DOI:
10.1039/C4RA09123J
(Paper)
RSC Adv., 2014,
4, 53149-53156
Comparative DFT- and DFT-D-based molecular dynamics studies of pressure effects in crystalline 1,3,5-triamino-2,4,6-trinitrobenzene at room temperature†
Received
23rd August 2014
, Accepted 25th September 2014
First published on 25th September 2014
Abstract
The crystal, molecular, and electronic structures of crystalline 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) in the pressure range of 0–100 GPa under room temperature have been studied by ab initio molecular dynamics (MD). The DFT- and DFT-D-based MD were used to investigate the effects of the vdW correction on the results. The lattice parameters and P–V isotherm of TATB under compression by the DFT-D agree well with the experimental results, whereas the DFT without the vdW correction either underestimates or overestimates the results evidently. The DFT-D results show that TATB is chemically stable in the entire investigated pressure range, in agreement with the experiments, but the DFT without the vdW correction misestimates that TATB decomposes at 50 GPa and polymerizes at 100 GPa. Both the intra- and intermolecular hydrogen bonding are strengthened with the increasing pressure from 5 to 50 GPa, consistent with the experimental results. The DFT without the vdW correction misestimates that TATB turns into a metal system at 100 GPa. The PBE0 with the vdW correction was used to study the band structures of TATB, and the obtained results up to 50 GPa are in agreement with the available experiment results.
Introduction
1,3,5-Triamino-2,4,6-trinitrobenzene (TATB, C6H6N6O6, Fig. 1)1 is a graphite-like layered molecular crystal, which has high detonation performance but displays an exceptional insensitivity to thermal, impact, friction, and shock insults, making it one of the most important insensitive explosives. It crystallizes in a triclinic lattice with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and contains two C6H6N6O6 molecules per unit cell.1 The strong two-dimensional hydrogen bonded network is thought to be responsible for its extremely low sensitivity and insolubility in most solvents.2,3 TATB has attracted considerable attention from both theoretical and experimental studies. Unfortunately, many fundamental problems of TATB are still not well understood, because it possesses a complex chemical behavior. It is known that explosives may detonate when subjected to an external stimulus. The detonation process generates a shock wave, which represents a condition of high pressure and high temperature. Therefore, the structure and properties of TATB under these extreme conditions would be of great importance in understanding its sensitivity and in designing novel insensitive high explosives.
space group and contains two C6H6N6O6 molecules per unit cell.1 The strong two-dimensional hydrogen bonded network is thought to be responsible for its extremely low sensitivity and insolubility in most solvents.2,3 TATB has attracted considerable attention from both theoretical and experimental studies. Unfortunately, many fundamental problems of TATB are still not well understood, because it possesses a complex chemical behavior. It is known that explosives may detonate when subjected to an external stimulus. The detonation process generates a shock wave, which represents a condition of high pressure and high temperature. Therefore, the structure and properties of TATB under these extreme conditions would be of great importance in understanding its sensitivity and in designing novel insensitive high explosives.
 |
| | Fig. 1 A molecule (a), a unit cell (b), a 1 × 1 × 2 supercell (c), a 1 × 2 × 1 supercell (d), and a 2 × 1 × 1 supercell (e) of TATB. Carbon, nitrogen, oxygen, and hydrogen atoms are represented by gray, blue, red, and white spheres, respectively. The four intermolecular hydrogen bonds are displayed by the pink dotted lines. | |
Several experiments4–9 have been conducted to investigate the behavior of TATB under different pressures in the past. Only two studies4,7 reported its cell volumes and cell parameters under high pressures less than 15 GPa. Also, other two studies8,9 extended the investigated pressure more than 20 GPa. The former collected the IR data up to 40 GPa and stated the strengthening of intermolecular hydrogen bonding with the increasing pressure without any apparent phase transitions, while the latter reported that TATB undergoes two phase transitions at 28 and 56 GPa, respectively, and then it is a chemically stable insulator until 150 GPa, but this study did not give any data of cell volumes, cell parameters, or electronic structure under high pressures.
In addition, some theoretical studies10–15 have been carried out to study the structure and properties of TATB under hydrostatic compression at 0 K by density functional theory (DFT). A DFT-LDA study on electronic structure of solid TATB under uniaxial compression reports that there is a metallization in TATB at a pressure of 120 GPa.10 Two studies predicted the P–V curves of TATB under hydrostatic compression from 0 to 7 GPa (ref. 11) and from 0 to 10 GPa (ref. 12) using different functionals and plane wave basis set sizes, but their obtained results are found to be different with experiments, which is due to the inadequate treatment of van der Waals (vdW) interactions. A latter study15 extended the investigated pressure up to 250 GPa using periodic DFT calculations without the vdW correction and indicated that TATB undergoes a displacive phase transition around 70 GPa and is unreacted within a wide pressure range. Since TATB is a layered molecular system, the vdW forces between the layers play a very important role in its crystal packing. The vdW correction should be considered while studying its structure and properties. This can be supported by two recent DFT studies,13,14 in which the results with the vdW correction were close to experimental results up to 7 (ref. 13) and 30 GPa,14 respectively. Overall, these studies either did not consider the influence of vdW forces and room temperature or considered narrow pressure ranges. Thus, further systematic studies on TATB at elevated pressures under room temperature are needed.
It is still a challenging task to investigate microscopic properties of the explosives. Theoretical calculations are an effective way to model the physical and chemical properties of the explosives under high pressure. Recently, ab initio molecular dynamics (MD) have been employed to study the structure and decomposition mechanisms of the explosives under different temperatures or shock loading successfully, for example, pentaerythritol tetranitrate (PETN),16 CL-20 (2,4,6,8,10,12-hexnitro-2,4,6,8,10,12-hexaazaisowurtzitane),17 and silver azide.18
In this work, we performed ab initio MD simulations to study the crystal, molecular, and electronic structures of crystalline TATB in the pressure of 0–100 GPa under room temperature. The effects of vdW correction on the results were investigated. Nonlocal exchange-correlation functionals were used to predict more accurate and reliable band gap of TATB. Our main purpose here was to examine the effects of different pressures on the microscopic properties of the crystal under room temperature.
Computational methods
Our ab initio MD simulations were performed within the framework of DFT based on CASTEP code19 using norm-conserving pseudopotentials20 and a plane-wave expansion of the wave functions. The Perdew–Burke–Ernzerhof21 (PBE) exchange-correlation function and a single k-point were employed. To correct vdW correction, we used two types of corrections, namely, the Grimme22 and the Tkatchenoko and Scheffer (TS),23 to the PBE functional. We utilized a plane wave cutoff of 500 eV for MD simulations and that of 830 eV for geometry and cell optimizations and electronic structure calculations. We controlled the ionic temperature and pressure using a Nosé thermostat24 and an Andersen barostat,25 respectively. A time step of 1.0 fs was used in time integration. Previous studies16 reported that a time step of 1.2 fs was used to study the thermal decomposition of PETN. A time step of 1.0 fs was also adopted in the studies of sodium26 and iron27 under extreme conditions of temperature and pressure using AIMD. Both the NVT and NPT ensembles were employed. The initial positions of the simulation supercell were taken from the experimentally determined X-ray crystal structure.1 We used bong-lengths criteria to identify the changes in geometry during the simulations. Each ensemble using NPT was equilibrated for 3 ps, following which simulations were performed for an additional 7 ps to collect data for statistical analysis.
Effects of vdW correction
Since the vdW forces between the TATB layers play a very important role along the c direction, 1 × 1 × 2 supercell (4 molecules and 96 atoms, Fig. 1) were built along this direction to study the effects of the vdW correction. Two models 1 × 1 × 2/DFT and 1 × 1 × 2/DFT-D stand for pure DFT- and vdW-corrected DFT-based MD simulations, respectively. First, the 1 × 1 × 2 supercell was equilibrated at room temperature (298.15 K) for 5 ps using a NVT ensemble. Then, based on this equilibrated system, ab initio MD simulations were carried out at 5, 10, 20, 30, 40, 50, 75, and 100 GPa under room temperature with or without the vdW correction for 10 ps using a NPT ensemble, respectively. In order to examine the effects of different input systems, 2 × 1 × 1 supercell (defined as 2 × 1 × 1/DFT-D) and 1 × 2 × 1 supercell (defined as 1 × 2 × 1/DFT-D) (Fig. 1) were built as input models to perform ab initio MD simulations at 5, 10, 20, 30, 40, and 50 GPa under room temperature with the vdW correction for 10 ps using a NPT ensemble, respectively. The two systems were also first equilibrated at room temperature for 5 ps using a NVT ensemble. The data for analyzing the effects of pressure on the hydrogen bonds were collected from our simulations. The time dependences of total potential energy, total kinetic energy, and temperature in the 5 ps of equilibration at 298.15 K using NVT for all four systems (112, 112-D, 121-D, and 211-D) are provided in the ESI.† It was found that the systems are stable and converged.
Electronic structure calculations
Standard DFT calculations were not adequate to predict accurate band gaps, even with the vdW correction, while nonlocal exchange-correlation functionals resulting from the generalized Kohn-Sham procedure would improve the description of band gaps in insulators and semiconductors compared with LDA or GGA calculations. However, this additional accuracy comes at the price of much more time consuming calculations.28 Thus, PBE0 and B3LYP hybrid functionals were employed to calculate electronic structure of solid TATB.
Results and discussion
Crystal structure
As a benchmark, different functionals with or without the vdW correction were used to fully relax TATB crystal at ambient pressure without any constraints. Table 1 lists the experimental and relaxed cell parameters of TATB crystal. It is found that the results by the functionals without the vdW correction both in this work (PBE) and two previous studies (PW91 (Perdew-Wang 91)11 and PBE15) are obviously higher than the experimental results at ambient conditions, especially for the c lattice parameter. However, this inconsistency can be corrected when using the DFT-D methods. For instance, the calculated c lattice parameters by the PBE with the Grimme (labeled as PBE-Grimme) and TS (labeled as PBE-TS) vdW corrections are only underestimated by 0.4% and 2.2%, respectively. This indicates that the vdW correction is extremely necessary while studying the TATB crystal. Thus, we employed PBE-Grimme method to investigate the structure and properties of TATB under high pressure.
Table 1 Comparison of experimental and relaxed cell parameters of TATB crystal at ambient conditionsa
| Parameter |
a |
b |
c |
α |
β |
γ |
Cell volume |
| Cell dimension in Å, angles in degree and cell volume in Å3 the values in parentheses correspond to the percentage differences relative to experimental data. Experimental X-ray from ref. 1. Current work. Calculated values of ref. 15. Calculated values of ref. 11. |
| Expt.b |
9.010 |
9.028 |
6.812 |
108.6 |
91.8 |
120.0 |
442.3 |
| PBE-Grimmec |
9.119 (1.2) |
9.134 (1.2) |
6.787 (−0.4) |
109.0 |
91.9 |
119.7 |
451.1 |
| PBE-TSc |
9.112 (1.1) |
9.126 (1.1) |
6.661 (−2.2) |
109.4 |
91.8 |
119.8 |
440.1 |
| PBEc |
9.328 (3.5) |
9.341 (3.5) |
7.870 (15.5) |
106.1 |
91.9 |
120.0 |
551.9 |
| PBEd |
9.219 (2.3) |
9.181 (1.7) |
7.161 (5.1) |
107.7 |
92.0 |
120.2 |
485.9 |
| PW91e |
9.364 (3.9) |
9.369 (3.8) |
7.505 (10.2) |
107.2 |
91.8 |
120.2 |
530.8 |
Fig. 2 displays the calculated pressure dependence of lattice parameters of TATB at room temperature along with the previous experimental results.7 It is seen that the three results by the DFT-D are very close each other, indicating that each of them can be used to study the structures of TATB under pressure. The lattice parameters decrease gradually with the increasing pressure. The DFT-D results are in good agreement with the experimental values up to 13.2 GPa,7 but the DFT results are obviously lower than the experimental ones. The c lattice parameter is significantly overestimated by the pure DFT calculations, while this disagreement can be effectively improved by the DFT-D. This suggests that the vdW forces between the layers play a very important role in crystal packing along the c direction. Table 2 lists the deviations of our calculated lattice parameters of TATB at 5 GPa and 10 GPa from available experiment results at 4.95 GPa and 10.17 GPa under room temperature, respectively. It is found that our DFT-D results are close to the experimental ones, while DFT results without the vdW correction are obviously different from the experiments, especially the c lattice parameter.
 |
| | Fig. 2 Calculated pressure dependence of lattice parameters of TATB at room temperature along with previous experimental results. | |
Table 2 The deviation of calculated lattice parameters (Å) of TATB at 5 GPa and 10 GPa under room temperature from the experiments. “−” and “+” signs indicate underestimation and overestimation in comparison with the experimental values, respectively
| System |
5 GPa |
10 GPa |
| a |
b |
c |
a |
b |
c |
| 1 × 1 × 2/DFT |
+0.20 |
−0.04 |
+1.13 |
−0.20 |
−0.38 |
+1.04 |
| 1 × 1 × 2/DFT-D |
+0.18 |
+0.09 |
+0.24 |
+0.08 |
−0.07 |
+0.30 |
| 1 × 2 × 1/DFT-D |
+0.17 |
+0.08 |
+0.23 |
+0.10 |
−0.06 |
+0.32 |
| 2 × 1 × 1/DFT-D |
+0.17 |
+0.08 |
+0.24 |
+0.10 |
−0.07 |
+0.32 |
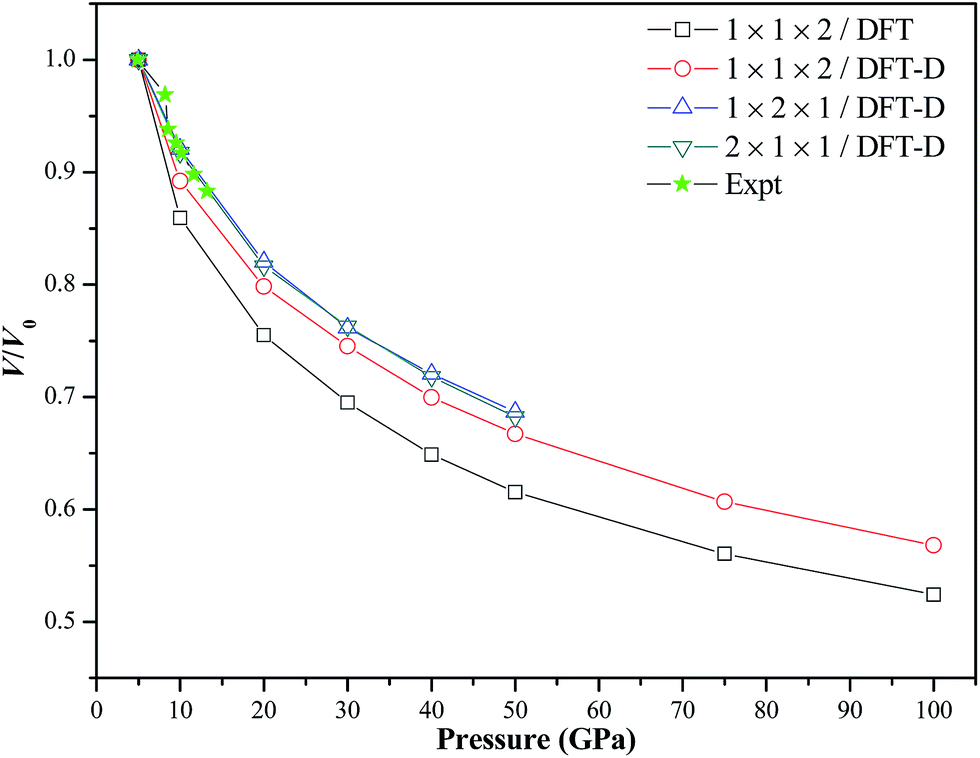
Fig. 3 shows the calculated P–V isotherm of TATB along with previous experimental results. It is seen that our three DFT-D results are in very good agreement with the experimental values up to 13.2 GPa. However, our DFT results without the vdW correction are obviously different from the experimental results. A similar phenomenon was also observed in the two previous pure DFT studies without the vdW correction on TATB,11,15 in which the calculated equation of state was noticeably stiffer than the experiments. This further confirms the necessity and importance of vdW corrections in studying the structure of TATB. Our DFT-D MD simulations indicate that TATB is compressed about 40% when the pressure is increased from 5 to 100 GPa.
 |
| | Fig. 3 Calculated P–V isotherm of TATB at room temperature along with previous experimental results. | |
Molecular structure
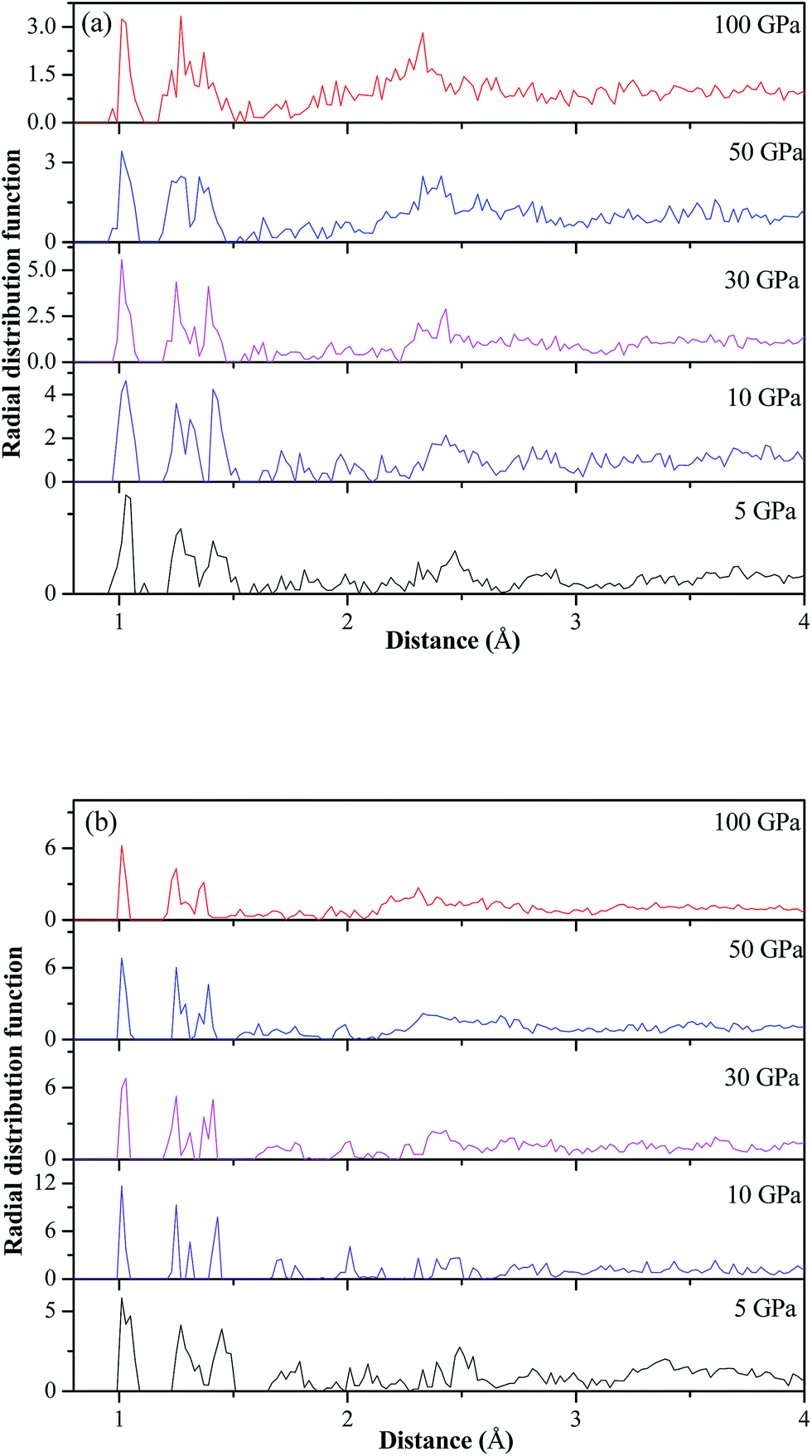
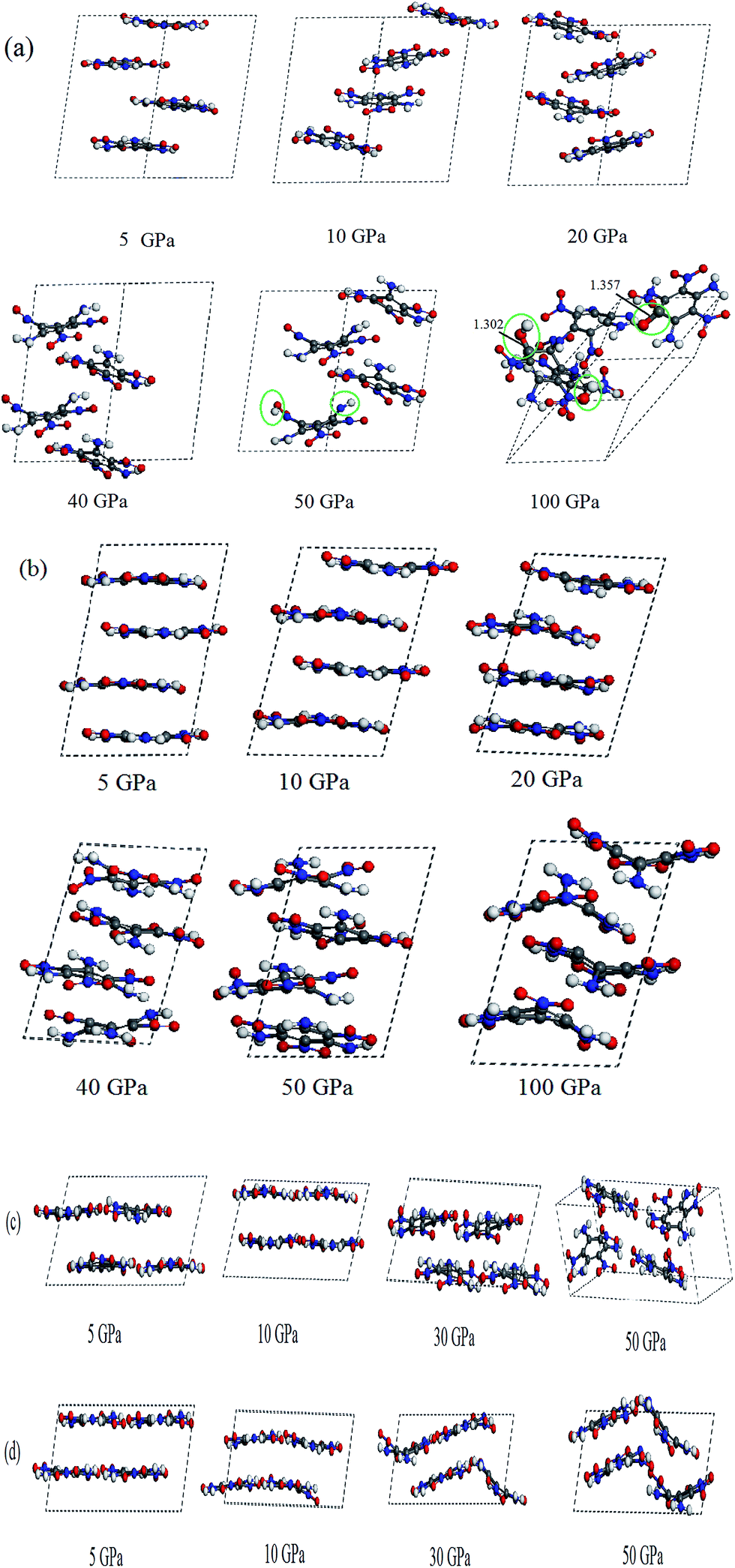
Fig. 4 displays the calculated pressure dependence of total energy (E) and total enthalpy (H) of TATB. As the pressure increases, E and H increase gradually, which are due to the compressive energy. The estimated E and H by the DFT without the vdW correction are obviously higher than those by the three DFT-D investigations, indicating that the DFT results without the vdW correction may misestimate the changing of molecular structures of TATB under high pressure like decomposition or polymerization. Fig. 5 presents the radial distribution functions (RDF) of TATB by the DFT without the vdW correction ((a) for system 1 × 1 × 2/DFT) and the DFT-D ((b) for system 1 × 1 × 2/DFT-D) at 5, 10, 30, 50, and 100 GPa. From Fig. 5(a), it is seen that the RDF changes noticeably at 50 and 100 GPa, suggesting that TATB may undergo significant structural changes here. However, it is seen in Fig. 5(b) that the RDF changes little in the pressure range of 5–100 GPa, indicating that no obvious structural changes took place. This conflicting conclusions can be confirmed by Fig. 6(a and b), which displays the perspective view of TATB at different pressures by the DFT ((a) for system 1 × 1 × 2/DFT) and DFT-D ((b–d) for systems 1 × 1 × 2/DFT-D, 1 × 2 × 1/DFT-D, and 2 × 1 × 1/DFT-D, respectively) simulations. From Fig. 6(a), it is found that TATB decomposes by intramolecular hydrogen transfer at 50 GPa. Then, at 100 GPa, TATB not only decomposes by intermolecular hydrogen transfer to form a new O–H covalent bond and by N–O breaking to form a new C–OH bond, but also polymerizes and forms a new C–O covalent bond. This is quite inconsistent to the previous experimental study,9 in which it was reported that TATB is chemically stable to 150 GPa. However, the three DFT-D results shown in Fig. 6(b–d) are in agreement with the experiments that TATB remains unreacted in all the investigated pressure ranges. This shows that the DFT without the vdW correction misestimates the decomposition and polymerization of TATB at 50 and 100 GPa, respectively. From Fig. 6(b), it is seen that the four molecules in TATB are parallel to each other at 5 GPa, and they all tilt gradually to become more and more close with the increase in pressure. The unimolecular planarity of TATB is damaged gradually with the increment of pressure but TATB is stable up to 100 GPa. This unbelievable high stability of TATB is mainly due to its strong intra- and inter-hydrogen bonding, which is also supported by Fig. 6(c and d) and 7. From Fig. 6(c and d), it is seen that though the planarity of unimolecule is destroyed gradually, but the layered structure is maintained when the pressure is increased from 5 to 50 GPa, which is due to the strengthening of hydrogen bonding under pressure.
 |
| | Fig. 4 Calculated pressure dependence of total energy (E) and total enthalpy (H) of TATB at room temperature. | |
 |
| | Fig. 5 Radial distribution functions (RDF) of TATB at 5, 10, 30, 50, and 100 GPa under room temperature by the DFT ((a) 1 × 1 × 2/DFT) and DFT-D ((b) 1 × 1 × 2/DFT-D). | |
 |
| | Fig. 6 Perspective view of TATB at different pressures by the pure DFT ((a) 1 × 1 × 2/DFT) and DFT-D ((b) 1 × 1 × 2/DFT-D, (c) 1 × 2 × 1/DFT-D, (d) 2 × 1 × 1/DFT-D). | |
 |
| | Fig. 7 Calculated pressure dependence of intra- and intermolecular hydrogen bond lengths of TATB under room temperature by the DFT-D. | |
Fig. 7 displays the calculated pressure dependence of intra- and intermolecular hydrogen bond lengths of TATB by the DFT-D simulations. The intra- and intermolecular hydrogen bond lengths are obtained by averaging the lengths of twenty four intramolecular hydrogen bonds and four intermolecular hydrogen bonds (see Fig. 1), respectively. It is found that both of them reduce gradually with the increase in pressure from 5 to 50 GPa, indicating the strengthening of intra- and intermolecular hydrogen bonding under compression, which is in good agreement with the previous experimental results9 up to 40 GPa. However, the lengths of intramolecular hydrogen bonds increase gradually when the pressure further increases from 50 GPa to 100 GPa, suggesting that the weakening of intramolecular hydrogen bonding destroys the planarity of TATB under high pressure. In this case, because of very close intermolecular distance, the hydrogen bonded network is strengthened by stronger intermolecular hydrogen bonding. Therefore, a complicated and intensive intermolecular hydrogen bonding would play a more important role in maintaining the stability of TATB under high pressure, as shown in Fig. 8. Since the short-range hydrogen bonds are more insensitive to the supercell configuration than the long-range hydrogen bonds, it can be seen from Fig. 7 that the hydrogen bonds are significantly longer for some supercells than others.
 |
| | Fig. 8 Hydrogen bonding in the 1 × 1 × 2 supercell of TATB at 75 and 100 GPa under room temperature by the DFT-D. The hydrogen bonding is presented by the blue-green dotted lines. | |
Electronic structure
Previous studies29–34 reported that the electronic structure of energetic compounds has a significant influence on their structure and properties. For instance, first-principles band gap criterion of impact sensitivity35 have reported that for energetic crystals with similar structure or with similar thermal decomposition mechanism, the smaller the band gap is, the easier is the electron transfer from the valence band to the conduction band, and the more they decompose and explode. Thus, it is necessary to investigate the electronic structure of the TATB crystal under different pressures. Several studies have been carried out to study the band gap of TATB at ambient conditions. One experimental band gap value36 of crystal TATB was estimated to be about 6.6 eV by two indirect methods using X-ray-absorption spectroscopy (XAS) and core-level and valence-band photo-electron spectroscopy (VBPES). The two methods are found to be unsuitable for TATB, and thus their inferred result has a much large source of uncertainty. The band gap of TATB was calculated to be around 10.8–11.6 eV by the Hartree–Fock (HF) method,10,37 which are obviously too large for an organic aromatic compound. Its DFT-LDA10,38 calculated band gap values are around 2.4–2.6 eV, which are too low and not in agreement with the experimental result that TATB is an insulator at ambient conditions. The CIS, CASSCF, and EOM-CCSD methods10 were used to calculate the band gaps of TATB. The former gave a band gap of about 6.0 eV, which is considered to have a large error, while the latter two gave values of around 4.9 eV, which are thought to be much more accurate and reliable than other results.
Nowadays, it is found that nonlocal exchange-correlation functionals resulting from the generalized Kohn-Sham procedure would improve the description of band gaps in insulators and semiconductors compared with the LDA or GGA calculations.28 However, this additional accuracy comes at the price of much more time consuming calculations. Here, PBE0 and B3LYP hybrid functional were employed to calculate the electronic structure of solid TATB. Table 3 lists the estimated band gaps of TATB obtained by different methods reported in this work and previous investigations. We employed a unit cell of crystal TATB (see Fig. 1) as an input structure. It is found that both the PBE and PBE-Grimme results are close to previous DFT results, and the calculated values are located in the range of semiconductors, showing that the standard DFT calculations are not adequate to predict the band gap of TATB, even with the vdW correction. The HF method gave a value of 9.9 eV, which is obviously too large and not reliable. The PBE0 and B3LYP methods estimated its band gap to be 4.1 and 3.8 eV, respectively. A similar overestimate was found in the band gap of another famous insensitive explosive 1,1-diamino-2,2-dinitroethelene (FOX-7). The band gap of FOX-7 increases from 2.3 eV (using the DFT-GGA) to 5.1 eV (using the Hybrid functional and GW approximation). Besides, the PBE0 and B3LYP results of TATB are close to those estimated by the CASSCF and EOM-CCSD methods. Thus, we used PBE0 to calculate the band gap of TATB in the pressure of 5–100 GPa, and the results are displayed in Fig. 9 along with the DFT and three DFT-D results.
Table 3 Band gaps of TATB crystal at ambient conditions
| Method |
Band gap (eV) |
| Current work. Calculated values of ref. 10. Calculated values of ref. 37. Experimental values of ref. 36. |
| PBEa |
2.6 |
| PBE-Grimmea |
2.5 |
| PBE0a |
4.1 |
| B3LYPa |
3.8 |
| HF (1)a |
9.9 |
| LDAb |
2.6 |
| CASSCF/EOM-CCSDb |
4.9 ± 0.3 |
| HF (2)b |
10.8 |
| HF (3)c |
10.8 |
| CISb |
6.0 |
| XAS + VBPESd |
6.6 |
 |
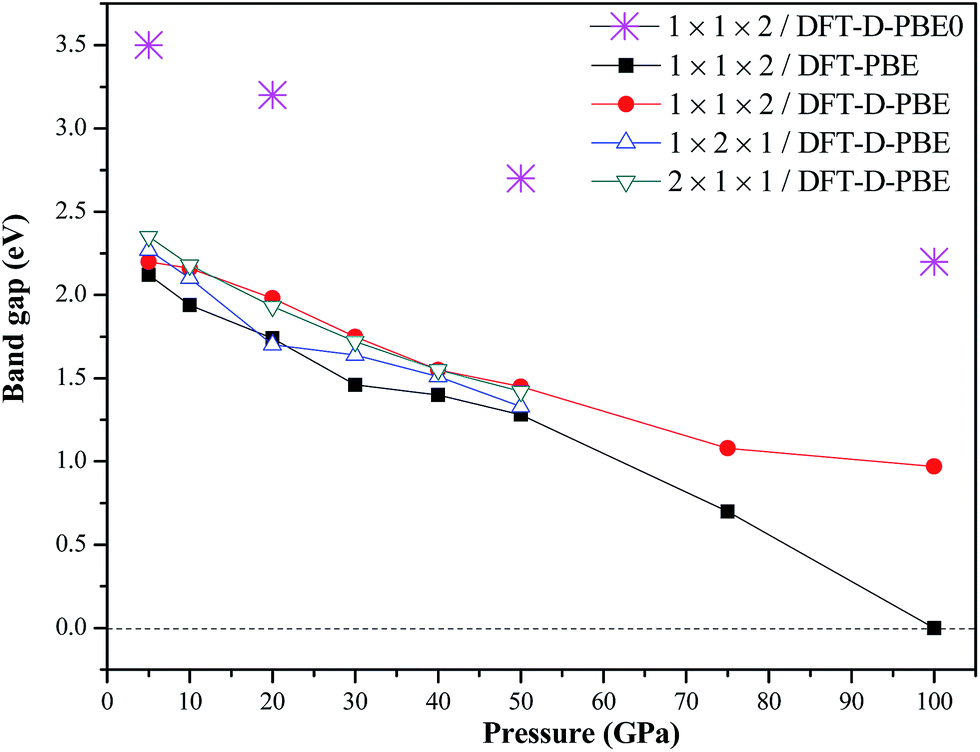
| | Fig. 9 Calculated band gaps of TATB in pressures of 5–100 GPa under room temperature by the DFT, DFT-D, and PBE0 methods. | |
From Fig. 9, it is seen that the band gap of TATB reduces gradually with the increasing pressure, suggesting that it becomes more and more sensitive. Both the DFT and DFT-D calculated band gaps decrease from about 2.6 to 1.0 eV when the pressure increases from 5 to 75 GPa, indicating that TATB is a semiconductor in the pressure range of 5–75 GPa. Furthermore, the DFT without the vdW correction misestimates the band gap of 0 eV for TATB at 100 GPa, indicating that TATB has metallic properties. A similar phenomenon was observed in previous DFT-LDA studies on TATB at 120 GPa and 0 K. These results are not in agreement with experimental results,9 indicating that the DFT without the vdW correction is not proper to compute the band gap of TATB under pressure. Finally, using the 1 × 1 × 2 supercell as an input and the DFT-D method, the PBE0 estimated the band gaps of TATB at 5 and 50 GPa to be 3.5 and 2.7 eV, respectively, suggesting that TATB is an insulator, which is well consistent with the experimental results.9 To the best of our knowledge, this is the first time that the theoretical studies estimate a matched result of band structure with experiments for TATB under high pressure. Note that our PBE0 calculation presents the band gap of 2.2 eV for TATB at 100 GPa. But previous experimental results9 inferred that TATB is still an insulator at 100 GPa, though without giving a specific value of band gap. Altogether, the PBE0 method can give much more reliable band gaps than the DFT without the vdW correction. The calculated band structure of TATB at 5 and 100 GPa by the PBE0 method are shown in Fig. 10. As the pressure increases, the conduction bands have a tendency to shift to lower energy. This leads to a reduction in the gap between the conduction and valence bands. In addition, the bandwidth of different band groups increase with the increase in pressure.
 |
| | Fig. 10 Calculated band structures of TATB at 5 and 100 GPa under room temperature by the PBE0. The input structures are the 1 × 1 × 2 supercell. | |
Conclusions
In this work, we performed ab initio MD simulations to investigate the crystal, molecular, and electronic structures of crystalline TATB in the pressure range of 0–100 GPa under room temperature. The DFT- and DFT-D-based MD were used to investigate the effects of the vdW correction on the results. The relaxed cell parameters of the TATB crystal at ambient conditions by the PBE-Grimme method are in agreement with the experimental values, while the DFT without the vdW correction critically overestimates the lattice parameter c. The lattice parameters and P–V isotherm of TATB under compression by the DFT-D agree well with the experimental results, whereas the DFT without the vdW correction either underestimates or overestimates the results evidently. The DFT-D results show that TATB is chemically stable in the entire investigated pressure range, in agreement with the experiments, but the DFT without the vdW correction misestimates that TATB decomposes at 50 GPa, whereas it decomposes and polymerizes at 100 GPa. These results indicate that it is essential to consider the vdW forces while studying TATB, both at ambient conditions and high pressures.
Both the intra- and intermolecular hydrogen bonding are strengthened with the increase in pressure from 5 to 50 GPa, consistent with the experimental results. The DFT without the vdW correction misestimates that TATB turns into a metal system at 100 GPa. The PBE0 with the vdW correction was used to study the band structures of TATB, and the obtained results up to 50 GPa are in agreement with the available experiment results.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21273115). Q. Wu thanks the Innovation Project for Postgraduates in University of Jiangsu Province (Grant no. CXZZ13_0199) for the partial financial support. Q. Wu thanks Y. Q. Wang for the help in collecting the coordinates.
References
- H. H. Cady and A. C. Larson, Acta Crystallogr., 1965, 18, 485–496 CrossRef CAS.
- J. R. Travis, TATB: The IHE Exempla, Report no. LA-UR-92-3883, 1992 Search PubMed.
- M. F. Foltz, D. L. Ornellas, P. F. Pagoria and A. R. Mitchell, J. Mater. Sci., 1996, 31, 1893–1901 CrossRef CAS.
- B. Olinger and H. Cady, The 6th Symposium (International) on Detonation, Coronado, California, USA, 1976, vol. 224, p. 24 Search PubMed.
- W. M. Trott and A. M. Renlund, J. Phys. Chem., 1988, 92, 5921–5925 CrossRef CAS.
- S. K. Satija, B. Swanson, J. Eckert and J. A. Gladstone, J. Phys. Chem., 1991, 95, 10103–10109 CrossRef CAS.
- L. L. Stevens, N. Velisavljevic, D. E. Hooks and D. M. Dattelbaum, Propellants, Explos., Pyrotech., 2008, 33, 286–295 CrossRef CAS.
- M. Pravica, B. Yulga, S. Tkachev and Z. Liu, J. Phys. Chem. B, 2009, 113, 9133–9137 CrossRef CAS PubMed.
- A. J. Davidson, R. P. Dias, D. M. Dattelbaum and C. Yoo, J. Chem. Phys., 2011, 135, 174507 CrossRef PubMed.
- C. J. Wu, L. H. Yang and L. E. Fried, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 235101 CrossRef.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. C, 2007, 111, 2787–2796 CAS.
- H. Liu, J. Zhao, J. Du, Z. Gong, G. Li and D. Wei, Phys. Lett. A, 2007, 367, 383–388 CrossRef CAS.
- D. C. Sorescu and B. M. Rice, J. Phys. Chem. C, 2010, 114, 6734–6748 CAS.
- M. M. Budzevich, A. C. Landerville, M. W. Conroy, Y. Lin, I. I. Oleynik and C. T. White, J. Appl. Phys., 2010, 107, 113524 CrossRef.
- M. R. Manaa and L. E. Fried, J. Phys. Chem. C, 2012, 116, 2116–2122 CAS.
- C. J. Wu, L. E. Freid, L. H. Yang, N. Goldman and S. Bastea, Nat. Chem., 2009, 1, 57–62 CrossRef CAS PubMed.
- O. Isayev, L. Gorb, M. Qasim and J. Leszczynski, J. Phys. Chem. B, 2008, 112, 11005–11013 CrossRef CAS PubMed.
- W. H. Zhu and H. M. Xiao, J. Phys. Chem. C, 2011, 115, 20782–20787 CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- J. Y. Raty, E. Schwegler and S. A. Bonev, Nature, 2007, 449, 448–451 CrossRef CAS PubMed.
- M. Pozzo, C. Davies, D. Gubbins and D. Alfè, Nature, 2012, 485, 355–358 CrossRef CAS PubMed.
- A. Seidl, A. Gorling, P. Vogl, J. A. Majewski and M. Levy, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 3764–3774 CrossRef CAS.
- W. H. Zhu, J. J. Xiao, G. F. Ji, F. Zhao and H. M. Xiao, J. Phys. Chem. B, 2007, 111, 12715–12722 CrossRef CAS PubMed.
- M. M. Kuklja, E. V. Stefanovich and A. B. Kunz, J. Chem. Phys., 2000, 112, 3417–3423 CrossRef CAS.
- T. Luty, P. Ordon and C. J. Eckhardt, J. Chem. Phys., 2002, 117, 1775–1785 CrossRef CAS.
- G. J. Gilman, Philos. Mag. Lett., 1998, 77, 79–82 CrossRef.
- Q. Wu, W. H. Zhu and H. M. Xiao, RSC Adv., 2014, 4, 15995–16004 RSC.
- Q. Wu, W. H. Zhu and H. M. Xiao, J. Phys. Chem. C, 2013, 117, 16830–16839 CAS.
- W. H. Zhu and H. M. Xiao, Struct. Chem., 2010, 21, 657–665 CrossRef CAS.
- S. Kakar, A. J. Nelson, R. Treusch, C. Heske, T. van Buuren, I. Jimenez, P. Pagoria and L. J. Terminello, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 15666–15672 CrossRef CAS.
- A. B. Kunz, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 9733–9738 CrossRef CAS.
- W. H. Zhu, X. W. Zhang, T. Wei and H. M. Xiao, J. Mol. Struct.: THEOCHEM, 2009, 900, 84–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09123j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.