
New cellulose–silica composite IMAC/C18 for the selective enrichment of phosphorylated molecules and the improved recovery of hydrophilic species†
Muhammad Najam-ul-Haq *,
Fahmida Jabeen,
Faiza Shafiq,
Salman Sajid and
Ambreen Saba
*,
Fahmida Jabeen,
Faiza Shafiq,
Salman Sajid and
Ambreen Saba
Division of Analytical Chemistry, Institute of Chemical Sciences, Bahauddin Zakariya University, Multan 60800, Pakistan. E-mail: najamulhaq@bzu.edu.pk; Tel: +92 306 7552653
First published on 17th November 2014
Abstract
Cellulose and silica are the traditional quality sorbents with a range of reported applications, particularly for hydrophilic biomolecules. With this in mind, cellulose and silica were brought into a composite form to bring their individual benefits on to a single platform. Cellulose and silica were chosen in order to obtain a completely hydrophilic composite, which was then derivatized for the selective enrichment of phosphopeptides and as a desalting material prior to mass spectrometric analysis. The composite was characterized by FT-IR, EDX and SEM. As a material for immobilized metal ion affinity chromatography (IMAC), the cellulose–silica composite enriches phosphopeptides from a complex mixture in which β-casein is spiked in de-phosphorylated HeLa cell extract. The inherent hydrophilic nature of the composite gives a higher selectivity of up to 2000 times the complexity level and sensitivity down to 1 femtomole for phosphopeptides. As well as the immobilized metal ions on the IMAC composite, the roles of the base materials in the composite, i.e. cellulose and silica were also tested in the enrichment which helped thereof in optimizing the best sample preparation protocol. With the introduction of successive elution conditions, phospholipids and phosphopeptides were enriched and identified from egg yolk digest using a single batch extraction method. The phosphopeptides were analyzed with MALDI-MS, whereas the low molecular weight phospholipids were analysed using newly designed gold nanoparticles and carbon based materials through LDI-MS. Phosphopeptides were also enriched from complex serum digest and discussed with relevance to prostate cancer. As a reverse-phase (RP) material, it provides a combination of the hydrophilic composite and the hydrophobic C18 chain. Efficient desalting of higher salt concentrations, improved recovery of hydrophilic peptides and high sequence coverage in comparison to commercial materials makes cellulose–silica C18 a better desalting tool with in-house economical synthesis.
Introduction
Affinity chromatography is developing along with the growing diversity of “Omics” technologies, such as genomics, proteomics and metabolomics.1,2 The breakthrough development of selective affinity chromatography has enabled researchers to explore fields such as protein–protein interactions, post-translational modifications3 and protein degradation that could not be examined previously.4 The coupling of reversed phase affinity chromatography with mass spectrometry has aided the discovery of protein biomarkers.5,6 For the past few decades polymer–inorganic composite materials have attracted interest in affinity chromatography, due to their synergistic and hybrid properties derived from the individual components.7,8 The properties of the composites are not only the sum of the individual contributions of both materials, but the role of the inner interfaces can be predominant and provide interaction sites for various analytes.9The specificity is related to the immobilized surface functionalities, however the properties of the support material must be chosen to limit non-specific bindings. Supports which have little or no non-specific bindings mimic the properties of the aqueous mobile phase. Therefore, chemically inert support materials are hydrophilic in nature.10 The chemical inertness of cellulose and silica enables the selective binding of molecules of interest with little or no non-specific bindings. In addition, cellulose and silica are chemically stable under normal operating conditions11 and have been utilized individually in phosphoproteomics studies.12,13 Therefore, they have enjoyed the attention of separation chemists for the development of selective enrichment tools in phosphoproteomics, glycoproteomics etc.
At the clinical level and proteomic research centers, there is a high demand for desalting materials. Commercial materials like Zip tip (Millipore), graphite spin columns (GC tip, GL Science) or Eppendorf GELoader have been in use in recent times. However, the loss of small, hydrophilic and some post-translationally modified peptides is still a limitation of the commercially available desalting materials14 and hence new materials are always in demand.
Knowing the uniqueness of cellulose and silica in proteomics, the present research focused on the synthesis of a cellulose–silica composite and the evaluation of the benefits of this combination in comparison to cellulose and silica individually. Through functionalization as IMAC and RP materials, their adsorptions can be modified as desired. In addition to considering the role of the immobilized metal ions on the IMAC composite, the roles of the base materials were tested to optimize the best enrichment strategy. A complete enrichment methodology for phosphopeptides and phospholipids was developed using standards and real samples. Cellulose–silica C18 was explored as a unique hydrophobic/hydrophilic desalting tool to overcome the limitations of commercial materials such as the loss of hydrophilic species.
Results and discussion
Characterization of the cellulose–silica composite
Cellulose–silica composites have previously been synthesized using different strategies and an overview of their synthetic procedures and applications is given in Table S1.† The current composite was synthesized using a new method, derivatized as IMAC and RP materials and applied in proteomics for the first time. The synthesis of the composite involved the oxidation of cellulose using periodate, amino modification of silica and finally composite fabrication (illustrated in Fig. S1†). Further derivatization of the composite is described in Fig. S2.† The surface morphology was tested by Scanning Electron Microscopy (SEM) and the purity and elemental composition were determined by Energy Dispersive X-ray (EDX) spectroscopy . The composite reveals the presence of micro-scale cellulose fibers and silica particles with heterogeneous morphology (Fig. S3A†). The elemental composition shows no impurities with a carbon content of 19.93%, an oxygen content of 52.72% and a silicon content of 27.34% (Fig. S3B†).Chemical functionalization like the oxidation of cellulose, aminopropyl bonded silica formation and formation of the final composite were confirmed by Fourier transform infrared (FT-IR) spectroscopy. During the oxidation of cellulose, there was a change in the region of the carbonyl group stretching vibration. In the case of the composite, the peak at 1630 cm−1 belongs to the carbonyl group of the amide, which shows the formation of the cellulose–silica composite. The peak at 3370 cm−1 represents the N–H stretch which also confirms the attachment of the cellulose to the amino-propyl bonded silica. The peak is broad due to the presence of OH groups. The peaks for Si–O and Si–C are observed in the regions of 1100 cm−1 and 800 cm−1 (Fig. S4†). For cellulose–silica IMAC, the C–N stretch is at 1056 cm−1, which indicates the attachment of the iminodiacetic acid (IDA) to the composite (Fig. S5†). Similarly, the functionalization as a RP material is confirmed by the signal at 3370 cm−1 for the N–H stretch and strong peaks for the C18 C–H stretch around 2900 cm−1 (Fig. S6†).
The commercially available silica used in this study has a particle size of 200–425 mesh, pore size of 60 Å, pore volume of 0.75 cm3 g−1 and surface area of 480 m2 g−1. The pore size and surface chemistry (silanol groups) affect the mass transfer and surface reactivity15 and the physical characteristics of silica with respect to separation can be determined by near infrared spectroscopy, as reported earlier.16 The higher surface area allows better derivatization of the silica with cellulose to form the composite. The enrichment efficiency of the composite also depends on the hydrophilicity in addition to the above-mentioned physical characteristics like porosity, particle size and surface area.
Cellulose–silica IMAC – metal ion immobilization
Four metal ions were immobilized onto cellulose–silica IMAC for phosphopeptide enrichment from β-casein digest. Fe3+ was chosen because of its traditional use in phosphoproteomics, however there is the problem of acidic peptides being enriched along with the phosphopeptides which hampers the selectivity and hence compromises the binding capacity (Fig. S7a†). Zr4+ shows better selectivity with five phosphopeptides (one tetra phosphorylated and 4 mono phosphorylated peptides) being enriched (Fig. S7b†). However, the focus in this study was placed on lanthanide ions due to their performance in phosphopeptide enrichment compared to transition metal ions. In the cases of cellulose–silica IMAC-La3+ (Fig. S7c†) and cellulose–silica IMAC-Sm3+ (Fig. S7d†), there was a difference of one mono-phosphopeptide at 3054.892. Sm3+ enriched the highest number of phosphopeptides and hence was immobilized onto the cellulose–silica IMAC for further studies.Selectivity of cellulose–silica IMAC-Sm3+ for phosphopeptides
Different complexity levels were used to determine the selectivity of the cellulose–silica IMAC-Sm3+ composite, i.e. the ability to bind the target molecules present in complex mixtures like serum or HeLa cell extract. The complexity levels were attained by mixing varying ratios of protein/peptide backgrounds. Selectivity depends on the nature of the affinity material as well as on the nature of the competing molecules. Phosphopeptides from β-casein digest were spiked in de-phosphorylated HeLa cell extract. Fig. 1 represents the selectivity measurements recorded for spiked mixtures in the ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100; (b) 1:500; (c) 1:1000; (d) 1:1500; and (e) 1:2000. Five phosphopeptides were detected with the amount of background increased up to 1500 times. Washing steps removed the non-phosphopeptides. For the sample 1:2000 (β-casein:de-HeLa cell), two phosphopeptides at m/z 2061 and 3122 were enriched. Both of these phosphopeptides are hydrophilic in nature; 2061 (FQSEEQQQTEDELQDK, 33-48, 1P) and 3122.9 (RELEELNVPGEIVESLSSSEESITRI, 26-41, 4P) with Krokhin_NET hydrophobicity values of 0.09920053 and 0.5137824 respectively. No methyl esterification was carried out as it does not derivatize 100% of the carboxylic acid groups and it can increase the complexity of the analysis due to signals originating from peptides with different degrees of O-methylation. Furthermore, the buffer used for O-methyl esterification can result in partial de-amidation forming glutamic acid from glutamine as well as aspartic acid from asparagine. These amino acid residues are subsequently O-methylated, adding further complexity to the analysis. In addition, O-methyl esterification requires lyophilisation of the sample, which causes adsorptive losses of phosphopeptides to the surfaces.
:100; (b) 1:500; (c) 1:1000; (d) 1:1500; and (e) 1:2000. Five phosphopeptides were detected with the amount of background increased up to 1500 times. Washing steps removed the non-phosphopeptides. For the sample 1:2000 (β-casein:de-HeLa cell), two phosphopeptides at m/z 2061 and 3122 were enriched. Both of these phosphopeptides are hydrophilic in nature; 2061 (FQSEEQQQTEDELQDK, 33-48, 1P) and 3122.9 (RELEELNVPGEIVESLSSSEESITRI, 26-41, 4P) with Krokhin_NET hydrophobicity values of 0.09920053 and 0.5137824 respectively. No methyl esterification was carried out as it does not derivatize 100% of the carboxylic acid groups and it can increase the complexity of the analysis due to signals originating from peptides with different degrees of O-methylation. Furthermore, the buffer used for O-methyl esterification can result in partial de-amidation forming glutamic acid from glutamine as well as aspartic acid from asparagine. These amino acid residues are subsequently O-methylated, adding further complexity to the analysis. In addition, O-methyl esterification requires lyophilisation of the sample, which causes adsorptive losses of phosphopeptides to the surfaces.
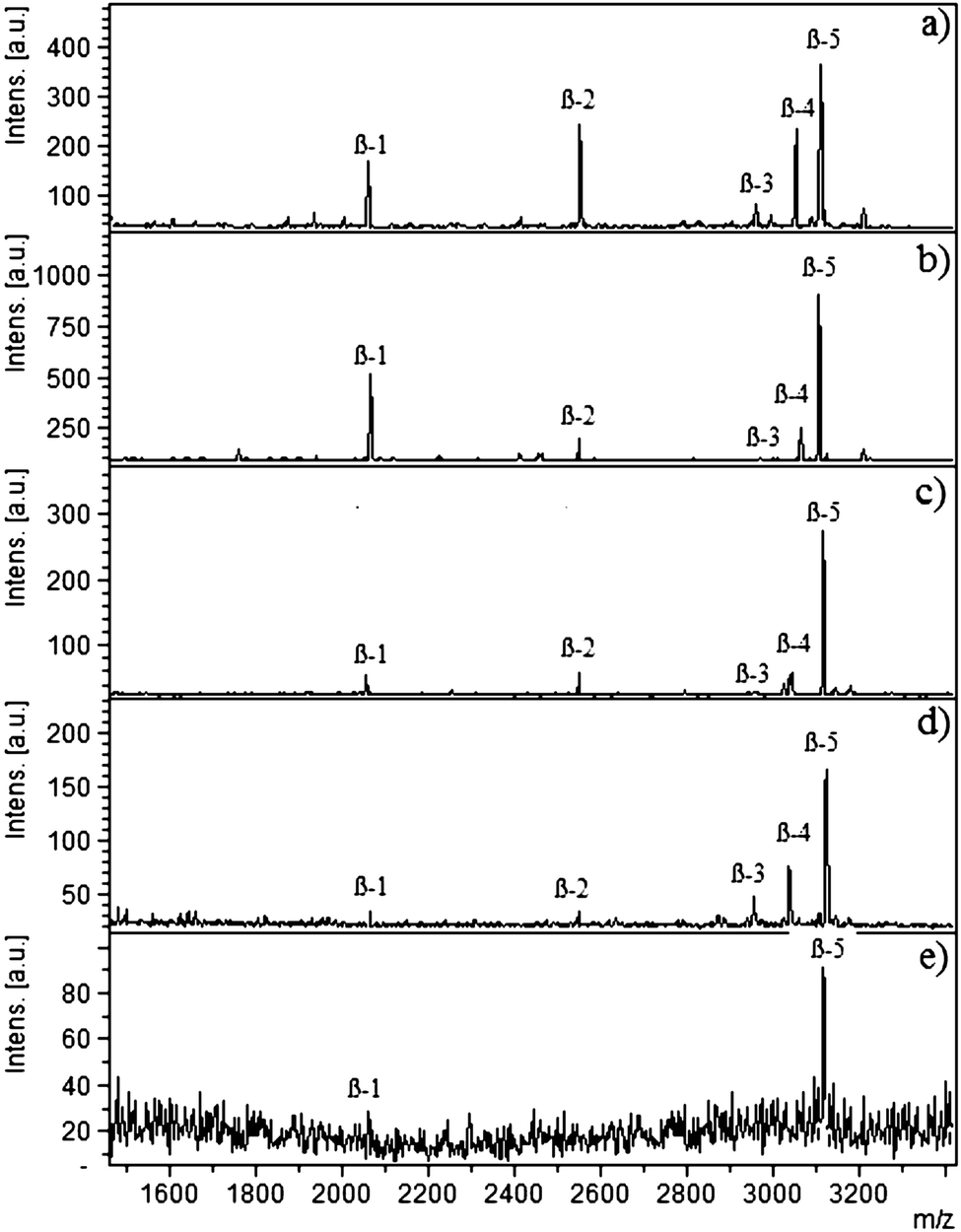 | ||
| Fig. 1 MALDI-MS spectra of tryptic β-casein digest spiked in dephosphorylated HeLa cell extract using cellulose–silica IMAC-Sm3+ in different ratios as: (a) 1:100; (b) 1:500; (c) 1:1000; (d) 1:1500; (e) 1:2000. The symbol β represents phosphopeptides derived from β-casein. | ||
Sensitivity of cellulose–silica IMAC-Sm3+
The ability of the composite to enrich the lower concentrations of phospho content was determined by applying varying concentrations. The complete study was designed with the starting concentration of 100 femtomole of β-casein and going down to 1 femtomole (Fig. S8†). Three phosphopeptides with one tetra phosphorylated peptide were evident in the case of the 1 femtomole sample. Usually multi-phosphopeptides are difficult to enrich at lower concentrations due to their easy loss during washing steps,17 however, Sm3+ showed high affinity which was enhanced by the hydrophilic base materials (cellulose and silica) and overall there was improved enrichment of the multi-phosphopeptides.Roles of the base materials (cellulose and silica)
Base materials play an important role in enrichments, as reported earlier for the MELDI technique.18 The consideration of only immobilized metal ions and ignoring the base materials leads to non-specific bindings. To observe the impact, β-casein digest was applied to cellulose, silica, cellulose–silica composite and end capped silica–lanthanum oxide19 (Fig. 2). In the cases of cellulose and silica, mostly the non-phosphopeptides were detected which are labelled as NPP (Fig. 2A). The binding of these peptides is linked to the hydrophilic nature and surface hydroxyl groups of cellulose and silica; depicted through the common bound peptides (NPP) because of the structural similarities. Phosphopeptides interact through their phosphorylated serine site whereas non-phosphopeptides (NPP) interact through the carboxylic group of acidic amino acids such as glutamic acid (Glu) and aspartic acid (Asp). In the case of cellulose, hydrophobic sites due to CH–O20 allow the binding of peptides with a relatively higher degree of hydrophobicity, contributed to the peptide backbone by alanine, valine, leucine, isoleucine and proline. For silica, silanol groups (Si–OH) are abundant on the surface and available for interaction with acidic and phosphorylated peptides. As a combination of both materials, cellulose–silica IMAC-Sm3+ has three interaction sites, namely cellulose, silica and Sm3+ ions. Nine phosphopeptides were detected in the MS spectrum of the cellulose–silica composite (Fig. 2A-c). The presence of weak signals for non-phosphopeptides at m/z 1069 and 2106 are because of cellulose and silica. Under similar washing steps, it can be seen that for the cellulose–silica composite, non-phosphopeptide interactions are weakened, whereas these washings may not be enough for individual cellulose and silica.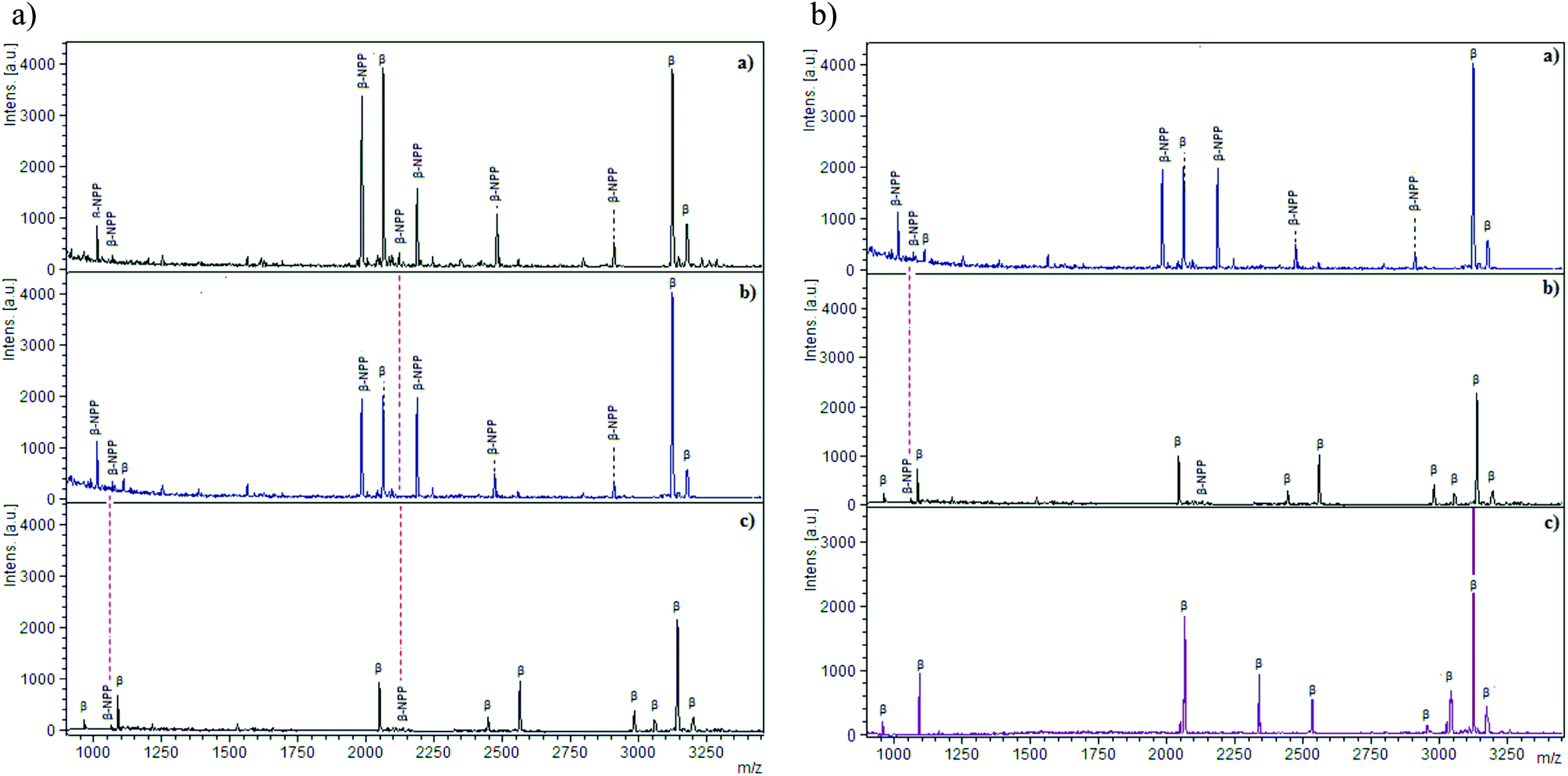 | ||
| Fig. 2 Comparison (A) MALDI-MS spectra recorded for (a) cellulose (powder, Sigma Aldrich); (b) silica gel (Sigma Aldrich mesh size 70–230); (c) cellulose–silica IMAC-Sm3+. Comparison (B) (a) silica gel (Sigma Aldrich mesh size 75–230); (b) cellulose–silica IMAC-Sm3+ (c) end-capped silica–lanthanum oxide composite. Tryptic digest of β-casein is applied (20 μL diluted in 0.1% TFA) following the optimized buffer conditions. Enriched phosphopeptides are labeled as β, whereas non-phosphopeptides are labeled as β-NPP. | ||
A second comparison is made among silica, cellulose–silica IMAC-Sm3+ and end-capped silica–lanthanum oxide composite (Fig. 2B). The presence of non-phosphopeptide at m/z 1069 because of silica is present in the MS spectra of both individual silica (Fig. 2B-a) and cellulose–silica IMAC-Sm3+ (Fig. 2B-b). The absence of non-phosphopeptides in silica–lanthanum oxide composite is because of the end-capping which blocks the un-derivatized silanol groups on silica (Fig. 2B-c). The details of the peptides detected during the comparison are given in Table 1. In conclusion, not only the metal ions immobilized on the IMAC material are taken into account while designing any material for enrichment, rather the base material also contributes to enhancing the enrichment efficiency. Proper consideration towards the role of the base material actually helps to optimize the sample preparation methodology.
| m/z | Sequences | Amino acid position | Nature of peptide | Enriched peptides | |||||
|---|---|---|---|---|---|---|---|---|---|
| Phospho-rylation | Acidic peptide (Glu/Asp) | Degree of hydrophobicity (Krokhin_NET) | Cellulose | Silica | Cellulose–silica IMAC | Silica–La2O3 | |||
| 975 | KFQS*EEQQQ | 47–48 | 1P | 2 Glu | 0.0445766 | — | — | * | * |
| 1013 | HKEMPFPK | 106–113 | 1 Glu | 0.140542 | * | * | — | — | |
| 1094 | PVLGPVRGPFPIIV | 215–224 | 0.4724251 | * | * | — | — | ||
| 1104 | KFQS*EEQQQT | 47–49 | 1P | 0.04997731 | — | * | * | * | |
| 1994 | LLYQEPVLGPVRGPFPIIV | 207–224 | Glu | 0.5902297 | * | * | — | — | |
| 2061 | FQS*EEQQQTEDELQDK | 33–48 | 1P | 0.09920053 | * | * | * | * | |
| 2106 | FLLQEPVLGPVRGPFPIIV | 205–224 | Glu | 0.6408923 | * | — | * | — | |
| 2187 | DMPIQAFLLYQEPVLGPVR | 184–202 | Asp | 0.5987214 | * | * | * | — | |
| 2352 | NVPGEIVES*LS*S*S*EESITR | 32–40 | 4P | 0.300066 | — | — | * | * | |
| 2556 | FQS*EEQQQTEDELQDKIHPF | 33–52 | 1P | 0.2779962 | — | — | * | * | |
| 2467 | WMHQPHQPLPPTVMFPPQSVL | 158–178 | 0.5060722 | * | * | — | — | ||
| 2909 | DMPIQAFLLQEPVLGPVRGPFPIIV | 200–224 | Asp/Glu | 0.7687585 | * | * | — | — | |
| 2965 | RELEELNVPGEIVES*LS*S*S*EESITR | 1–24 | 4P | 0.476857 | — | — | * | * | |
| 3054 | KIEK FQS*EEQQQTEDELQDKIHPF | 29–52 | 1P | 0.04226933 | — | — | * | * | |
| 3124 | RELEELNVPGEIVES*LS*S*S*EESITRI | 1–25 | 4P | 0.5137824 | * | * | * | * | |
| 3180 | RELEELNVPGEIVES*LS*S*S*EESIT | 26–39 | 4P | 0.4447257 | * | * | * | * | |
Reproducibility
Development of the enrichment strategy relies on the reproducibility of the material and method independent of time and space. Fig. S9† is representative of the reproducible results using serum digest. The intensity difference can be attributed to the sample preparation, the co-crystallization procedure and instrumental factors such as laser power and laser intensity. For cellulose–silica IMAC-Sm3+, statistical analysis was applied to the reproducibility study. The highest standard deviation (SD) found was 0.379 and the lowest was 0.150 (Table S2†).Cellulose–silica IMAC-Sm3+ for phosphoproteins/phosphopeptides
The developed enrichment strategy for cellulose–silica IMAC-Sm3+ was tested on a protein mixture containing phosphoproteins like α- and β-casein with non-phosphoproteins such as lysozyme, myoglobin, cytochrome c and BSA. The non-phosphoproteins were selected considering the range from hydrophobic to hydrophilic. Phosphoproteins α- and β-casein having a mass in the range of 24 kDa to 26 kDa were successfully enriched from the protein mixture with no trace of non-phosphoproteins (Fig. S10†). Both α-casein variants, αS1 and αS2 including the β-casein, were detected.The complexity level was further enhanced and a fraction of this protein mixture (200 μL) was digested to provide a high complexity background in the peptide range. Fig. 3a shows the MS spectrum of the direct analysis of the peptide mixture after digestion. Only four phosphopeptides can be detected with a high number of non-phosphopeptides. However, after the enrichment, 22 phosphopeptides were detected with 13 mono-phosphorylated, three di-phosphorylated, one tri-phosphorylated and five tetra-phosphorylated peptides derived from α- and β-casein.
 | ||
| Fig. 3 Enrichment of the α- and β-casein phosphopeptides from the protein mixture digest (containing α-, β-casein, myoglobin, lysozyme, cytochrome c, BSA, 1 mg mL−1) (a) before enrichment; (b) after enrichment. Symbols α and β represent the phosphopeptides, with position and number of phosphorylated serine also given.. | ||
Cellulose–silica IMAC-Sm3+ for phospholipid analysis (from standard)
The separation strategy for phospholipids is based on Folich’s extraction method, which is time consuming. An IMAC based strategy is reported here for the first time to enrich phospholipids without any pre-fractionation and extraction. The composite-IMAC-Sm3+ and the optimized protocol simultaneously enrich both phospho-based classes of compounds, i.e. phospholipids and phosphopeptides, in a single step. They are successively eluted with no cross contamination. Phospholipids, due to their low mass/size, bind to IMAC materials and occupy/compete for the binding sites which would otherwise be used for phosphopeptides. Therefore, cellulose–silica IMAC-Sm3+ was also tried for the analysis of phospholipids with little modification in the enrichment protocol. As phospholipid has lipid as its counterpart, the bound phospholipids can be eluted using 5% ammonium hydroxide in a methanol–dichloromethane mixture (1:9, v/v). It is important to consider that the phosphopeptides are not eluted because of the hydrophobic nature of the optimized elution buffer. A phospholipids mixture (Sigma Aldrich) was applied to the cellulose–silica IMAC-Sm3+. As they are low mass species, gold nanoparticles in 0.1% TFA and a carbon based LDI matrix were used for their MS analysis to avoid matrix interferences (Fig. S11a and b†). All phospholipids present in the mixture were enriched with no background from the materials used as the LDI matrix.
Enrichment of phospholipids/phosphopeptides from egg yolk digest
Hen egg yolk consists of phosphatidylcholine (PC, 73%), lysophosphatidylcholine (LPC, 5.8%), sphingomyelin (SM, 2.5%), phosphatidylethanolamine (PE, 15.0%), lysophosphatidylethanolamine (LPE, 2.1%) and phosphatidylinositol (PI, 0.6%). This composition can only be used as a rough guide, because the phospholipids and acyl composition of egg are influenced by many parameters, including the season, foods eaten by the hen, etc. However, at least six different PLs are present in egg yolk extract. Among the egg yolk proteins, phosvitin (PV, 7%) and lipovitellin (LP, 69%) are phosphorylated. Single batch extraction was applied to the cellulose–silica IMAC-Sm3+ for egg yolk digest. The stepwise elution of phospholipids and phosphopeptides was carried out. In the first elution, enriched phospholipids were recovered. This was followed by an extra methanol and water wash step to remove any residual traces. They were analysed by LDI-MS. In the second eluted fraction, with 2% aqueous ammonium hydroxide, phosphopeptides were released, spotted with 2,5-dihydroxybenzoic acid (DHB) buffer and recorded by MALDI-MS. Fig. 4 shows the MS spectra of the tryptic digest of the egg yolk before and after the enrichment. Only one phosphopeptide was identified prior to enrichment (Fig. 4a) whereas, after the enrichment by cellulose–silica IMAC-Sm3+, 15 phosphopeptides were detected (Fig. 4b). Fig. 5 (3 expanded mass ranges) shows phospholipids detected in the eluted fraction belonging to different phospholipid classes. Phosphopeptides identified from phosvitin belong to Domain-III, position 1301–1322 SGHLEDDSSSSSSSSVLSKIWG (PV 190–211) with 6–10 phosphorylations. A highly phosphorylated peptide from phosvitin with 10P was detected at m/z 1894.98 SGHLEDDSSSSSSSSVLSK (10P). For lipovitellin, Domain IV, position 1056-1076 IITEVNPESEEEDESSPYEDI, three phosphopeptides were detected. | ||
| Fig. 4 MALDI-MS analysis of phosphopeptides derived from phosvitin (P02845) and lipovitellin (P87498) in egg yolk digest (a) before enrichment; (b) after applying cellulose–silica IMAC-Sm3+. Phosphopeptides are labeled as PV for phosvitin and LP for lipovitellin with position numbers. | ||
 | ||
| Fig. 5 MALDI-MS spectra of the phospholipids in the egg yolk digest employing two batches of cellulose–silica IMAC-Sm3+ using (Batch A) gold nanoparticles in 0.1% TFA as the LDI matrix; (Batch B) a carbon based LDI matrix. Abbreviations used: PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI, phosphatidylinositol; SM, sphingomyelin; LPC, lyso-phosphatidylcholine; LPE, lyso-phosphatidylethanolamine. | ||
Phosphorylation and prostate cancer
Phosphoproteins of clinical value are reported for prostate cancer. Their phosphorylated peptides in the serum digest are enriched by cellulose–silica IMAC-Sm3+ (Table S3†). The same phosphorylation sites can also be used to enrich the whole phosphoprotein (as shown in Fig. S10†). Thus, the developed enrichment material and protocol can enrich the phosphopeptides/phosphoproteins of clinical significance. Phosphopeptides associated with prostate cancer are highlighted in Table S3.† To understand the importance, some of the phosphorylated proteins with relevance to prostate cancer are discussed here. CCDC124 (TIEDAIAVLSVAEEAADR, peak 26, inset Fig. S12†) is a novel factor operating both for proper progression of the late cytokinetic stages in eukaryotes and for the establishment of Rap2 signaling dependent cellular functions proximal to the abscission site.21 Down-regulation of phosphodiesterase 4B (PDE4B, ALDPQSSPGLGRIMQAPVPHSQR, peak 36, inset Fig. S12†) activates protein kinase A and contributes to the progression of prostate cancer.22 In clinical prostate cancer specimens, the expression of the uncoupling protein 1 (UCP1, SRQTMDCAT, peak 18, NNILADDVPCHLVSALIAGFCATAMSSPVDVVK, peak 45, inset Fig. S12†), a brown fat-specific marker, is enhanced with the level of expression correlated to disease progression from primary to bone metastatic cancers.23 Aminopeptidase N (GASVLRMLSSFLSEDVFK, peak 29, inset Fig. S12†) has been associated with the growth of prostate cancer and has been suggested as a suitable target for anti-cancerous therapy.24 Ang-III and related converting enzymes contribute to the cell proliferation of prostate cancer, and may be implicated in cancer progression.25 Functional PDC can form in mitochondria outside of the matrix in prostate cancer cells and PDHK1 (SFSSDSGSSPASER, peak 27, LFNY*MY*STAPRPRVETSR, peak 35, inset Fig. S12†) is commonly tyrosine phosphorylated by diverse oncogenic tyrosine kinases localized to different mitochondrial compartments. Expression of phosphorylation-deficient, catalytic hypomorph PDHK1 mutants in cancer cells leads to decreased cell proliferation under hypoxia and increased oxidative phosphorylation with enhanced mitochondrial utilization of pyruvate and reduced tumor growth in xenograft nude mice. Together, tyrosine phosphorylation activates PDHK1 to promote the Warburg effect and tumor growth.26 The expression patterns of the HOX network in human prostate cell phenotypes, representing different stages of prostate physiology and prostate cancer progression, make it possible to discriminate between different human prostate cell lines and to identify loci and paralogous groups harboring the HOX mostly involved in prostate organogenesis and cancerogenesis.27 The amount of cross-linked fibrinogen gamma-chain dimer in plasma may correlate with tumor-associated fibrin deposition. The tumor-biological relevance of this potential marker protein needs to be explored.28 Mitochondrial ribosomal protein L54 has been shown to result in prostate cellular differentiation and cell growth arrest. Up-regulation is identified in malignant prostate cancer cell lines and in prostate cancer tissue samples.29 This information can be used along with the PSA test for prostate cancer to diagnose with a high confidence level.According to the information provided by the National Cancer Institute, screening tests for prostate cancer need a method with a high sensitivity and specificity. After optimization of the protocol for the synthesized composite in different formats with commercial perspective like packed in-tip or in column, it can be used for prostate cancer screening.
Cellulose–silica C18 – desalting and recovery of hydrophilic molecules
Pre-concentration of peptide mixtures by desalting materials is preferred prior to mass spectrometric analysis for increased signal-to-noise ratio, sensitivity and consequently the sequence coverage. A large proportion of peptides, however, remain undetected by MS presumably because of their loss during sample preparation, or suppression in the ionization process. Cellulose–silica C18 is synthesized as an alternative to the conventional desalting materials which suffer from a loss of hydrophilic species. As a RP material, it provides a combination of the hydrophilic composite and the hydrophobic C18 chain. This contrasting combination of cellulose–silica-C18 helps to retain moieties of both hydrophilic and hydrophobic origin and hence leads to a better recovery of proteins/peptides from a range of samples. Cellulose–silica C18 was applied to a casein mixture (α- and β-) with different salt concentrations (Fig. S13†). The high salt concentration of 8 M urea produced no adducts with all of the peptides enriched and identified. The issue of loss of hydrophilic species by most desalting materials was also verified. During the protein digestion procedures, the peptides containing methionine are oxidized which makes the peptides more hydrophilic. For the tryptic digest of α-casein, the styrene divinyl benzene (SDB) tips (GL Sciences®) and Zip tip C18 (Millipore®) show a loss of peptides (Fig. S14a and b†). For the graphite tips (GL Sciences®), there is a high number of peptides in the mass range 1500 to 3200 Da (Fig. S14c†). Cellulose–silica C18 shows a distinctive peptide mass range with a total of 32 peptides identified. The highlighted region shows the major difference between the GL tips and cellulose–silica C18 (Fig. S14d†). The difference in relative ion intensity of peptides observed also indicates that hydrophilic peptides are favoured by cellulose–silica C18.Another study with non-fat milk digest was carried out for comparison and improvement of the sequence coverage. Peptides enriched by cellulose–silica C18 are shown in Fig. S15.† Peptides from milk caseins were identified using the SwissProt database and sequence coverage analysis was also carried out for these milk proteins (Table S4†). For α-casein, sequence coverage of 26%, 38%, 65.8% and 84.8% was shown by SDB tip, Zip tip C18, GC tip and cellulose–silica C18 respectively. For β-casein, the comparison is distinctive with the highest sequence coverage shown by cellulose–silica C18 (98.4%) and the lowest by Zip tip C18 (30.2%) and SDB tip (32.9%). The efficiency difference shows that hydrophobicity due to the C18 chains is not enough for desalting, rather contribution also comes from the hydrophilic base of cellulose and silica.
Experimental
Materials
The chemicals and reagents used in this study are given in the ESI.†Synthesis of the cellulose–silica composite
The derivatization of silica with aminopropyl silane, the oxidation of cellulose and the chlorination are given in the ESI.† Aminopropyl bonded silica was refluxed with cellulose acetyl chloride in 10 mL of anhydrous tetrahydrofuran (THF) for 24 h at 66 °C. The resulting composite was purified by washing with THF followed by methanol–water (1:1, v/v) to hydrolyze the unreacted acetyl chloride.
Functionalization of the cellulose–silica composite as IMAC
Non-selective oxidation of the composite was carried out in a suspension with 300 mL of a 0.1 M aqueous solution of perchlorate (HClO4) for 24 h at 25 °C. The subsequent chlorination involved a reflux reaction in 10 mL of anhydrous thionyl chloride for 24 h at 76 °C. After the removal of thionyl chloride and vacuum drying, the cellulose–silica composite with acid chloride functionalities was suspended in 20 mL of anhydrous THF, refluxed with 1 g of iminodiacetic acid (IDA) and 1 mL of triethylamine (TEA) for 72 h at 66 °C. The product was washed with dichloromethane followed by ethanol and water.Metal immobilization
Different metal ions (Fe3+, Zr4+, La3+, Sm3+) were immobilized using their respective salt solutions. In general, 1 mg of cellulose–silica IMAC was incubated with 100 μL of the 0.1 M salt solution for 45 min at 28 °C. The material was washed twice with water followed by the activation buffer (100 μL), 80% acetonitrile (ACN) in 0.1% (v/v) trifluoroacetic acid (TFA).Functionalization as a desalting material
The synthesis of cellulose–silica C18 was carried out by suspending cellulose–silica acid chloride in 25 mL of ACN and mixed with 2 g of octadecylamine (ODA). The mixture was refluxed for 72 h at 80 °C. The product was washed with ethanol.Sample preparation for enrichment/desalting
The digestion of standards and biological fluids (non-fat milk, egg yolk and serum for prostate cancer), preparation of the dephosphorylated HeLa cell sample, and details of the selectivity, sensitivity and use as a desalting material tests are provided in the ESI.†Enrichment protocol for phosphopeptides/phospholipids on cellulose–silica IMAC-Sm3+
Phosphopeptides enrichment was carried out by incubating 20 μL of tryptic digest containing 0.1% TFA with cellulose–silica IMAC-Sm3+ at room temperature under gentle shaking for 30 min. The material was washed twice with 100 μL of 80% ACN in 0.1% TFA followed by a wash with 110 mg of DHB30 dissolved in 10% ACN/0.2% TFA. After a wash with water, 20 μL of 2% ammonium hydroxide (pH 10) was used for the elution of bound phosphopeptides. DHB could serve as a displacer for the non-phosphorylated peptides which thus significantly prevented the binding of non-phosphorylated peptides. The idea of using DHB in the washing solvent was supported by Larsen et al.31 During the current study, it was optimized that 20 mg mL−1 DHB in 2% ACN/0.1% TFA was efficient in removing the non-phosphorylated compounds.For the phospholipids, the phospholipid mixture from Sigma Aldrich (40 μL) was used as the standard. The sample was loaded on activated cellulose–silica IMAC-Sm3+ and incubated for half an hour at room temperature. The material was washed with 800 μL of 1% CH3COOH in CHCl3 and CH3OH (10:30) followed by 80% ACN in 0.2% TFA. For the elution of the phospholipids, 20 μL of 2% ammonia in CH3OH (70:30) was used.
In the case of the egg yolk and serum, successive elution of the phospholipids followed by the phosphopeptides was carried out. First the phospholipids were eluted using 2% ammonia in CH3OH (70:30) followed by elution of the phosphopeptides using 1.5% ammonium hydroxide solution.
Desalting by cellulose–silica C18
Desalting was carried out for different samples like β-casein in different salt concentrations (1 M urea, 4 M urea and 8 M urea) for desalting efficiency, α-casein for hydrophilic recovery and milk digest for improved sequence coverage. A comparison is made with the commercial desalting materials. Cellulose–silica C18 (1 mg) was activated by 100 μL of 80% ACN in 0.1% TFA. The sample (20 μL) was incubated for 10 min and the supernatant was removed. The material was washed with 100 μL of 5% ACN in 0.1% TFA followed by a wash with water. Phosphopeptides were eluted using 10 μL of 50% ACN in 0.1% TFA.Mass spectrometric analysis
The MS analysis details are provided in the ESI.†Conclusion
A new methodology has been developed to synthesize a cellulose–silica composite and its functionalization as an IMAC and RP material. A FT-IR spectral study confirmed the synthesis by showing the characteristic peak of the carbonyl amide at 1630 cm−1 and the C–N stretch at 1056 cm−1. A cellulose–silica IMAC-Sm3+ composite has been developed to enrich phosphopeptides/phospholipids in a single step followed by their two-step successive elution. Phosphopeptides were recorded with MALDI-MS whereas the low molecular weight phospholipids were analyzed by the newly designed LDI-MS methodology. The role of metal ions on the designed IMAC composite was compared to the contribution coming from the base materials, i.e. cellulose and silica. High selectivity of 1:2000 and sensitivity down to 1 fmol allowed the detection of low concentrated phosphopeptides from protein mixture digest and human serum. The material and the sample preparation protocol was optimized to the extent that the low concentrated phosphopeptide biomarkers relevant to prostate cancer were enriched from serum digest. Efficient enrichment from high salt concentrations, recovery of hydrophilic species and the improved sequence coverage makes cellulose–silica C18 a competent desalting tool compared to SDB tip, GC Tip from GL Sciences and Zip tip C18.
Acknowledgements
This work is supported by the Higher Education Commission (HEC) of Pakistan. The authors are thankful to the Institute of Analytical Chemistry and Radiochemistry, Leopold-Franzens University, Innsbruck, Austria for providing access to laboratories and the MALDI-MS instrument. Furthermore, the authors declare that they have no conflict of interest.References
- H. Ma, J. R. McLean, L. F. Chao, S. Mana-Capelli, M. Paramasivam, K. A. Hagstrom, K. L. Gould and D. McCollum, Mol. Cell. Proteomics, 2012, 11, 501–511 Search PubMed.
- J. Zhu and G. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 925–932 CAS.
- A. Fleitz, E. Nieves, C. Madrid-Aliste, S. J. Fentress, L. D. Sibley, L. M. Weiss, R. H. Angeletti and F. Y. Che, Anal. Chem., 2013, 85, 8566–8576 CrossRef CAS PubMed.
- M. Ndao, Methods Mol. Biol., 2012, 818, 67–79 CAS.
- A. Venugopal, R. Chaerkady and A. Pandey, Ann. Indian Acad. Neurol., 2009, 12, 3–11 Search PubMed.
- V. Guryča, D. Roeder, P. Piraino, J. Lamerz, A. Ducret, H. Langen and P. Cutler, Biology, 2014, 3, 205–219 CrossRef PubMed.
- A. Khan, A. M. Asiri, M. A. Rub, N. Azum, A. A. P. Khan, I. Khan and P. J. Mondal, Int. J. Electrochem. Sci., 2012, 7, 3854–3902 CAS.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS PubMed.
- A. A. Taha, Y. Wu, H. Wang and F. Li, J. Environ. Manage., 2012, 112, 10–16 CrossRef CAS PubMed.
- A. Tolonen, W. Haas, A. C. Chilaka, J. Aach, S. P. Gygi and G. M. Church, Mol. Syst. Biol., 2011, 7, 461–472 CrossRef PubMed.
- T. Kimuraa, Phys. Chem. Chem. Phys., 2013, 15, 15056–15061 RSC.
- I. Feuerstein, S. Morandell, G. Stecher, C. W. Huck, T. Stasyk, H. L. Huang, D. Teis, L. A. Huber and G. K. Bonn, Proteomics, 2005, 5, 46–54 CrossRef CAS PubMed.
- L. Trojer, G. Stecher, I. Feuerstein, S. Lubbad and G. K. Bonn, J. Chromatogr. A, 2005, 1079, 197–207 CrossRef CAS PubMed.
- J. Duan, H. Wang and Q. Cheng, Anal. Chem., 2010, 82, 9211–9220 CrossRef CAS PubMed.
- T. Witoon and M. Chareonpanich, Songklanakarin J. Sci. Technol., 2012, 34, 403–407 CAS.
- C. W. Huck, R. Ohmacht, Z. Szabo and G. K. Bonn, J. Near Infrared Spectrosc., 2006, 14, 51–57 CrossRef CAS.
- J. Fíla and J. Honys, Amino Acids, 2012, 43, 1025–1047 CrossRef PubMed.
- M. Najam-ul-Haq, M. Rainer, L. Trojer, I. Feuerstein, R. M. Vallant, C. W. Huck, R. Bakry and G. K. Bonn, Expert Rev. Proteomics, 2007, 4, 447–452 CrossRef CAS PubMed.
- F. Jabeen, D. Hussain, B. Fatima, S. G. Musharraf, C. W. Huck, G. K. Bonn and M. Najam-ul-Haq, Anal. Chem., 2012, 84, 10180–10185 CrossRef CAS PubMed.
- Q. Li and S. Renneckar, Biomacromolecules, 2011, 12, 650–659 CrossRef CAS PubMed.
- P. Telkoparan, S. Erkek, E. Yaman, H. Alotaibi, D. Bayık and U. H. Tazebay, PLoS One, 2013, 8, e69289 CAS.
- E. Kashiwagi, M. Shiota, A. Yokomizo, M. Itsumi, J. Inokuchi, T. Uchiumi and S. Naito, Prostate, 2012, 72, 741–751 CrossRef CAS PubMed.
- H. E. Zhau, H. He, C. Y. Wang, M. Zayzafoon, C. Morrissey, R. L. Vessella, F. F. Marshall, L. W. Chung and R. Wang, Clin. Cancer. Res., 2011, 17, 2159–2169 CrossRef CAS PubMed.
- M. Wickström, R. Larsson, P. Nygren and J. Gullbo, Cancer Sci., 2011, 102, 501–508 CrossRef PubMed.
- J. I. Teranishi, H. Ishiguro, K. Hoshino, K. Noguchi, Y. Kubota and H. Uemura, Prostate, 2008, 68, 1666–1673 CrossRef CAS PubMed.
- T. Hitosugi, J. Fan, T. W. Chung, K. Lythgoe, X. Wang, J. Xie, Q. Ge, T. L. Gu, R. D. Polakiewicz, J. L. Roesel, G. Z. Chen, T. J. Boggon, S. Lonial, H. Fu, F. R. Khuri, S. Kang and J. Chen, Mol. Cell, 2011, 44, 864–877 CrossRef CAS PubMed.
- M. Cantile, A. Kisslinger, L. Cindolo, G. Schiavo, V. D’antò, R. Franco, V. Altieri, A. Gallo, A. Villacci, D. Tramontano and C. Cillo, J. Cell. Physiol., 2005, 205, 202–210 CrossRef CAS PubMed.
- C. Gerner, W. Steinkellner, K. Holzmann, A. Gsur, R. Grimm, C. Ensinger, P. Obrist and G. Sauermann, Thromb. Haemostasis, 2001, 85, 494–501 CAS.
- J. L. Goodin and C. L. Rutherford, Mol. Biol. Rep., 2002, 29, 301–316 CrossRef CAS.
- M. R. Mirza, M. Rainer, Y. Güzel, M. I. Choudhary and G. K. Bonn, Anal. Bioanal. Chem., 2012, 404, 853–862 CrossRef CAS PubMed.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. D. Jørgensen, Mol. Cell. Proteomics, 2005, 4, 873–886 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10254a |
| This journal is © The Royal Society of Chemistry 2015 |