
2′-N-Guanidino,4′-C-ethylene bridged thymidine (GENA-T) modified oligonucleotide exhibits triplex formation with excellent enzymatic stability†
Jharna Barmana,
Deepanjali Guravbc,
Oommen P. Oommenb and
Oommen P. Varghese*b
aAgricultural and Ecological Research Unit, Biological Science Division, Indian Statistical Institute, Kolkata 700108, India
bPolymer Chemistry Division, Department of Chemistry, The Ångström Laboratory, Uppsala University, Box 538, 751 21 Uppsala, Sweden. E-mail: oommen.varghese@kemi.uu.se; Fax: +46 18 471 3477
cDepartment of Chemistry, SavitriBai Phule Pune University, India
First published on 14th January 2015
Abstract
Here we present the synthesis and characterization of a new 2′-N-Guanidino,4′-C-ethylene bridged thymidine (GENA-T) modified oligonucleotide possessing North-locked sugar conformation. Incorporation of GENA-T nucleotide though did not change the thermal stability of the oligonucleotides toward the complementary RNA; it significantly increased the stability of the parallel triplex at pH 7. The melting temperature of the triplex was increased by +9.5 °C as compared to that of the isosequential unmodified sequence. Moreover this modification imparted exceptional nuclease stability to the oligonucleotides for over 33 h. This study clearly demonstrates that GENA-T modified oligonucleotides could improve triplex formation with phenomenal enzymatic stability and could be used for various biomedical applications.
Chemically modified nucleotides have generated enormous interest in recent years as they improve the enzymatic stability and efficacy of oligonucleotides, transforming them into drug molecules for various therapeutic applications such as antisense, antigene, siRNA, microRNA, aptamer etc.1 Of particular interest has been North-type (C3′-endo) conformationally locked nucleotides, namely, Locked/Bridged Nucleic Acid (LNA/BNA). These modified nucleotides have an extra ring fused to the furanose structure at the 2′–4′ position.2,3 These types of 2′–4′ fused LNA analogues increase oligonucleotide (ON) rigidity which enhances the binding toward complimentary sequence by either preorganization or improved stacking (entropy or enthalpy).4,5 This stabilization can also be attributed to the presence of heteroatom at the 2′ position which influences the pseudoaromatic character of the nucleobase, as evident from the change in nucleobase pKa.6 LNA analogues have been used for different gene silencing applications such as antisense,7 siRNA8 and miRNA9 therapeutics in vivo. These modifications are also known to favour triplex formation when incorporated into triplex forming oligonucleotides (TFOs) for gene-targeted mutagenesis as in antigene approach.10 DNA TFOs bind purine-rich strand of a target double stranded DNA (dsDNA) either by parallel motif, as in pyrimidine-rich TFOs, or by antiparallel motif, as in polypurine TFOs. Though this technology is very promising, one challenge that remains elusive is to make these therapeutic ONs more resilient towards enzymatic degradation without altering the backbone structure or compromising their efficacy. Enhancing nuclease resistance of ONs by changing the phosphodiester backbone to phosphorothioates11 or N3′ → P5′ phosphoramidates12 is well established but often at the cost of non-specific binding to several proteins.13 We have earlier reported synthesis and properties of 2′-N,4′-C ethylene bridged thymidine analogue (aza-ENA-T) which showed significant improvement in enzymatic stability.14 Since the amino group (positive charge) near phosphate offers extra enzymatic stability (as observed in azetidine vs. oxetane comparison),15 we anticipated that converting amino residue of aza-ENA to guanidino group could impart interesting features to the existing properties of aza-ENA.
The unique ability of the guanidinium moiety to remain protonated at physiological pH (pKa ≈ 12.5) imparts interesting biological function when conjugated to ONs. For example, Sugimoto et al. demonstrated that incorporation of four guanidinium conjugated nucleobases in a 20mer oligonucleotide significantly enhanced cellular uptake.16 In another study, by Prakash et al., the 2′-amino and also corresponding guanidinium modified ONs have displayed the ability to bind dsDNA to form triplexes through the pyrimidine motif by Hoogsteen hydrogen bonding.17 Though TFOs are not approved for commercial use as drugs yet, these oligonucleotides have demonstrated great potential for therapeutic and biotechnological applications.18 Apart from the aforementioned problem of nuclease stability, TFOs face additional problem of intrinsic stability at physiological condition because of ionic repulsion of the three negatively charged DNA strands and the need for N3 protonation of cytosine nucleobase for G·C Hoogsteen H-bond formation.19 Only purine rich TFOs form stable triplexes at physiological pH.18 Since the presence of North-sugar puckering is known to favour Hoogsteen H-bonding,20 we envisaged that TFOs modified with GENA could demonstrate good triplex formation at physiological pH along with enhanced enzymatic stability.
Synthesis of GENA derivative was initiated from the previously synthesized aza-ENA intermediate 1.14 This intermediate was synthesized following a modified strategy of our previously reported protocol (incorporation of cyano intermediate) as described in the ESI.† Trifluoroacetyl group was deprotected using aqueous CH3NH2 in methanol and the product obtained was directly used for the guanylation reaction (Scheme 1). We initially attempted the guanylation reaction using PAC (phenoxyacetyl) protected SMe isothiourea. With this reagent guanylation reaction proceeded smoothly with 76% yield (as a mixture of rotamers). However, the corresponding phosphoramidate reaction was unsuccessful. This is presumably due to highly electrophilic imine bond of guanidine (due to amide protection), which undergo nucleophilic addition reaction with neighbouring trivalent P having non-bonding electrons. We circumvented this problem by using the carbamate protected SMe isothiourea, 2 having N-(2-(cyano)ethoxycarbonyl) or CEOC protecting group as reported by Manoharan et al.17 With this reagent the guanylation reaction was sluggish (48% in two steps after 4 days as mixture of rotamers). However, the phosphitylation reaction went smoothly under standard conditions to give the desired phosphoramidite building block with 66% yield. This highlights the significance of carbamate protecting group in phosphoramidite chemistry. Oligonucleotide sequences were constructed by solid phase synthesis using phosphoramidite approach and its mass was confirmed by MALDI TOF spectroscopy (see Tables 1 and 2). The CEOC protecting group was removed from ONs using 50% aqueous piperidine to avoid side reactions.21
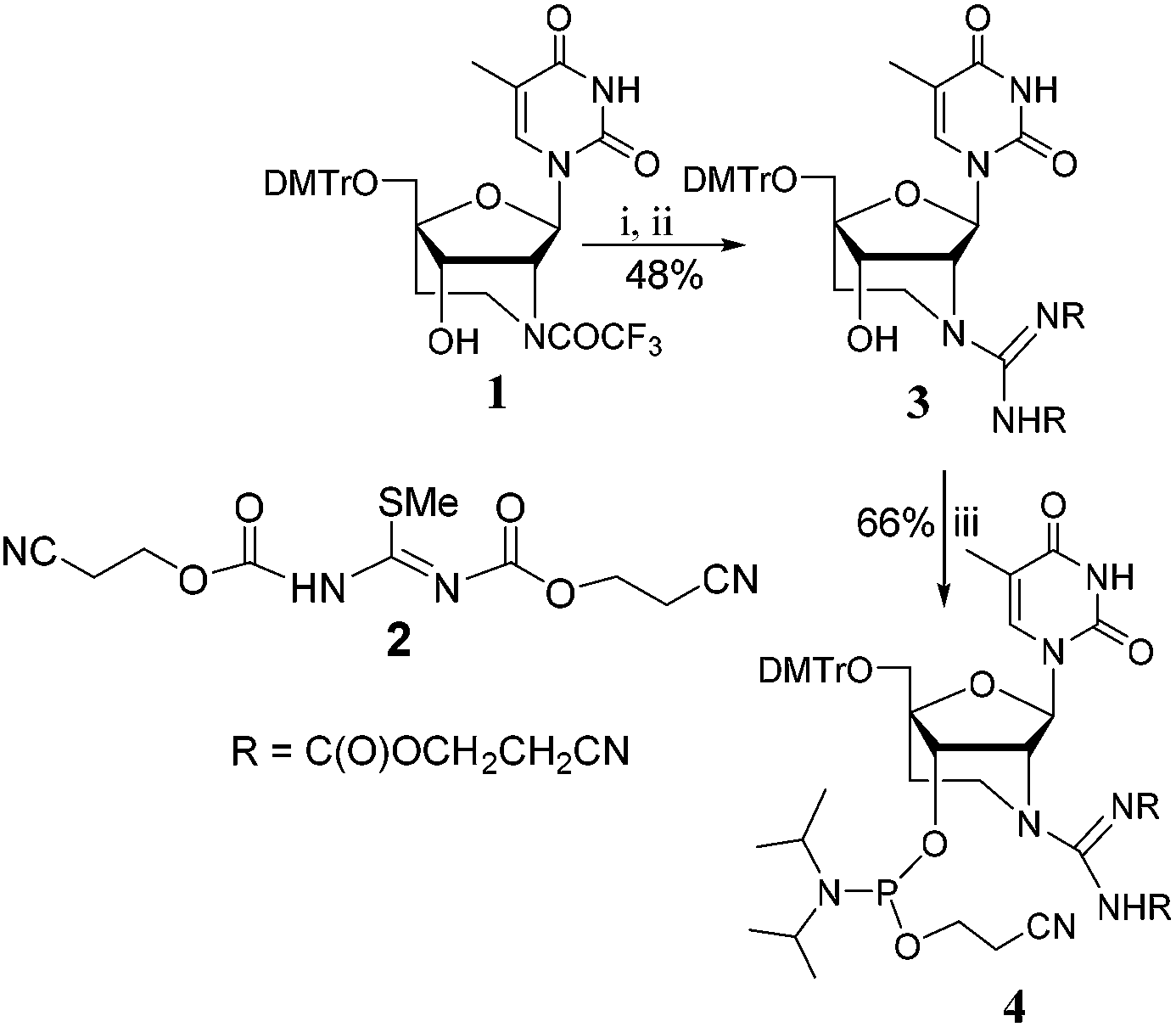 | ||
| Scheme 1 Synthesis of GENA-T phosphoramidite. Reagents: (i) 40% aqueous CH3NH2, CH3OH, 3 h, rt; (ii) dry CH2Cl2, 2, Et3N, 4 days, rt, (iii) N,N diisopropylphosphoramido chloridite, DIPEA, dry CH2Cl2, 2 h, rt. | ||
| ON | Sequences (3′…5′) | Tm/°C with RNA | ΔTm (°C) | MALDI-MS calc./found [M + H]+ |
|---|---|---|---|---|
a ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) = aza-ENA and = aza-ENA and ![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif) = GENA. Buffer and condition: standard deviation in all the melting experiments were below ±0.4 °C. Buffer and conditions: 60 mM Tris–HCl pH 7.5, 60 mM KCl, 0.8 mM MgCl2, and 2 mM DTT; strand concentration = 1 μM. = GENA. Buffer and condition: standard deviation in all the melting experiments were below ±0.4 °C. Buffer and conditions: 60 mM Tris–HCl pH 7.5, 60 mM KCl, 0.8 mM MgCl2, and 2 mM DTT; strand concentration = 1 μM. |
||||
| 1 | 3′-d(CTTCTTTTTTACTTC) | 44 | 4448.6/4448.7 | |
| 2 | 3′-d(CTTCTTTTTACTTC) |
46.5 | +2.5 | 4489.7/4490.7 |
| 3 | 3′-d(CTTCTTTTTACTTC) |
44 | 0 | 4533.9/4533.8 |
| 4 | 3′-d(CTTCTTTTTACTTC) |
47.5 | +3.5 | 4489.7/4490.7 |
| 5 | 3′-d(CTTCTTTTTACTTC) |
44 | 0 | 4533.9/4534.0 |
| 6 | 3′-d(CTTCTTTTTACTTC) |
48 | +4 | 4489.7/4490.8 |
| 7 | 3′-d(CTTCTTTTTACTTC) |
45 | +1 | 4533.9/4533.9 |
| ON | Sequence (5′…3′) | Tm/°C with dsDNA | ΔTm (°C) | MALDI-MS calc./found [M + H]+ |
|---|---|---|---|---|
a Target duplex: 5′-d(GCT![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) TCG)-3′/-3'-d(CGATTTTTCTTTCTCTCTAGC)-5′; underlined portion indicates the target site for triplex formation. = aza-ENA, = GENA and mC = 5-methylcytidine; standard deviation in all the melting experiments were below ±0.4 °C. Conditions: 7.5 mM Na2HPO4 buffer pH 7, containing 140 mM KCl; strand concentration = 1 μM; scan rate 0.5 °C min−1.b M + 2H. TCG)-3′/-3'-d(CGATTTTTCTTTCTCTCTAGC)-5′; underlined portion indicates the target site for triplex formation. = aza-ENA, = GENA and mC = 5-methylcytidine; standard deviation in all the melting experiments were below ±0.4 °C. Conditions: 7.5 mM Na2HPO4 buffer pH 7, containing 140 mM KCl; strand concentration = 1 μM; scan rate 0.5 °C min−1.b M + 2H. |
||||
| 1 | TTTTTmCTTTmCTmCTmCT | 33 | 4497.9/4497.8 | |
| 2 | TTTTTmCTTmCTmCTmCT |
35.5 | +2.5 | 4539.3/4539.4 |
| 3 | TTTTTmCTTmCTmCTmCT |
42.5 | +9.5 | 4581.9/4582b |
We evaluated the hybridization properties of GENA incorporated oligonucleotide with complementary ssRNA using UV melting analysis. To our surprise, the UV melting temperature with complementary ssRNA showed a moderate decrease of ≈3 °C compared to that of the isosequential aza-ENA modified oligonucleotide (Table 1). This was however, a marginal increase (≈+1 °C) with respect to the unmodified sequence. This decrease may be due to the additional steric crowding because of the presence of guanidinium group at the minor groove of DNA–RNA hybrid duplex. Furthermore, we examined the influence of these North-locked aza-ENA and GENA on triplex formation with dsDNA, when incorporated into oligo-DNA. The triplex forming property of the modified ONs were determined using 21mer DNA duplex having purine rich triplex binding site (Table 2).
The UV melting studies with the third strand showed a two-phase dissociation curve with a low triplex melting temperature (33 °C for unmodified TFO) and a high duplex melting temperature (55 °C) which is in agreement with previous reports.10b Interestingly, the GENA containing TFO displayed a triplex thermal stabilization of 42.5 °C at pH 7, which is an increase of +9.5 °C as compared to the unmodified TFO. The aza-ENA modification on the other hand showed a thermal stabilization of 35.5 °C, which corresponds to an increase of +2.5 °C with respect to that of the unmodified TFO. It has been reported previously that 2′-O-[2-(guanidino)ethyl] modification in isosequential TFO showed a maximum increase of ΔTm = +4 °C per modification in triplex melting temperature.17 This clearly suggests that both cationic nature of the guanidinium group near phosphate (forming salt-bridge with neighbouring strand) and the North-conformation of bridged sugar play critical roles in triplex stabilization. Thus high triplex stabilization at neutral pH toward efficient pyrimidine-motif triplex formation was achieved with minimal modification on a TFO (single modification).
We further investigated the enzymatic stability of such modified ON in human serum and with snake venom phosphodiestrase (SVPDE). Human serum mainly comprises of 3′-exonucleases along with some endonucleases and 5′-exonucleases whereas, the SVPDE is a 3′-exonuclease. We anticipated that similar to aza-ENA, GENA could significantly improve the 3′-exonuclease stability of the ON with single modification near the 3′-end. We therefore performed enzymatic digestion studies with such a sequence and compared with those of the unmodified and aza-ENA-modified ONs. Comparison of enzymatic stability studies with single aza-ENA and GENA modifications incorporated at second position from the 3′-end showed great improvements in stability as compared to the unmodified sequence. Interestingly, the guanidine-residue of GENA seemed to have higher impact on stability of the modified ON as compared to that of the amine residue of aza-ENA (Fig. 1). The unmodified deoxy-ON sequence was completely degraded in less than 2 h while the aza-ENA modified sequence was degraded in less than 24 h, when incubated with SVPDE. On the other hand, the GENA modified sequence was still present to some extent even after 33 h. It is noteworthy that both aza-ENA and GENA provided stability to one extra nucleotide towards 3′-end (3′-terminal nucleotide in this case) and prevented degradation of the full-length sequence (Fig. 1a). The human serum digestion footprints also indicated similar results as with SVPDE, with the exception that the n − 1 band (where ‘n’ is the full length oligonucleotide) was visible after some time (Fig. 1b). The stability of the ONs in human serum could not be monitored after 24 h presumably due to the presence of 5′-phosphodiestease in the serum, which cleaves the 5′-32P label.22 Degradation of 5′-32P label is evident from lack of the label in mononucleotide degradation products at higher time points (Fig. 1b). Thus our result unequivocally shows that protonated species in the vicinity of 3′-phosphate indeed impart extra stability towards 3′-exonuclease.
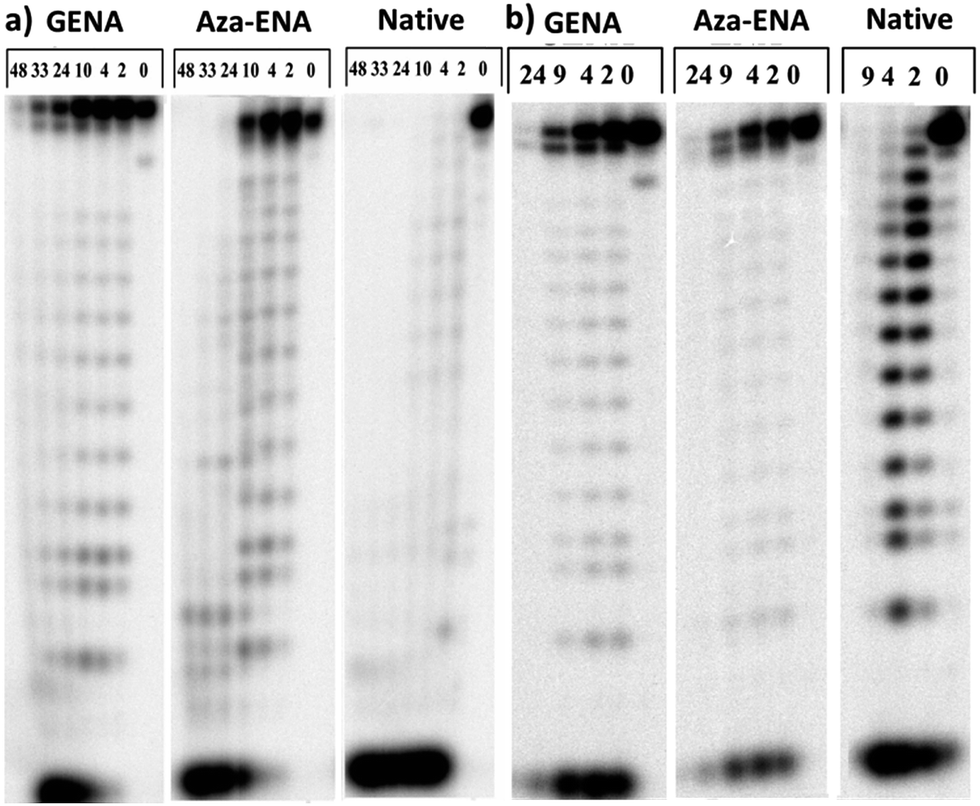 | ||
| Fig. 1 Autoradiograms of 20% denaturing PAGE, showing the degradation pattern of 5′-32P-labeled deoxy-ONs. Sequence: 3′-d(CTCTTTTTTACTTC)-5′ where is either, GENA, aza-ENA or T. Incubation was done at 21 °C (a) with SVPDE at 0, 2, 4, 10, 24, 33 and 48 h; (b) with human serum at 0, 2, 4, 9, 24 h. | ||
In conclusion, we present the synthesis of North-conformationally constrained GENA nucleoside, which upon incorporation into ON showed marginal increase in Tm with complementary RNA (ΔTm ≈ +1 °C) as compared to that of the unmodified ON. However, such modified ON displayed high affinity for target dsDNA to form triplex with ΔTm = +9.5 °C and +7 °C, as compared to the unmodified and aza-ENA modified ONs respectively. A single GENA modification near 3′-end of an ON could impart great stability towards 3′-exonucease (SVPDE and human serum). Further evaluation of such modifications to target genomic DNA without the aid of any intracellular delivery agents is currently underway.
Acknowledgements
The present work was supported by Swedish Strategic Research grant ‘StemTherapy’ and EU Framework Program-7 project ‘Biodesign’.Notes and references
- B. Vester and J. Wengel, Biochemistry, 2004, 43, 13233–13241 CrossRef CAS PubMed.
- C. Zhou and J. Chattopadhyaya, Chem. Rev., 2012, 112, 3808–3832 CrossRef CAS PubMed.
- M. Koizumi, Biol. Pharm. Bull., 2004, 27, 453–456 CAS.
- P. M. McTigue, R. J. Peterson and J. D. Kahn, Biochemistry, 2004, 43, 5388 CrossRef CAS PubMed.
- J. Bhattacharyya, S. Maiti, S. Muhuri, S. Nakano, D. Miyoshi and N. Sugimoto, Biochemistry, 2011, 50, 7414–7425 CrossRef CAS PubMed.
- S. Chatterjee, W. Pathmasiri, O. Plashkevych, D. Honcharenko, O. P. Varghese, M. Maiti and J. Chattopadhyaya, Org. Biomol. Chem., 2006, 4, 1675–1686 CAS.
- T. Yamamoto, M. Harada-Shiba, M. Nakatani, S. Wada, H. Ya-suhara, K. Narukawa, K. Sasaki, M.-A. Shibata, H. Torigoe, T. Yamaoka, T. Imanishi and S. Obika, Mol. Ther.–Nucleic Acids, 2012, 1, e22 CrossRef PubMed.
- (a) O. R. Mook, F. Baas, M. B. de Wissel and K. Fluiter, Mol. Cancer Ther., 2007, 6, 833–843 CrossRef CAS PubMed; (b) M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680–5689 RSC.
- S. Obad, C. O. dos Santos, A. Petri, M. Heidenblad, O. Broom, C. Ruse, C. Fu, M. Lindow, J. Stenvang, E. M. Straarup, H. F. Hansen, T. Koch, D. Pappin, G. J. Hannon and S. Kauppinen, Nat. Genet., 2011, 43, 371–378 CrossRef CAS PubMed.
- (a) T. Hojland, S. Kumar, B. R. Babu, T. Umemoto, N. Albæk, P. K. Sharma, P. Nielsen and J. Wengel, Org. Biomol. Chem., 2007, 5, 2375 RSC; (b) S. M. A. Rahman, S. Seki, S. Obika, S. Haitani, K. Miyashita and T. Imanishi, Angew. Chem., Int. Ed., 2007, 46, 4306–4309 CrossRef CAS PubMed.
- B. P. Monia, J. F. Johnston, H. Sasmor and L. L. Cummins, J. Biol. Chem., 1996, 271, 14533–14540 CrossRef CAS PubMed.
- S. M. Gryaznov, Chem. Biodiversity, 2010, 7, 477–493 CAS.
- P. Hawley, J. S. Nelson, K. L. Fearon, G. Zon and I. Gibson, Antisense Nucleic Acid Drug Dev., 1999, 9, 61–69 CrossRef CAS PubMed.
- O. P. Varghese, J. Barman, W. Pathmasiri, O. Plashkevych, D. Honcharenko and J. Chattopadhyaya, J. Am. Chem. Soc., 2006, 128, 15173–15187 CrossRef CAS PubMed.
- D. Honcharenko, J. Barman, O. P. Varghese and J. Chattopadhyaya, Biochemistry, 2007, 46, 5635–5646 CrossRef CAS PubMed.
- T. Ohmichi, M. Kuwahara, N. Sasaki, M. Hasegawa, T. Nishikata, H. Sawai and N. Sugimoto, Angew. Chem., Int. Ed., 2005, 44, 6682–6685 CrossRef CAS PubMed.
- T. P. Prakash, A. Püschl, E. Lesnik, V. Mohan, V. Tereshko, M. Egli and M. Manoharan, Org. Lett., 2004, 6, 1971–1974 CrossRef CAS PubMed.
- (a) M. Duca, P. Vekhoff, K. Oussedik, L. Halby and P. B. Arimondo, Nucleic Acids Res., 2008, 36, 5123–5138 CrossRef CAS PubMed; (b) J. Y. Chin, F. Reza and P. M. Glazer, Mol. Ther., 2013, 21, 580–587 CrossRef CAS PubMed.
- S. F. Singleton and P. B. Dervan, Biochemistry, 1992, 31, 10995–11003 CrossRef CAS PubMed.
- (a) W. Roberts and D. M. Crothers, Science, 1992, 258, 1463 Search PubMed; (b) S. M. Gryaznov, D. H. Lloyd, J.-K. Chen, R. G. Schultz, L. A. Dedionisio, L. Ratmeyer and W. D. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5798–5802 CrossRef CAS PubMed.
- T. P. Prakash, A. Puschl and M. Manoharan, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 149–159 CAS.
- J. Liithje and A. Ogilvie, Clin. Chim. Acta, 1987, 164, 275–284 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and purification of oligos, 32P labelling experiments, enzymatic digestion studies and UV melting experiments. See DOI: 10.1039/c4ra14721a |
| This journal is © The Royal Society of Chemistry 2015 |