
Practical aqueous reactions leading to skeletally diverse carbohydrate-derived ketones†
Hongming Liu,
Xiaoxing Liu,
Lei Liu,
Xixi Zhang and
Chunbao Li*
Department of Chemistry, School of Science, Tianjin University, Tianjin, 300072, P. R. China. E-mail: lichunbao@tju.edu.cn; Fax: +862227403475; Tel: +86-022-27892351
First published on 12th January 2015
Abstract
Four types of skeletally diverse compounds have been synthesized from protected aldosyl hemiacetals and methyl ketones using cheap catalysts in water in one pot. Among the four skeletons, two of them are not accessible by current methods. The reactions are operationally simple, high yielding and scalable, which opens a practical channel for utilizing carbohydrates to produce chemical and pharmaceutical intermediates and products.
Introduction
Carbohydrates are annually renewable and the quantities produced exceed those for other renewable organic compounds. Therefore utilizing carbohydrates for chemical, energy and material supplies to replace fossil fuels is an important area for sustainable development.1 Partially modified carbohydrates are important drugs, such as iminosugars which are used to treat diabetes.1f,2 When carbohydrates are dehydrated and cyclized, they form hydroxymethyl furfural, which is a potential green fuel, and can then be transformed into a series of chemicals.3 In the synthesis of natural products and other chemicals, carbohydrates are often transformed into basic building blocks.1c,d,4 In these processes, expensive reagents, harsh reaction conditions, prolonged reaction times or organic solvents are often required, which make the manufacture of these carbohydrate-derived products impractical due to high costs and adverse environmental effects.2–4 Therefore, it is beneficial to develop operationally simple and economical procedures to produce carbohydrate-derived chemicals using green media and cheap catalysts. Herein, the synthesis of dienediketones, enetriketones, furanyl-substituted α,β-unsaturated ketones and furanyl-substituted diketones from protected carbohydrates and methyl ketones using inexpensive catalysts in water is reported.Results and discussion
Previously, we synthesized (S,Z)-3-(1,3-bis(benzyloxy)-4-hydroxy-1-butenyl)-1,5-diphenyl-1,5-pentanedione (Scheme 2, Ia) from a reaction between O-benzyl-protected D-xylosyl hemiacetal and acetophenone catalyzed by 2.0 equiv. of LiOH mediated by water.5 In that work, the 1,5-pentanedione product was isolated and purified after TLC indicated that the aldol reaction was complete. In this work, after the completion of the aldol reaction, the reaction mixture was treated with 2.5 equiv. of 10% HCl, in which 2.0 equiv. of HCl was used to neutralize the 2.0 equiv. of LiOH and the remaining 0.5 equiv. of HCl was used to catalyze the hydrolysis of the enolether. The reaction was then continued at 60 °C for 8 h, to give furanyl-substituted diketone Ia (Scheme 1) in 83% yield after in-flask extraction and filtration through a silica gel pad. | ||
| Scheme 1 One-pot synthesis of furanyl-substituted diketones 1 and furanyl-substituted α,β-unsaturated ketones 2 from aldosyl hemiacetals and methyl ketonesa,b. aReaction conditions: (1) aldosyl hemiacetal (0.25 mmol), methyl ketone (6.0 equiv. for 1a–1h, 3.0 equiv. for 2a–2d), LiOH (2.0 equiv.), TBAB (1.0 equiv.), water (0.5 mL), 60 °C, 15 min for 1 and 2. (2) 10% HCl aq (2.5 equiv.) 8 h for 1a–1f, 32 h for 1g and 1h, 2 h for 2a, 2c and 2e, 20 h for 2b and 2d. bIsolated yields. c3β-hydroxyandrost-5-en-17-one (0.25 mmol), aldosyl hemiacetal (2.2 equiv.). | ||
To test the scope of the reaction, a series of substituted acetophenones were used for the condensation reactions with O-benzyl-protected D-glucosyl and D-xylosyl hemiacetals, followed by one-pot hydrolysis. Using different substituents on the phenyl rings had little impact on the reaction yields and rates (Scheme 1, 1a–1g) showing that the scope of usable substrates is wide. The reaction yields varied from 73–87% and the reaction times from 8–32 h.
Subjecting aliphatic ketones and carbohydrates to a similar reaction led to another type of product, α,β-unsaturated ketones (Scheme 1, 2a–e). The reaction yields ranged from 76–95% and the reaction times were 2–20 h. In the synthesis of both 1 and 2, the reaction times for O-benzyl-protected D-glucosyl hemiacetals were much longer than those of O-benzyl-protected D-xylosyl hemiacetals (1g, 32 h and 2b, 20 h; 1a, 8 h and 2a, 2 h). When more catalyst was used (3.0 equiv. of HCl), the reaction times for 1g and 2b were shortened from 32 h and 20 h to 3 h and 2 h respectively. The reactions are easily scalable since similar yields of both 1a and 2b were produced when the amounts of aldosyl hemiacetal starting materials were raised from 0.1 g to 10 g.
These furanyl-substituted products are also accessible staring from furfurals and methyl ketones via aldol reactions.6 The method reported here provides an alternative to known methods with advantages of using carbohydrates instead of furfurals and using water as the solvent. The better performance of the water-mediated aldol reaction than the organic solvent-mediated one is attributed to the heteroatom effects.5 The furan-forming step could be achieved in organic solvents and in water.7
A possible pathway for the one-pot reaction is shown in Scheme 2. First, the acidic hydrolysis of the enolether group of the aldol product Ia condensed from two methyl ketones and a carbohydrate to give IIa, which eliminated benzyl alcohol, yielding the trans-unsaturated ketone IIIa.8 The cis-unsaturated ketone IVa is in equilibrium with the trans isomer IIIa under the acidic conditions. Only the former is capable of cyclizing to form furan Va. A bulkier substituent on the γ position of IIIa results in a lesser amount of cis-unsaturated ketone IVa in the equilibrium.7 Therefore the reactions starting from xylose (Scheme 1, 1a, 2a) were much faster than those starting from glucose (Scheme 1, 1g, 2b). The aldol reaction product Ib was transformed into Vb via a pathway that is similar to that for Ia.
 | ||
| Scheme 2 A possible pathway for the one-pot synthesis of furanyl-substituted diketones and furanyl-substituted α,β-unsaturated ketones. | ||
To obtain useful chemical intermediates as well as to prove the reaction mechanism, the reaction conditions used in Scheme 1 were modified by treating the aldol condensation reaction mixture from the acetophenone and O-benzyl-protected D-xylosyl hemiacetal with less HCl (2.1 equiv. instead of 2.5 equiv. of Scheme 1). The reaction took 10 min at 60 °C to produce triketone 3a (Scheme 3) in 86% yield. Further transformation of 3a into furan 1a was successfully avoided by a timely workup.
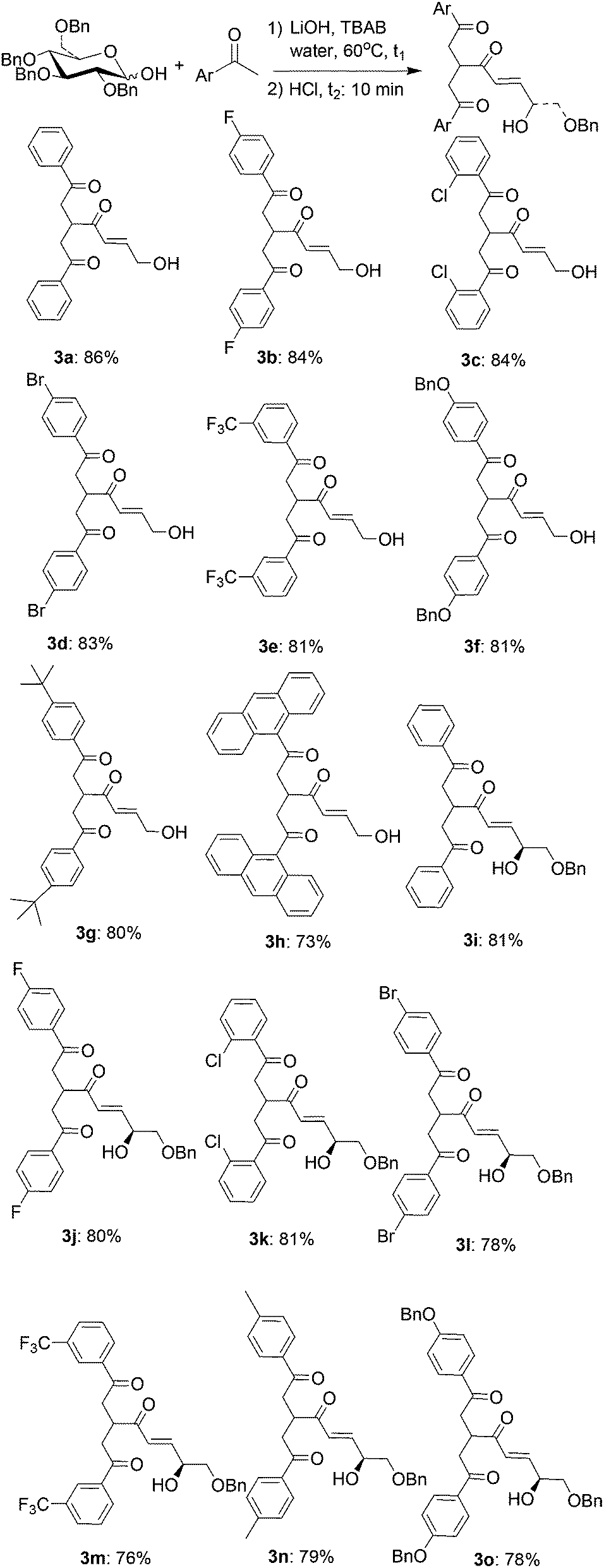 | ||
| Scheme 3 One-pot synthesis of enetriketones from aldosyl hemiacetals and methyl ketones. aReaction conditions: (1) aldosyl hemiacetal (0.25 mmol), methyl ketone (6.0 equiv.), LiOH (2.0 equiv.), TBAB (1.0 equiv.), water (0.5 mL), 60 °C, 15 min except for 3f (3 h), 3j (1 h), 3n (0.5 h), 3o (13 h). (2) 10% HCl aq (2.1 equiv.), 10 min. bIsolated yields. | ||
A series of substituted aryl methyl ketones were then reacted with O-benzyl-protected D-glucosyl and D-xylosyl hemiacetals under these conditions to produce triketones 3a–3o (Scheme 3). The substituents on the aromatic ring of the methyl ketones included electron-withdrawing (F3C– on 3e, 3m and F– on 3b, 3g), electron-neutral (H– on 3a, 3i) and electron-donating (BnO– on 3f, 3o) groups. All the reactions proceeded expeditiously and gave good to excellent yields. Enetriketone 3a was prepared starting with 10 g of O-benzyl-protected D-xylosyl hemiacetal with a similar reaction rate and yield as those obtained with 0.1 g of the hemiacetal.
Several methods to prepare triketones have been reported in the literature. A four-component radical coupling reaction of iodide, CO, α,β-unsaturated ketones and tin enolates leading to triketones was reported by Hosomi and Ryu et al.9 Stetter and Simons reported that a thiazolium salt-catalyzed addition of aldehydes to 2-methylene-l,4-ketoketals produces triketones.10 However, the enetriketone skeleton reported here has not been described in any references. Extending the known methods to build this type of skeleton would be expected to be quite difficult in regards to achieving the desired chemoselectivities and with respect to the availability of starting materials. Furthermore, the known methods use volatile organic solvents, expensive reagents or high pressures. Our one-pot procedure employs water as the reaction media and the phase transfer catalyst (TBAB) and catalyst (HCl) are cheap and water-soluble. The hydroxyl and benzyloxy functional groups on the enetriketone skeleton are additional advantages of our approach.
Encouraged by the successful synthesis of 3a–3o (Scheme 3), we attempted to prepare dienediketone 4a (Scheme 4) via a similar reaction. However, reacting crude aldol product (S,5E,7Z)-7,9-bis(benzyloxy)-10-hydroxy-2-methyl-5,7-decadien-4-one (Scheme 2, Ib)5 from the reaction between 4-methyl-2-pentanone and O-benzyl-protected D-xylosyl hemiacetal with less HCl (≤2.5 equiv.) at temperatures ranging from 0 to 60 °C led to the formation of furan 2a (Scheme 1), and in all cases 4a was not formed.
 | ||
| Scheme 4 Synthesis of dienediketones from aldosyl hemiacetals and methyl ketones. aReaction conditions: (1) aldosyl hemiacetal (0.5 mmol), methyl ketone (3.0 equiv.) except for (4h, 4i), LiOH (2.0 equiv.), TBAB (1.0 equiv.), water (1 mL), 60 °C, 15 min. (2) FeCl3·6H2O (1.5 equiv.) for 4a, 4c, 4e, 4g, 4h, (1.0 equiv.) for 4b, 4d, 4f, 4i, ethyl acetate (2 mL), rt. bIsolated yields. cFor 4h and 4i 3β-hydroxyandrost-5-en-17-one (0.25 mmol), aldosyl hemiacetal (2.2 equiv.). | ||
So the crude aldol product was subjected to in-flask extraction and concentration, which was then treated with different acids (AcOH, ZnCl2, H2SO4, TsOH, AlCl3, BF3 or FeCl3·6H2O) in different solvents (DMSO, toluene, CHCl3, CH2Cl2, methanol, 1,4-dioxane or EtOAc). The reactions catalyzed by all the acids except FeCl3·6H2O led to furan 2a as the major product. The reaction catalyzed by FeCl3·6H2O did not occur in DMSO and was sluggish (>10 h) in methanol and 1,4-dioxane. The reaction went to completion in toluene, CHCl3 and CH2Cl2 but the products were contaminated with substantial amounts of furan 2a. The reaction product was pure only when mediated by ethyl acetate. The amount of FeCl3·6H2O was optimized by screening catalyst loads from 0.1 to 2.0 equiv. and 1.5 equiv. was found to be optimal. If less than 1.5 equiv. of FeCl3·6H2O were used, the reaction was slow and the side product 2a formed. The better performance of the ethyl acetate-mediated reaction is probably due to the chelation of the lone paris of ethyl acetate oxygen atoms with FeCl3, which does not occur for toluene, CHCl3 and CH2Cl2. The ethyl acetate chelates with FeCl3 which prevents it from further catalyzing reactions that lead to the side product furan.
Altogether nine dienediketones (Scheme 4, 4a–i) were obtained in good to excellent yields using these conditions (FeCl3·6H2O catalyst in ethyl acetate). There was a slight difference in the method used to synthesize the glucose-derived dienediketones and that for the xylose-derived dienediketones. For the glucose-derived dienediketones (4b, 4d, 4f, 4i), 1.0 equiv. of FeCl3·6H2O was used, and the yields were good to excellent (81–91%). For the xylose-derived dienediketones (4a, 4c, 4e, 4g, 4h), the amount of catalyst was increased to 1.5 equiv. in order to increase the reaction rate. This decreased the production of the side product furans. These reaction conditions are tolerant to ketone (4a–i), ketal (4c, 4d), olefin (4g), small rings (4e, 4f), hydroxyl (4h, 4i) and ether (4g) groups. In addition the reaction was scalable and the amount of O-benzyl-protected D-glucosyl hemiacetal starting material could be increased from 0.1 g to 10 g for the preparation of 4b with the same yield and reaction rate.
Followings are two relevant research reports in the literature. On the nonenzymatic generation of oxidized lipids, it was reported that a six-step procedure starting from 1-hexyne and substituted furans leads to 2,4-diene-1,4-diketones in less than 30% yield.11 In the mechanism research for the transformation of hexose to hydroxymethyl furfural, 3,4-deoxyglucosene was proposed to be an intermediate.12 The dienediketones synthesized here are an advanced skeleton, which are not accessible via extending the known methods.
Experimental
General experimental information
All of the chemicals were obtained from commercial sources or prepared according to standard methods. NMR spectra were recorded with a 600 MHz spectrometer for 1H NMR, 151 MHz for 13C NMR using TMS as an internal standard. Chemical shifts (δ) are reported relative to TMS (1H) or CDCl3 (13C). Multiplicities are reported as follows: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), dd (doublet of doublets) and dt (doublet of triplets). Coupling constants were reported in Hertz (Hz). Melting points were recorded with a micro melting point apparatus. Infrared analyses (KBr pellet) were performed by FT-IR. High resolution spectra (HRMS) were recorded on a QTOF mass analyzer with electrospray ionization (ESI†).General procedure for the synthesis of furanyl-substituted diketone 1
To a 10 mL test tube were added aldosyl hemiacetal (0.25 mmol), methyl ketone (6 equiv.), LiOH (2 equiv.), TBAB (1 equiv.) and water (0.5 mL). The mixture was stirred and heated at 60 °C for 15 min and TLC indicated completion of the first step. Then 10% HCl aq. (2.5 equiv.) was added to the test tube, which was kept at 60 °C until TLC indicated completion of the reaction (1a–f 8 h, 1g, 1h 32 h). The reaction was stopped and in-tube extracted with ethyl acetate (3 × 2 mL), dried over Na2SO4 and purified on a silica gel pad (eluted with petroleum ether/ethyl acetate) to give products 1a–1h.The synthesis of 1a starting from 10 g of O-benzyl-protected D-xylosyl hemiacetal was performed in a 100 mL round-bottom flask in the same conditions yielding 1a (6.5 g, 86%, t1: 0.33 h, t2: 9 h).
General procedure for the synthesis of furanyl-substituted α,β-unsaturated ketones 2
To a 10 mL test tube were added aldosyl hemiacetal (0.25 mmol), methyl ketone (3 equiv.), LiOH (2 equiv.), TBAB (1 equiv.) and water (0.5 mL). The mixture was stirred and heated at 60 °C for 15 min and TLC indicated completion of the first step. Then 10% HCl aq. (2.5 equiv.) was added to the test tube, which was kept at 60 °C until TLC indicated completion of the reaction (2a, 2c 2 h, 2b, 2d 20 h). The reaction was stopped and in-tube extracted with ethyl acetate (3 × 2 mL), dried over Na2SO4 and purified on a silica gel pad (eluted with petroleum ether/ethyl acetate) to give products 2a–2d.The synthesis of 2b starting from 10 g of O-benzyl-protected D-glucosyl hemiacetal was performed in a 100 mL round-bottom flask in the same conditions yielding 2b (4.63 g, 84%, t1: 0.33 h, t2: 22 h).
To a 10 mL test tube were added 3β-hydroxyandrost-5-en-17-one (0.25 mmol), aldosyl hemiacetal (1.2 equiv.), LiOH (2 equiv.), TBAB (1 equiv.) and water (0.5 mL). The mixture was stirred and heated at 60 °C for 1.5 h. Then more aldosyl hemiacetal (1 equiv.) was added. After 0.5 h, TLC indicated completion of the first step. Then 10% HCl aq. (2.5 equiv.) was added and the reaction was stirred at 60 °C until TLC indicated completion of the reaction (2 h). The reaction was stopped and in-tube extracted with ethyl acetate (3 × 2 mL), dried over Na2SO4 and purified on a silica gel pad (eluted with petroleum ether/ethyl acetate) to give product 2e.
General procedure for the synthesis of enetriketone 3
To a 10 mL test tube were added aldosyl hemiacetal (0.25 mmol), methyl ketone (6 equiv.), LiOH (2 equiv.), TBAB (1 equiv.) and water (0.5 mL). The mixture was stirred and heated at 60 °C for 15 min and TLC indicated completion of the first step. Then 10% HCl aq. (2.1 equiv.) was added to the test tube, which was kept at 60 °C for 10 min. The reaction was stopped and in-tube extracted with ethyl acetate (3 × 2 mL), dried over Na2SO4 and purified on a silica gel pad (eluted with petroleum ether/ethyl acetate) to give products 3a–3o.The synthesis of 3a starting from 10 g of O-benzyl-protected D-xylosyl hemiacetal was performed in a 100 mL round-bottom flask in the same conditions yielding 3a (7.04 g, 88%, t1: 0.33 h, t2: 0.25 h).
General procedure for the synthesis of dienediketone 4
To a 10 mL test tube were added aldosyl hemiacetal (0.25 mmol), methyl ketone (3 equiv.), LiOH (2 equiv.), TBAB (1 equiv.) and water (0.5 mL). The mixture was heated and stirred at 60 °C for 15 min and TLC indicated completion of the reaction. The reaction was in-tube extracted with ethyl acetate (3 × 2 mL), dried over Na2SO4. After concentration to about 2 mL, FeCl3·6H2O (1.5 equiv. for 4a, 4c, 4e, 4g, 1.0 equiv. for 4b, 4d, 4f) was added to the ethyl acetate solution, which was stirred at rt for a certain period (4a 1.5 h, 4b 3.5 h, 4c 2 h, 4d 5 h, 4e 1.5 h, 4f 4 h, 4g 0.25 h). The reaction was stopped by addition of Na2CO3 (5 equiv.) under stirring. Then the solution was filtered and the filtrate was purified on a silica gel pad (eluted with petroleum ether/ethyl acetate) to give products 4a–4g.The synthesis of 4b starting from 10 g of O-benzyl-protected D-glucosyl hemiacetal was performed in a 100 mL round-bottom flask in the same conditions yielding 4b (5.09 g, 87%, t1: 0.33 h, t2: 4 h).
To a 10 mL test tube were added 3β-hydroxyandrost-5-en-17-one (0.25 mmol), aldosyl hemiacetal (1.2 equiv.), LiOH (2 equiv.), TBAB (1 equiv.) and water (0.5 mL). The mixture was heated and stirred at 60 °C for a certain period (4h 1.5 h, 4i 2 h), then more aldosyl hemiacetal (1 equiv.) was added. After 0.5 h, TLC indicated completion of the reaction, which was in-tube extracted with ethyl acetate (3 × 2 mL), dried over Na2SO4. After concentration to about 2 mL, FeCl3·6H2O (1.5 equiv. for 4h, 1.0 equiv. for 4i) was added to the ethyl acetate solution, which was stirred at rt for a certain period (4h 2 h, 4i 4 h). The reaction was stopped by addition of Na2CO3 (5 equiv.) under stirring. Then the solution was filtered and the filtrate was purified on a silica gel pad (eluted with petroleum ether/ethyl acetate) to give products 4h and 4i.
Conclusions
In conclusion, one-pot methods to synthesize four types of skeletally diverse compounds starting from carbohydrates and methyl ketones have been developed. Two of these skeletons are not accessible via current methods. Inexpensive catalysts were used in all cases and in most cases, aqueous media were used. The reaction yields were good to excellent and the operations simple. The four types of ketones have all been scaled up to using 10 g of carbohydrate starting material. The products should be useful intermediates for chemical and pharmaceutical industries. In his paper on the principles of utilizing carbohydrates to produce chemicals, Lichtenthaler et al. stated that: “Unlike fossil resources, which essentially are devoid of oxygen, carbohydrates are overfunctionalized with hydroxyl groups, thus requiring efficient methodologies for the simultaneous reduction of their oxygen content and the introduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO unsaturations”.4a,1 This work meets these requirements. Research on transforming these CO and CC containing compounds into other chemicals is currently underway in our laboratory.
C and CO unsaturations”.4a,1 This work meets these requirements. Research on transforming these CO and CC containing compounds into other chemicals is currently underway in our laboratory.
Notes and references
- (a) F. W. Lichtenthaler, Carbohydr. Res., 1998, 313, 69–89 CrossRef CAS; (b) N. Kardos and J. Luche, Carbohydr. Res., 2001, 332, 115–131 CrossRef CAS; (c) S. M. Andersen, I. Lundt, J. Marcussen and S. Yu, Carbohydr. Res., 2002, 337, 873–890 CrossRef CAS; (d) P. Gallezot, Catal. Today, 2007, 121, 76–91 CrossRef CAS PubMed; (e) P. Gallezot, Green Chem., 2007, 9, 295–302 RSC; (f) S. Jarosz, M. Magdycz and B. Lewandowski, Carbohydr. Chem., 2009, 35, 232–258 CAS; (g) J. Dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017–1034 CrossRef PubMed; (h) J. O. Metzger, ChemCatChem, 2013, 5, 680–682 CrossRef CAS.
- For reviews on iminosugars see: (a) V. H. Lillelund, H. H. Jensen, X. Liang and M. Bols, Chem. Rev., 2002, 102, 517 CrossRef PubMed; (b) M. S. M. Pearson, M. Mathé-Allainmat, V. Fargeas and J. Lebreton, Eur. J. Org. Chem., 2005, 2159–2191 CrossRef CAS; (c) B. G. Winchester, Tetrahedron, 2009, 20, 645–651 CrossRef CAS PubMed; (d) B. L. Stocker, E. M. Dangerfield, A. L. Win-Mason, G. W. Haslett and M. S. M. Timmer, Eur. J. Org. Chem., 2010, 1615–1637 CrossRef CAS.
- For reviews on HMF see: (a) A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC; (b) R. Putten, J. C. Waal, E. Jong, C. B. Rasrendra, H. J. Heeres and J. G. Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef PubMed.
- (a) F. W. Lichtenthaler, A. Brust and E. Cuny, Green Chem., 2001, 3, 201–209 RSC; (b) K. C. Nicolaou and H. J. Mitchell, Angew. Chem., Int. Ed., 2001, 40, 1576–1624 CrossRef CAS; (c) F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728–737 CrossRef CAS PubMed; (d) D. Martin and F. W. Lichtenthaler, Tetrahedron, 2006, 17, 756–762 CrossRef CAS PubMed.
- (a) B. Li and C. Li, J. Org. Chem., 2014, 79, 2242–2254 CrossRef CAS PubMed; (b) B. Li and C. Li, J. Org. Chem., 2014, 79, 8271–8277 CrossRef CAS PubMed.
- (a) V. G. Kharchenko, E. V. Burov and V. A. Sedavkina, Chem. Heterocycl. Compd., 1981, 17, 1168–1170 CrossRef; (b) Y. Xia, Z. Liu, Q. Xiao, P. Qu, R. Ge, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 5714–5717 CrossRef CAS PubMed; (c) X. Huang, Q. Feng, Y. Sun, Q. He and Y. Wang, Lett. Org. Chem., 2012, 9, 280–286 CrossRef CAS.
- (a) T. D. Avery, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2000, 65, 5531–5546 CrossRef CAS PubMed; (b) B. W. Greatrex, M. C. Kimber, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2003, 68, 4239–4246 CrossRef CAS PubMed.
- (a) A. Sarah, Z. Jakov and F. Benzion, Tetrahedron Lett., 1980, 21, 2351–2354 CrossRef; (b) G. D. McAllister and R. J. K. Taylor, Tetrahedron Lett., 2001, 42, 1197–1200 CrossRef CAS.
- K. Miura, M. Tojino, N. Fujisawa, A. Hosomi and I. Ryu, Angew. Chem., Int. Ed., 2004, 43, 2423–2425 CrossRef CAS PubMed.
- H. Stetter and L. Simons, Chem. Ber., 1985, 118, 3172–3187 CrossRef CAS.
- W. Zhang, M. Sun and R. G. Salomon, J. Org. Chem., 2006, 71, 5607–5615 CrossRef CAS PubMed.
- M. L. Wolfrom, E. G. Wallace and E. A. Metcalf, J. Am. Chem. Soc., 1942, 64, 265 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental information, general procedure for all products, spectral data for all products, references for known compounds, NMR spectra of the products, See DOI: 10.1039/c4ra14457k |
| This journal is © The Royal Society of Chemistry 2015 |