Spaulding, Science, 1946, 103, 399 Search PubMed .
- National Committee for Clinical Laboratory Standards, Performance Standards for Antimicrobial Susceptibility Testing, 8th Informational Supplement. M100 S12, National Committee for Clinical Laboratory Standards, Villanova, Pa, 2002.
- J. N. Soni and S. S. Soman, Eur. J. Med. Chem., 2014, 75, 77 CrossRef CAS PubMed .
- V. K. Tandon, S. Kumar, N. N. Mishra and P. K. Shukla, Eur. J. Med. Chem., 2012, 56, 375 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and CIF files. See CCDC reference numbers 986339 for 1; 986337 for 2; 986340 for 3; 986336 for 4 and 986338 for 7. CCDC 986336–986340. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02295a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.