
Hierarchical structure of hollow thorn-like polypyrrole microtubes with enhanced electrochemical performance
Mei Li*abc,
Lanlan Yanga and
Yunqiang Zhanga
aSchool of Materials Science and Engineering, Qilu University of Technology, Jinan 250353, P.R. China. E-mail: lim@qlu.edu.cn
bShandong Provincial Key Laboratory of Processing and Testing Technology of Glass and Functional Ceramics, Jinan 250353, P.R. China
cKey Laboratory of Amorphous and Polycrystalline Materials, Qilu University of Technology, Jinan 250353, P.R. China
First published on 20th November 2014
Abstract
A hollow thorn-like polypyrrole (PPy) microtube structure (HTPMT) has been prepared by in situ chemical oxidation polymerization using Methyl Orange (MO) and sodium dodecylbenzene sulfonate (SDBS) as double soft templates, in which the MO and SDBS performed both as surfactant and dopant. The hierarchical structure of HTPMT formed in the presence of MO and SDBS was different from that of polypyrrole microtubes (PMT) or nanoparticles (PNP) using MO and SDBS separately. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images showed that the morphology of the HTPMT had the appearance of thorns growing on the surface of the PMT. The TEM images of the MO and SDBS solutions revealed that PPy had polymerized on the surface of the end-closed MO tubes and in the vesicular SDBS micelles. Electrostatic attraction between the anionic SDBS micelles and cationic pyrrole radicals during the polymerization assisted the formation of the HTPMT. The HTPMT exhibited improved electrochemical performance and thermal stability compared with PMT or PNP, illustrating that the morphology and structure of the conductive polymer influenced the electrochemical performance of the electrode materials.
1. Introduction
Since the discovery of carbon nanotubes in 1991,1 conductive polymers such as nanotubes and nanofibres have aroused considerable interest within the scientific community due to their unique properties and technological application. Among the conducting polymers, polypyrrole (PPy) is a particularly promising material on account of its high energy storage capacity, good electrical conductivity, low cost, stability in air and its potential applications.2–4 Recently, micro- and nanotubes of PPy have been obtained using hard and soft templates. Hard templates have often been used in the polymer matrix to give precise control over size and shape, but the removal of a hard template requires harsh conditions, which not only complicates the fabrication process but may also damage the polymer matrix. From the aspect of safety and environmental hazards, the soft template is more suitable for preparing conductive polymeric micro- and nanotubes since conductive polymers can be grown in tubular form, and the soft template is easily removed following polymerization.5–7Following its introduction by Yang et al., Methyl Orange (MO) has been successfully used in the preparation of PPy nanotubes.8 We ourselves have synthesized a number of PPy nanotubes of different length and tube diameter by changing the oxidizing agent used in the presence of MO.9 Meanwhile, PPy nanoparticles have been prepared, using sodium dodecylbenzene sulfonate (SDBS) as a soft template, by in situ emulsion polymerization. Castagno et al. have reported that with SDBS as stabilizer PPy particles were formed.10 Xing et al. have demonstrated that the presence of an anionic surfactant such as SDBS during the preparation of PPy can strongly influence its morphology, thermal stability and its other properties.11 However, no previous work has been reported regarding the preparation of PPy using MO and SDBS as double templates and stabilizers.
In the present paper MO tubes and SDBS micelles were used as double templates in the synthesis of hollow thorn-like polypyrrole microtubes (HTPMTs) with a hierarchical structure. Compared to polypyrrole microtubes (PMT) prepared from MO and polypyrrole nanoparticles (PNP) using SDBS as the only template, HTPMT had the appearance of a number of PPy thorns growing on the PMT. Many hollow MO tubes could be observed, with exterior diameter in the region of 20–30 nm. The SDBS micelles were vesicular and their hydrophilic groups pointed outwards and were negatively charged. The electrostatic absorption between the anionic SDBS micelles and the cationic pyrrole radicals during polymerization led to the formation of HTPMTs, which had not previously been reported. Interestingly, HTPMTs exhibit better thermal stability and improved electrochemical performance compared with either PMT or PNP, which gives HTPMT wider applicability in supercapacitors.
2. Experimental
2.1 Preparation of HTPMT, PMT and PNP
0.098 g MO and 0.1045 g SDBS were dissolved in 150 mL deionized water to produce a 4.0 mmol L−1 solution of the double micelles. 0.5 mL (7.20 mmol) pyrrole monomer was added to the solution and the mixture stirred at room temperature 1 h. An oxidant solution containing 50 mL ammonium peroxodisulfate (APS) (1.648 g, 7.20 mmol) was slowly added to the mixture over 0.5 h. The polymerization reaction was carried out at 0 °C for 24 h with continuous mechanical stirring. The resulting black precipitate was filtered off and washed six times with a mixture of equal parts of deionized water and ethanol until the filtrate was colourless and neutral. The final product, HTPMT, was dried under vacuum at 40 °C for 24 h.PMT and PNP were similarly prepared using a 2.0 mmol L−1 concentration of MO or SDBS solution, respectively.
2.2 Characterization
The morphology of the product was directly observed by scanning electron microscopy (SEM; FEIco-Holland, JSM-6700F) and transmission electron microscopy (TEM; JEOL, JEM-1011). Fourier transform infrared spectroscopy (FTIR) spectra of the samples were obtained in KBr pellets using a Shimadzu FTIR-8400s spectrophotometer over the range 4000–500 cm−1. Thermogravimetric analysis (TG) was conducted under nitrogen with a PerkinElmer TG7 at a scanning rate of 10 °C min−1. Differential scanning calorimetry (DSC) curves were recorded on a PerkinElmer DSC Q10 at a heating rate of 10 °C min−1 over a temperature range from 40 to 300 °C. Two-probe X-ray photoelectron spectroscopy (XPS) measurements were conducted on an Escalab 250 spectrometer (Thermo-Fisher Scientific) with an Mg Kα X-ray source (1253.6 eV photons). An X-ray diffraction (XRD) pattern was recorded using a Bruker D8 Advance XRD at a scanning speed of 10 °C min−1 between 20 and 80 °C.2.3 Electrochemical measurements
The electrochemical performance of the samples was evaluated by means of a Parstat 2263 electrochemical workstation under computer control at room temperature. Cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS) were measured in a conventional three-electrode electrochemical cell with platinum foil and saturated calomel electrodes. The working electrodes were prepared by mixing the samples under test, poly(tetrafluoroethylene) (PTFE) and carbon black in the mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, coating the mixtures on a 1 cm × 1 cm nickel foam current collector, and dried at 60 °C for 8 h. The electrical conductivity was determined after compacting into pellets 15 mm in diameter and about 0.5 mm in thickness under a pressure of 10 MPadat room temperature using an RTS-8 four-probe tester (Guangzhou Technology Ltd).
:1:1, coating the mixtures on a 1 cm × 1 cm nickel foam current collector, and dried at 60 °C for 8 h. The electrical conductivity was determined after compacting into pellets 15 mm in diameter and about 0.5 mm in thickness under a pressure of 10 MPadat room temperature using an RTS-8 four-probe tester (Guangzhou Technology Ltd).
3. Results and discussion
3.1 Morphology of HTPMT, PMT and PNP
Fig. 1(A) and (a) confirm the microtube morphology of the PMT, both ends of the PMT being closed. The microtubes were about 100–200 nm in diameter, with a wall thickness of about 30–50 nm. SEM and TEM images of PMT are illustrated. Fig. 1(B) and (b) are the corresponding images for PNP, and Fig. 1(C) and (c) for HTPMT. The PNP was polymerized in SDBS micelles and appeared in Fig. 1(B) and (b) as irregular particles, agglomerated into 300–400 nm lumps. The images in Fig. 1(C) and (c) indicated that HTPMT had a novel hierarchical structure with the appearance of a number of thorns growing on the surface of the hollow PPy microtubes. The diameter of the HTPMT was about 200–300 nm and the wall thickness of the tube about 50 nm, slightly thicker than that of PMT. The obvious differences in morphology between PMT and HTPMT were a result of the addition of SDBS. | ||
| Fig. 1 SEM and TEM images of PMT (A and a), PNP (B and b) and HTPMT (C and c), respectively. | ||
The TEM images of the MO solution and SDBS micelles illustrated the polymerization process clearly, and are shown in Fig. 2. There were a number of hollow tubes in the MO solution, the outer diameter of which was about 20–30 nm (Fig. 2(a)). Both ends of the MO tubes were closed, and the diameter of the PMT tubes was about 100–200 nm, much larger than that of the MO tubes. It can therefore be deduced that PPy had been deposited gradually on the surface of the MO tubes as a result of the addition of APS during the PPy polymerization process. Fig. 2(b) shows that a large number of vesicular micelles were present in the SDBS solution, in which the organophilic groups faced inwards and the hydrophilic groups outwards. These SDBS vesicular micelles and MO tubes were randomly scattered in the mixed MO and SDBS solutions, as shown in Fig. 2(c), implying that there had been no obvious reaction between the MO tubes and SDBS micelles. With the addition of Py and APS, it appears that the SDBS micelles became attracted to the surface of the PMT, in which the monomer was polymerized into the thorn-like PPy attachments, and the hierarchical structure of HTPMT was finally obtained.
 | ||
| Fig. 2 TEM images of (a) MO tubes, (b) SDBS micelles and (c) the mixed solution of MO and SDBS. | ||
The mechanism of the formation of HTPMT is proposed, according to the shape of the soft templates and the cationic radical polymerization characteristics of PPy in Fig. 3. The hydrophilic sulfonate groups of the SDBS were negatively charged and were absorbed by the cationic pyrrole radicals produced during the polymerization of PPy.12 The electrostatic absorption between oppositely charged ions induced the so-called thorns to become attached to the surface of the PMT. This mechanism exactly describes the process of formation of PMT, since the sulfonate groups in MO solution were oriented outwards towards the water, causing the cationic pyrrole radicals to become deposited on the surface of the hollow MO tubes.
 | ||
| Fig. 3 Formation mechanism of the HTPMT. | ||
3.2 The chemical structure and thermal stability of the samples
The chemical structure and the thermal stability of the three samples were evaluated by FTIR, XRD, TG and DSC, and the results are illustrated in Fig. 4. Fig. 4(a) shows the characteristic absorption peaks of the samples, the main peaks assigned to HTPMT being identical to those of PMT and PNP. The characteristic absorption of the PPy at 1553, 1471, 1040 and 910 cm−1 was due to stretching of the Py ring and to the conjugated C–N stretching mode.13,14 The N–H stretching band at 1402 cm−1 in the most oxidized form of PPy appeared in the HTPMT curve, and was not obvious in the spectra of PMT or PNP.15,16 The absorption peak at 3118 cm−1 in the HTPMT spectrum may have been due to hydrogen-bonded N–H.17 In addition, the peaks at 1184, 630 cm−1 correspond to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the –SO3− group, suggesting that the MO and SDBS anions had entered the PPy chains as dopant.18,19
O stretching vibration of the –SO3− group, suggesting that the MO and SDBS anions had entered the PPy chains as dopant.18,19
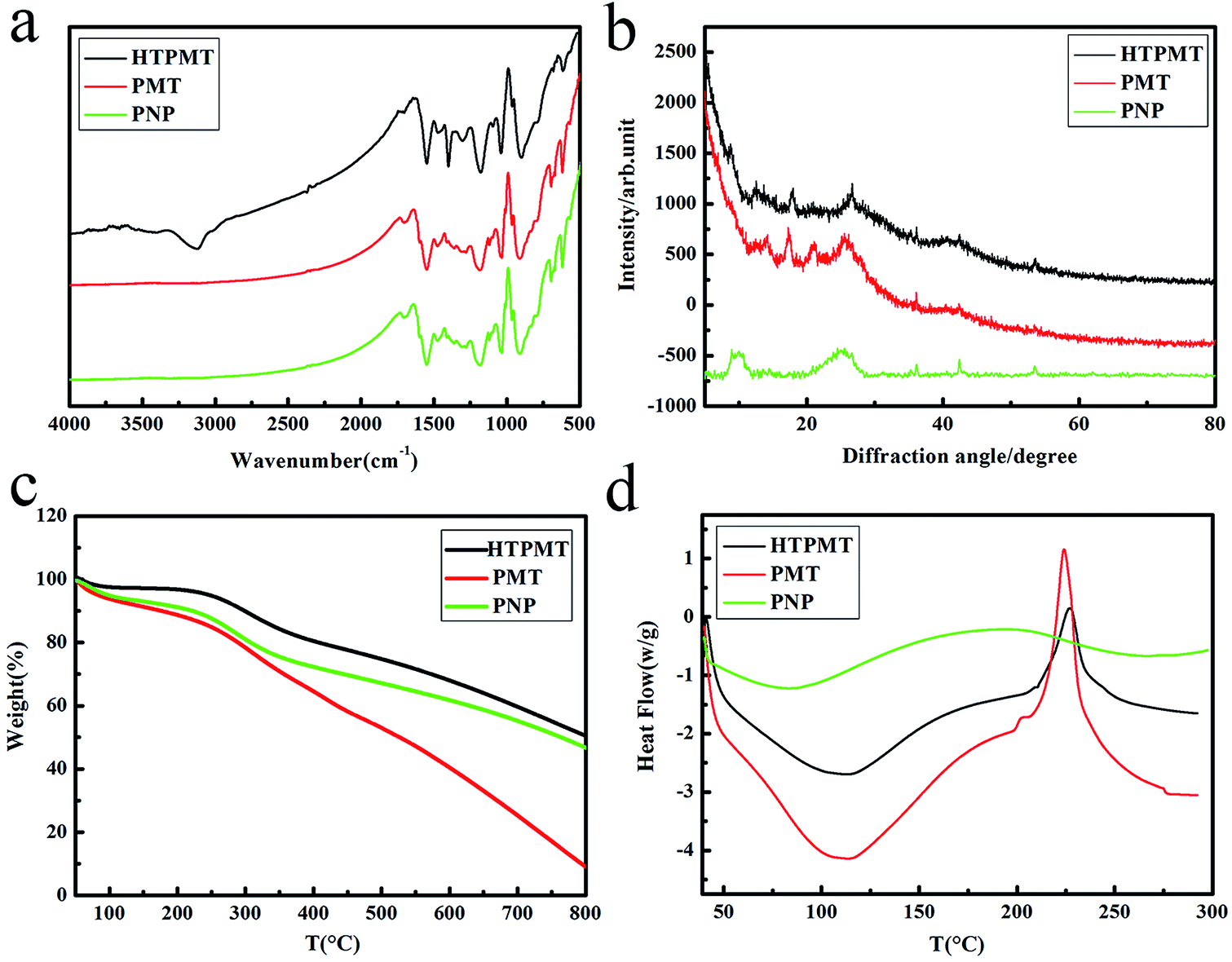 | ||
| Fig. 4 (a) FTIR, (b) XRD, (c) TG and (d) DSC spectra of the PNP, PMT and HTPMT samples. | ||
Fig. 4(b) shows the XRD spectra of HTPMT, PMT and PNP. There was a broad diffraction peak at about 26° which indicated that the PPy samples prepared were amorphous.20 Other sharp peaks at about 18° and 41° in the HTPMT curve corresponded to the diffraction peaks of PMT and PNP, further confirming that MO and SDBS act as soft templates and their anions entered the conductive polymer as dopant.21
The thermal stability of the samples was measured by TG and DSC, and obvious differences can be observed in Fig. 4(c) and (d). A weight loss was seen when the temperature exceeded 100 °C, due to the volatilization of absorbed water and low molecular weight oligomers, with a corresponding endothermic peak in DSC spectra centred around 110 °C.22 However, when the temperature exceeded 150 °C the weight loss for PMT was the largest, that for PNP was intermediate and that for HTPMT the lowest. When the temperature reached at 225 °C, the weight loss became more obvious, as the PPy chain began to decompose.23 As shown in Fig. 4(d), there was an exothermic peak in the DSC spectrum at about 225 °C, which is in accordance with the TG results. The intensity of the exothermic peak of PMT was clearly stronger than that of HTPMT, indicating that the PPy chain in PMT had decomposed rapidly. The exothermic peak of the PNP was broad between 200 °C and 320 °C, and not as sharp as that of PMT or HTPMT, illustrating the gradual decomposition of the PPy chain. The TG and DSC results confirmed that the thermal stability of HTPMT was the best of the three samples. HTPMT may thus exhibit ideal electrochemical performance since its good thermo-mechanical characteristics ensure good cycling stability during the repeated charge–discharge process.24
The XPS spectra were used to further investigate the structure of the three samples shown in Fig. 5. The characteristic binding energies at about 286.24 eV (C1s), 400.72 eV (N1s) and 533.5 eV (O1s) were similar to the XPS survey scans shown in Fig. 5(d). The broad N1s band could be fitted into the three Gaussian peaks of PMT in Fig. 5(a), with binding energies of 397.4 eV (N–, from MO), 398.8 eV (–NH–) and 401.0 eV (N+–), and the two peaks of PNP were about 399.76 eV (–NH–) and 400.97 eV (N+–) in Fig. 5(b).25,26 However, the N1s peak could be resolved into the three constituents of HTPMT in Fig. 5(c), with binding energies of 397.8 eV (N–), 399.4 eV (–NH–) and 401.4 eV (N+–), which suggested that HTPMT was in a higher oxidation state.27,28
 | ||
| Fig. 5 N1s XPS core level spectra of (a) PMT, (b) PNP and (c) HTPMT, and (d) XPS spectra of PMT, PNP and HTPMT. | ||
3.3 Electrochemical performance of the samples
The electrochemical performance of the samples was characterized in a 1 M KCl electrolyte using a three-electrode cell system. Cyclic voltammograms, with potential ranging from −0.1 V to −0.7 V at a scan rate of 5 mV s−1, are shown in Fig. 6(a). The CV curves for the samples exhibited quasi-rectangular voltammetry characteristics, indicating that the capacitance resulted specifically from electrical double-layer capacitors. The CV curve for HTPMT occupied a larger area than that for PMT and PNP, implying better capacitance characteristics. | ||
| Fig. 6 The electrochemical performance of the samples: (a) CV curves at a scan rate of 5 mV s−1, (b) GCD curves at a current density of 0.2 A g−1, (c) the specific capacitances at different current densities, and (d) Nyquist plots. | ||
The GCD behaviour of the different materials was investigated by chronopotentiometry in 1.0 M KCl solution at different current densities, and the corresponding results are shown in Fig. 6(b) and (c). The specific capacitance values (Cg) of the electrode materials were calculated from the galvanostatic discharge process, according to eqn (1):
| Cg = IΔt/(mΔV),29 | (1) |
As shown in Fig. 6(c), the Cg of the samples tended to gradually decrease with increasing current density, due to the fact that at low current densities the process of accumulation was slow. This allowed complete access to the active sites on the electrode, resulting in higher specific capacitance.30 When the current density was 5 A g−1, the Cg of HTPMT, PNP and PMT were respectively 269.8, 154.84 and 73.6 F g−1. The conservation rates of Cg for HTPMT, PNP and PMT were 71, 49 and 41%, respectively, indicating that the HTPMT electrode had the best rate capacity of the three.
The ion-transport kinetics and electrode conductivity of the samples were further evaluated by EIS within the frequency range 105 to 0.01 Hz with an AC voltage amplitude of 5 mV.
The characteristic Nyquist plots for HTPMT, PNP and PMT are shown in Fig. 6(d). In the low-frequency region, the slope of the plot of the HTPMT was steepest, and those of PMT and PNP were almost equal, confirming that HTPMT had the best capacitive behaviour (an ideal capacitor would give a vertical line). In the high-frequency region the real axis intercept is shown in the inset to Fig. 6(d), and is equivalent to the series resistance, including the resistance of the electrolyte, the intrinsic resistance of the active materials and the contact resistance at the interface of the active materials and current collector. The radius of the semicircle plotted is indicative of the inter-particle resistance and charge transfer impedance.31,32 The radius of the semicircle of HTPMT was clearly smaller than that of HTPMT or PMT, also indicating the higher capacity of the HTPMT. In addition, the resistance of the conducting polymer is related to the anion doping level of the conducting PPy.33,34 Elemental analysis showed that the S:N molar ratio (although a trace of N originated from MO) was in the range 0.25–0.30, as shown in Table 1. The ratio of the S/N of HTPMT was greatest, which meant that more anions entered the PPy chains. Since the electrical conductivity of a polymer is closely related to its doping level, its oxidation state and its conjugation length,35 the conductivity of HTPMT was higher than that of PMT or PNP, which is in accordance with the EIS results.
| Sample | C | H | N | S | S:N |
σ (S cm–1) |
|---|---|---|---|---|---|---|
| PMT | 51.59 | 4.24 | 14.53 | 3.59 | 0.25 | 0.62 |
| HTPMT | 52.22 | 3.97 | 15.12 | 4.48 | 0.30 | 1.89 |
| PNP | 52.60 | 4.11 | 14.78 | 4.09 | 0.28 | 1.06 |
Cycling stability is crucial to the practical application of supercapacitors. Fig. 7 illustrates the relative cycling performance of the electrodes composed of HTPMT, PMT or PNP, investigated by charging and discharging the capacitor for 1000 cycles at a current density of 1 A g−1. Again, HTPMT exhibited the best cycling stability and Cg still retained 80% of its initial value. This result further illustrated that the cycling stability of the electrodes was influenced by the structure and morphology of the samples obtained, and that the unique hierarchical structure of HTPMT improved the electrochemical performance of the electrode.
 | ||
| Fig. 7 Cycling performance of HTPMT, PMT and PNP. | ||
4. Conclusions
HTPMT, with a hierarchical structure, was successfully prepared by simple in situ chemical oxidation polymerization, with MO and SDBS as double templates. The pyrrole was polymerized both on the surface of the hollow MO tubes and within the SDBS micelles. The electrostatic absorption between the anionic SDBS micelles and the cationic pyrrole radicals during polymerization induced the formation of HTPMT, which had the appearance of thorns growing on the surface of the PPy microtubes. The HTPMT exhibited improved electrochemical performance, and this promises its wider potential in supercapacitors.Acknowledgements
The study was supported by the College Scientific Plan Fund of Shandong Education Department (J10LD23) and the Doctoral Startup Foundation of Qilu University of Technology (12042826).References
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, W. Song and Z. Liu, J. Phys. Chem. B, 2005, 110, 1158–1165 CrossRef PubMed.
- X. Zhang, J. Zhang, Z. Liu and C. Robinson, Chem. Commun., 2004, 1852–1853 RSC.
- T. Dai and Y. Lu, Macromol. Rapid Commun., 2007, 28, 629–633 CrossRef CAS.
- D. Tingyang, Y. Xiaoming and L. Yun, Nanotechnology, 2006, 17, 3028 CrossRef.
- J. Kopecká, D. Kopecký, M. Vrňata, P. Fitl, J. Stejskal, M. Trchová, P. Bober, Z. Morávková, J. Prokeš and I. Sapurina, RSC Adv., 2014, 4, 1551 RSC.
- X. Yang, Z. Zhu, T. Dai and Y. Lu, Macromol. Rapid Commun., 2005, 26, 1736–1740 CrossRef CAS.
- M. Li, W. Li, J. Liu and J. Yao, J. Mater. Sci.: Mater. Electron., 2012, 24, 906–910 CrossRef.
- K. R. L. Castagno, V. Dalmoro and D. S. Azambuja, Mater. Chem. Phys., 2011, 130, 721–726 CrossRef CAS PubMed.
- S. Xing and G. Zhao, Polym. Bull., 2006, 57, 933–943 CrossRef CAS.
- K. Leonavicius, A. Ramanaviciene and A. Ramanavicius, Langmuir, 2011, 27, 10970–10976 CrossRef CAS PubMed.
- Q.-G. Shao, W.-M. Chen, Z.-H. Wang, L. Qie, L.-X. Yuan, W.-X. Zhang, X.-L. Hu and Y.-H. Huang, Electrochem. Commun., 2011, 13, 1431–1434 CrossRef CAS PubMed.
- L. Cui, J. Shen, F. Cheng, Z. Tao and J. Chen, J. Power Sources, 2011, 196, 2195–2201 CrossRef CAS PubMed.
- Q.-F. Wu, K.-X. He, H.-Y. Mi and X.-G. Zhang, Mater. Chem. Phys., 2007, 101, 367–371 CrossRef CAS PubMed.
- C. Zhou, S. Kumar, C. D. Doyle and J. M. Tour, Chem. Mater., 2005, 17, 1997–2002 CrossRef CAS.
- R. Akinyeye, I. Michira, M. Sekota, A. Al-Ahmed, P. Baker and E. Iwuoha, Electroanalysis, 2006, 18, 2441–2450 CrossRef CAS.
- A. D. W. Carswell, E. A. O'Rea and B. P. Grady, J. Am. Chem. Soc., 2003, 125, 14793–14800 CrossRef CAS PubMed.
- J. Upadhyay and A. Kumar, Mater. Sci. Eng., B, 2013, 178, 982–989 CrossRef CAS PubMed.
- J. Liu and M. Wan, J. Mater. Chem., 2001, 11, 404–407 RSC.
- T. Dai and Y. Lu, J. Mater. Chem., 2007, 17, 4797–4802 RSC.
- W. G. Liu and K. D. Yao, Polymer, 2001, 42, 3943–3947 CrossRef CAS.
- Z. Bai, L. Yang, L. Li, J. Lv, K. Wang and J. Zhang, J. Phys. Chem. C, 2009, 113, 10568–10573 CAS.
- R. K. Sharma, A. C. Rastogi and S. B. Desu, Electrochim. Acta, 2008, 53, 7690–7695 CrossRef CAS PubMed.
- C. Pirvu, C. C. Manole, A. B. Stoian and I. Demetrescu, Electrochim. Acta, 2011, 56, 9893–9903 CrossRef CAS PubMed.
- B. J. West, T. F. Otero, B. Shapiro and E. Smela, J. Phys. Chem. B, 2009, 113, 1277–1293 CrossRef CAS PubMed.
- X. Feng, Z. Sun, W. Hou and J.-J. Zhu, Nanotechnology, 2007, 18, 195603 CrossRef.
- C. Shen, Y. Sun, W. Yao and Y. Lu, Polymer, 2014, 55, 2817–2824 CrossRef CAS PubMed.
- J. Zhang, Y. Yu, L. Liu and Y. Wu, Nanoscale, 2013, 5, 3052–3057 RSC.
- W. Lei, P. He, Y. Wang, S. Zhang, F. Dong and H. Liu, Electrochim. Acta, 2014, 132, 112–117 CrossRef CAS PubMed.
- J. Liu, J. An, Y. Ma, M. Li and R. Ma, J. Electrochem. Soc., 2012, 159, A828 CrossRef CAS PubMed.
- D. Zhang, X. Zhang, Y. Chen, P. Yu, C. Wang and Y. Ma, J. Power Sources, 2011, 196, 5990–5996 CrossRef CAS PubMed.
- Y. Li and Y. Fan, Synth. Met., 1996, 79, 225–227 CrossRef CAS.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- X. Wang and E. Smela, J. Phys. Chem. C, 2008, 113, 359–368 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |