
Pillarene functionalized polymer monolithic column for the solid-phase microextraction preconcentration of parabens
Dan Liua,
Nan Songa,
Ye-Chun Chengb,
Dai-Xiong Chena,
Qiong Jia*a and
Ying-Wei Yang *a
*a
aCollege of Chemistry, State Key Laboratory of Supramolecular Structure and Materials, International Joint Research Laboratory of Nano-Micro Architecture Chemistry (NMAC), Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: jiaqiong@jlu.edu.cn; ywyang@jlu.edu.cn
bCollege of Life Science, Jilin University, Changchun 130012, P. R. China
First published on 26th September 2014
Abstract
Poly(glycidyl methacrylate-co-ethylene dimethacrylate) modified with a new supramolecular macrocycle, carboxylatopillar[5]arene, was successfully prepared inside a capillary. The functionalized polymer monolith material was characterized by scanning electron microscopy, X-ray photoelectron spectroscopy, thermogravimetric analysis, and Fourier-transformed infrared spectra. The experimental results showed that this type of polymer possess a good network skeleton structure and excellent extraction properties for the enrichment of parabens by polymer monolith solid-phase microextraction coupled with high performance liquid chromatography. Compared with other pre-treatment methods, the simple operation and inexpensive instrumentation characteristics of the developed polymer monolith SPME method make it more suitable for the extraction of parabens in red wine samples with great potential for further broadening its applications in the enrichment of other real samples.
Introduction
As esters of p-hydroxybenzoic acid, parabens are widely employed as ideal bactericides and preservatives in the formulation of personal care, pharmaceutical, and food products.1 The extensive applications of parabens, including methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), and butyl paraben (BP), are due to their low toxicity, neutral pH, no perceptible odour, and thermal stability.2 Usually, two or more parabens are commonly used simultaneously to increase the activity of the system to withstand microbial contamination.3 However, parabens may modulate or disrupt the endocrine system, and thus may have harmful consequences on human health.1 Therefore, it is desirable to develop selective and efficient analytical methods for the simultaneous determination of parabens in real samples.4Recently, different methods have been reported for the analysis of parabens including spectrophotometry,5 gas chromatography (GC),6,7 high performance liquid chromatography (HPLC),3,4 capillary electrophoresis (CE),8 and micellar electrokinetic chromatography.1,9 Among these methods, HPLC has been extensively performed in many laboratories for the determination of parabens.3,4,10,11 Considering the effects of complex matrices, pretreatment methods are often required for the feasible analysis of real samples, which can increase the sample signal intensity. Various sample pretreatment methods have been reported for the analysis of parabens including cloud point extraction,10 stir-bar sorptive extraction,3,7 solid-phase extraction,1,4 solid-phase microextraction (SPME),6 and dispersive liquid-phase microextraction.12
As one of the most promising SPME methods, polymer monolith SPME using polymer monolithic column as the extraction material has become an interesting and efficient technique in recent years.13,14 The development and application of polymer have received much attention.15–20 Generally, polymer monoliths are fabricated with unsaturated functional monomers and cross-linkers inside the activated capillary and anchored to the wall through chemical bonding by in situ polymerization. Porous polymer monolithic columns have shown high permeability, which enables fast analysis and high-throughput screening.21 Moreover, with the interesting properties of high mechanical strength, little consumption of solvent and sample, high porosity and surface area, low flow-resistance, and high binding capacity, monolithic materials have been more and more attractive and widely used in many fields.22,23 Various functional monomers have been used for the preparation of polymers and the adjustment of surface properties of monolithic materials.24 The most commonly used monomers to synthesize organic polymer monoliths include styrene,25,26 acrylates,27,28 and acrylamides.29 Glycidyl methacrylate (GMA), a typical kind of acrylate, has been employed for the preparation of monolith because the GMA epoxy groups can undergo nucleophilic reaction to provide the chemical stability and surface property.30,31 In our previous research, poly(GMA-co-EDMA) monolithic column was successfully synthesized and modified with graphene (GN) to prepare GN@poly(GMA-co-EDMA) monoliths, which exhibited excellent performance for the isolation of sarcosine from human urine samples.32
On the other hand, synthetic macrocyclic host compounds,33–38 such as crown ethers, cyclodextrins, calixarenes, and cucurbiturils, play important roles in supramolecular chemistry and nanoscience, and receive considerable attention as adsorption materials in recent years.39–42 Pillarenes (or pillararenes),43 a new class of supramolecular hosts which were firstly synthesized in 2008,44 have become one of the most popular topics in supramolecular chemistry.45–58 Recently, we successfully employed carboxylatopillar[5]arenes (CP[5]As)59 as sorbent materials, either by functionalizing them on the surface of Fe3O4 magnetic nanoparticles via covalent bonds for the extraction and detection of pesticides60 or by packing them in a glass microcolumn for the separation and preconcentration of trace gold and palladium via flow injection flame atomic absorption spectrometry.61 Both systems illustrated the significant role of CP[5]A in the area of separation and preconcentration due to the existence of host–guest binding interactions between CP[5]A and analytes, such as π–π interactions and electrostatic interactions.60
In this work, we developed a CP[5]A@poly(GMA-co-EDMA) monolithic column as the polymer monolith SPME materials for the extraction and analysis of parabens coupled with HPLC. The experimental conditions affecting the extraction and desorption of parabens were optimized. To demonstrate the validation of the developed method, the detection limit, linearity, and precision were investigated.
Experimental section
Materials and methods
Methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), butyl paraben (BP), glycidyl methacrylate (GMA), ethylene dimethacrylate (EDMA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysulfosuccinimide (NHS), cyclohexanol, 1-dodecanol, and γ-methacryloxypropyltrimethoxy-silane (γ-MAPS) were purchased from Aladdin Reagent (Shanghai, China). N,N′-Dimethylformamide (DMF) and azobisisobutyronitrile (AIBN) were obtained from Tianjin Guangfu Fine Chemical Research Institute (Tianjin, China). Methanol (MeOH) and acetonitrile (ACN) were purchased from Fisher Scientific (Shanghai, China). CP[5]A was synthesized according to our previously reported procedure.59 All other reagents were obtained from various commercial sources and were of analytical or HPLC grade. Red wine samples were purchased from local supermarkets and stored in a refrigerator.Silica capillaries (530 μm i.d. × 690 μm o.d.) were purchased from Hebei Yongnian Optical Conductive Fiber Plant (Handan, China). An LSP01-1A programmable syringe pump (Baoding Longer Precision Pump Co., Ltd., Hebei, China) equipped with a polyethylene syringe was employed for the delivery of solutions. An LD5-2A centrifuge (Beijing Jingli centrifuge Co., Ltd., China) was used for centrifuging. A PB-10 digital pH meter (Shanghai Rex Instruments Factory, China) was used for pH measurements of sample solutions. A Milli-Q SP system (Millipore, Milford, MA, USA) was applied to prepare ultra-pure water.
Scanning electron microscope (SEM, JSM 6700 F, JEOL Company, Japan) was used to characterize the microscopic morphology of the monolith. Fourier-transformed infrared spectra (FTIR) was obtained using Thermo Nicolet 670 FTIR instrument (Thermo Nicolet Corporation, USA). X-ray photoelectron spectroscopy (XPS, Thermo Scientific Escalab 250, UK) was used to obtain XPS data. The thermogravimetric analysis (TGA) experiments were performed on a Q500 thermal gravimetric analyzer (TA, USA).
HPLC analysis was performed on a Waters 2489 series LC system (Waters, USA), equipped with binary pumps (Waters 1525), a column oven, and a dual-wavelength UV/Visible detector (Waters 2489). Breeze software was used for acquiring and processing data. An RP Symmetry C18 column (4.6 mm × 75 mm, 3.5 μm) was used for the separation of the target analytes, which was protected by a phenomenex C18 security guard column (4.0 mm × 3.0 mm, Phenomenex, Torrance, Canada). Isocratic elution was carried out with a mobile phase containing MeOH–water (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35, v/v) at a flow rate of 0.8 mL min−1. The injection volume was set at 5 μL and the detection wavelength was 256 nm.
:35, v/v) at a flow rate of 0.8 mL min−1. The injection volume was set at 5 μL and the detection wavelength was 256 nm.
Sample preparation
MP, EP, PP, and BP stock solutions were directly dissolved in mixed solvents of MeOH–water (40:60, v/v). Working solutions were prepared by appropriate dilution of the stock solutions with ultra-pure water. All solutions were stored in dark-glass flasks at 4 °C.
All the red wine samples were diluted ten times with 0.02 mol L−1 phosphate buffer solutions (pH 5.0). All the sample solutions were spiked with MP, EP, PP, and BP standard solutions at different concentration levels to assess the matrix effects.
Synthesis of poly(GMA-co-EDMA) and CP[5]A@poly(GMA-co-EDMA) monolithic columns
The poly(GMA-co-EDMA) column was prepared according to our published procedure.32 Subsequently, ethylenediamine solution was pumped through the poly(GMA-co-EDMA) column at a flow rate of 0.1 mL min−1 for 30 min at room temperature. The column was sealed with silicon rubber at both ends, and the reaction was performed in an oven at 70 °C for 4 h. The column was then rinsed with ultra-pure water until neutral pH. The monoliths containing amine groups can thus be obtained.A certain amount (1.0 mg) of CP[5]A was dispersed in DMF (1.0 mL) and ultrasonicated for 10 min, and then EDC (10.0 mg) and NHS (8.0 mg) were added into the above solutions and stirred for 30 min to activate the carboxyl groups of CP[5]A. Then the mixed solutions were passed through the monoliths containing amine groups at a flow rate of 0.01 mL min−1. The obtained CP[5]A@poly(GMA-co-EDMA) monolithic column was rinsed with MeOH and ultra-pure water to remove excess reagents. The procedure for the preparation of CP[5]A@poly(GMA-co-EDMA) monolith was illustrated in Fig. 1.
 | ||
| Fig. 1 Illustration of the procedure for the preparation of CP[5]A@poly(GMA-co-EDMA) monolith. | ||
Polymer monolith SPME procedure
The pretreatment device consisted of a syringe pump and a plastic syringe (5 mL). The critical part of polymer monolith SPME was the initial pinhead that was replaced by a polymer monolithic column. All the solvents were filtrated through a 0.22 μm Millipore filter before analysis.The polymer monolith SPME procedure was as follows: firstly, MeOH (0.2 mL) was passed through the polymer monolithic column at a flow rate of 0.05 mL min−1, and then phosphate solution (0.2 mL, pH 3) was injected under the same conditions. Thereafter the sorption of analytes was realized by injecting sample solution (1.0 mL) at a flow rate of 0.05 mL min−1. Subsequently, an empty and clean syringe was employed for driving out the residual solution in the polymer monolithic column. For the desorption step, the parabens adsorbed on the polymer monolithic column were eluted with MeOH–water (0.05 mL, 60:40, v/v) at a flow rate of 0.03 mL min−1, and the eluent was collected into a vial for HPLC analysis.
Results and discussion
Characterization of CP[5]A@poly(GMA-co-EDMA) column
The morphology of poly(GMA-co-EDMA) monolith and CP[5]A@poly(GMA-co-EDMA) monoliths with different contents of CP[5]As by SEM (Fig. 2) showed that poly(GMA-co-EDMA) monolithic column had many pores in the network skeleton, which guaranteed fast dynamic transport and high efficient enrichment in applications. The porous structure also existed in the network skeleton when the monolithic column was modified with CP[5]A (1.0 mg). However, the polymer surface became more rough and the permeability became lower when the contents of CP[5]A increased. In this experiment, 1.0 mg of CP[5]A was chosen to prepare the CP[5]A@poly(GMA-co-EDMA) monolithic column. | ||
| Fig. 2 SEM micrographs of (a) poly(GMA-co-EDMA) monolith and CP[5]A@poly(GMA-co-EDMA) monoliths with (b) 1.0 mg of CP[5]A and (c) 4.0 mg of CP[5]A. | ||
The FT-IR spectra of the monoliths (Fig. 3) has been used to monitor the modification of poly(GMA-co-EDMA) monolith with CP[5]A. Obvious differences can be readily identified from Fig. 3a of (poly(GMA-co-EDMA) monolith) and Fig. 3c of (CP[5]A@poly(GMA-co-EDMA) monolith). The apparent peaks at 1500 cm−1 and 1450 cm−1 in Fig. 3b and c could be identified as the skeleton stretching vibration of aromatic rings. The peak at 1660 cm−1 in Fig. 3c might be attributed to the generated amido bond on the column. In conclusion, these results indicated that the CP[5]A macrocycles were successfully grafted on the poly(GMA-co-EDMA) monolithic column.
 | ||
| Fig. 3 FTIR spectra of (a) poly(GMA-co-EDMA) monolith, (b) CP[5]A, and (c) CP[5]A@poly(GMA-co-EDMA) monolith. | ||
Poly(GMA-co-EDMA), amine-modified poly(GMA-co-EDMA), and CP[5]A@poly(GMA-co-EDMA) monoliths were characterized by XPS in order to explore the O, N, and C elements. Results were shown in Fig. 4. Typical O1s, N1s, and C1s XPS peaks appeared at 532 eV, 399 eV, and 284 eV, respectively. In Table 1, the elemental compositions and relative contents of poly(GMA-co-EDMA), amine-modified poly(GMA-co-EDMA), and CP[5]A@poly(GMA-co-EDMA) monoliths were listed. The N content of the amine-modified poly(GMA-co-EDMA) monolith increased significantly compared with that of the poly(GMA-co-EDMA) column, while C and O contents decreased. However, the relative contents of C and O of CP[5]A@poly(GMA-co-EDMA) monoliths were higher than those of amine-modified poly(GMA-co-EDMA) monoliths. These results certified that amine groups were successfully introduced into poly(GMA-co-EDMA) monolith and CP[5]A was successfully modified on the column.
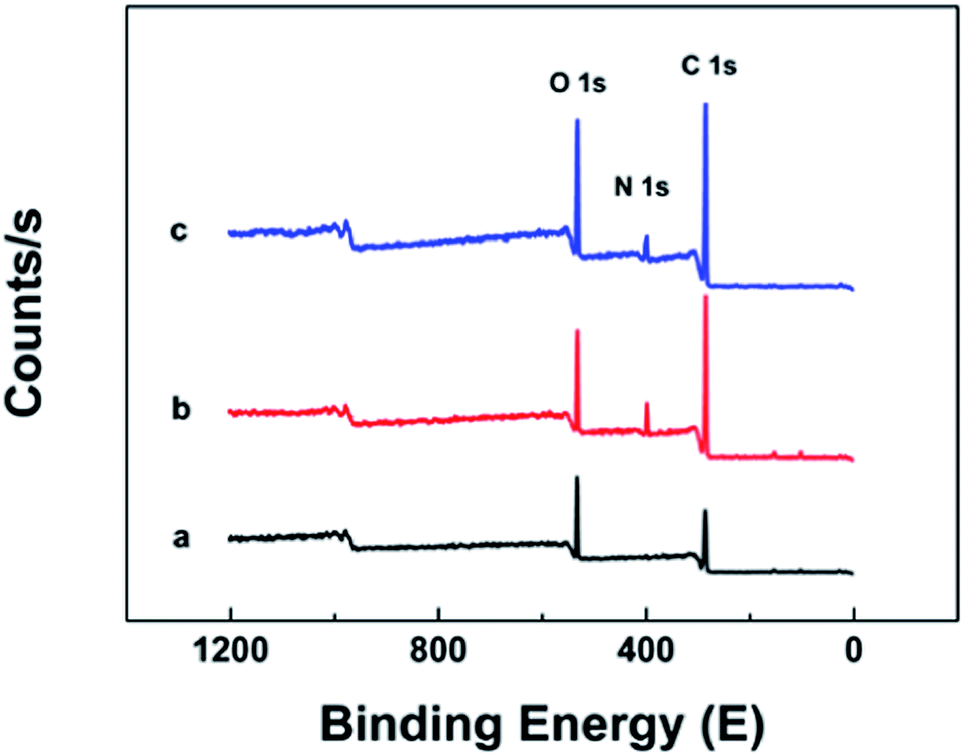 | ||
| Fig. 4 XPS spectra for elemental compositions of (a) poly(GMA-co-EDMA) monolith, (b) amine-modified poly(GMA-co-EDMA) monolith, and (c) CP[5]A@poly(GMA-co-EDMA) monolith. | ||
| Monolith | Elemental compositions | ||
|---|---|---|---|
| C | O | N | |
| Poly(GMA-co-EDMA) | 74.6% | 25.0% | 0.4% |
| Amine-modified poly(GMA-co-EDMA) | 75.4% | 19.0% | 5.6% |
| CP[5]A@poly(GMA-co-EDMA) | 77.5% | 20.3% | 2.2% |
Thermal properties of poly(GMA-co-EDMA) and CP[5]A@poly(GMA-co-EDMA) monoliths were analyzed by TGA measurement. It can be seen from Fig. 5 that the weight loss of the material began at 30 °C, which might be due to the decomposition of CP[5]A. TGA curve tended to be constant when the temperature was higher than 450 °C, indicating the complete decomposition of the monolithic material.
 | ||
| Fig. 5 TGA curves of poly(GMA-co-EDMA) and CP[5]A@poly(GMA-co-EDMA) monoliths. | ||
Optimization of polymer monolith SPME conditions
In order to obtain high extraction efficiency of parabens, conditions of polymer monolith SPME process, including sample pH, type of eluent, sample volume, sample flow rate, and eluent flow rate were investigated. The peak area of analytes as the HPLC response was used to evaluate the extraction efficiency under various experimental conditions.To achieve efficient desorption of analytes from monolith, various organic solvents, including ACN, ACN–water (60:40, v/v), MeOH, and MeOH–water (60:40, v/v), were adopted to investigate the desorption efficiency, as shown in Fig. 6a. The peak areas using ACN as the elution solvent was slightly higher than those of the other three. However, taking the effect of peak shape and separation degree into consideration, MeOH–water (60:40, v/v) was selected as the optimized elution solvent in the following work.
 | ||
| Fig. 6 Effects of (a) type of eluent; (b) sample pH; (c) sample volume, (d) sample flow rate, and (e) eluent flow rate. Standards: 2.0 μg mL−1. | ||
Sample pH plays an important role for the adsorption of target analytes by affecting the existing form of analytes. To investigate the sample pH effect, pH value was adjusted from 2.0 to 8.0 due to the hydrolysis of the analytes in alkaline solution. Results were shown in Fig. 6b. The extraction efficiency of polymer monolith SPME reached a better level at pH 3.0, because the pKa values of the target analytes are approximately equal to 8.3 and they exist in protonated forms at pH < 8.0. The protonation reaction led to easier interactions between parabens and carboxyl groups of CP[5]As. Moreover, hydrophobic and π–π interactions also exist in the extraction process. Hence, pH 3.0 was selected as the optimum value in subsequent experiments.
The influence of sample volume was evaluated in the range of 0.2–1.6 mL. Results were demonstrated in Fig. 6c, illustrating that a significant increasing tendency of the peak areas was observed when the sample volume increased from 0.2 to 1.6 mL and the extraction equilibrium did not reach a maximum even after 1.6 mL sample volume was employed. It could be expected that the peak areas would stop increasing after the sample volume reached a certain value.6,7 Taking the time of experiment into consideration, 1.0 mL sample volume was selected for further experiments.
In order to study the effects of sample flow rates, different sample flow rates ranging from 0.02–0.2 mL min−1 were investigated. It could be concluded from Fig. 6d that a decrease of the extraction efficiency occurred with increasing sample flow rate. It may be interpreted that higher flow rate resulted in shorter contact time, which did not benefit the interactions between the analytes and extraction materials. Thus, 0.05 mL min−1 was chosen as the sample flow rate in this experiment.
The influence of eluent flow rate on the polymer monolith SPME progress was also investigated by varying the flow rate from 0.01 to 0.07 mL min−1 with a total eluent volume of 0.05 mL. Fig. 6e indicated that the highest elution efficiency was obtained when the eluent flow rate was 0.03 mL min−1, after which the efficiency decreased because high flow rate generally led to a loss of contact time between the analytes and the eluent. Finally, 0.03 mL min−1 was employed as the eluent flow rate.
Method performance
Under the optimal experimental conditions, the linearity was tested from 0.05 to 5 μg mL−1 for the target parabens. The correlation coefficients (R2) were found to be in the range of 0.9957–0.9986. LOD and LOQ, calculated on the basis of S/N ratios of 3 and 10, were determined in the range of 0.008–0.033 and 0.027–0.109 μg mL−1, respectively. The intra-day and inter-day relative standard deviation (RSD) values for MP, EP, PP, and BP were based on the intensity of five replicate determinations. The intra-day and inter-day RSDs of the target parabens were determined in the range of 2.5–7.8% and 1.7–9.0%, respectively. In addition, in order to investigate the reproducibility of monolith, the intra-batch and inter-batch relative standard deviations were also important for the performance evaluation. The results of intra-batch and inter-batch RSDs for the target parabens were determined to be lower than 8.4% and 9.3%, respectively.The LOD comparison of polymer monolith SPME and other pretreatment methods are listed in Table 2. Although it could not provide the best LOD level, the present method is suitable for parabens analysis considering the improvement of the analysis in complex matrices. In addition, the developed polymer monolith SPME method is very simple to operate and does not need any expensive instruments and complex preparation steps. Moreover, the solvent-free characteristics of polymer monolith SPME method prevents itself from consuming a large volume of organic solvents, which make it environmental friendly. The comparable advantages entitle the polymer monolith SPME technique to search further applications in other real samples.
| Extraction method | Analytes | Instrument | LOD (μg mL−1) | Ref. |
|---|---|---|---|---|
| a CPE = cloud point extraction.b DLLME = dispersive liquid–liquid microextraction.c SPE = solid-phase extraction.d SBSE = stir bar sorptive extraction. | ||||
| CPEa | MP, EP, PP, AP | HPLC | 0.14–0.29 | 10 |
| DLLMEb | MP, EP, PP | GC | 0.005–0.015 | 12 |
| SPEc | MP, EP, PP | CE | 0.07–0.1 | 1 |
| SPEc | MP, EP, PP | HPLC | 0.01–0.02 | 4 |
| SPME | MP, EP, PP, BP | GC-MS | 4.0–17 × 10−6 | 6 |
| SBSEd | MP, EP, PP, BP | HPLC | 0.03–0.2 | 3 |
| SBSEd | MP, PP, BP | GC-MS | 0.64–4.12 | 7 |
| polymer monolith SPME | MP, EP, PP, BP | HPLC | 0.008–0.033 | Present work |
Under the optimal experimental conditions, the analytical performance after the enrichment using CP[5]A@poly(GMA-co-EDMA) column was compared with poly(GMA-co-EDMA) column enrichment and direct HPLC analysis. Results were shown in Fig. 7.
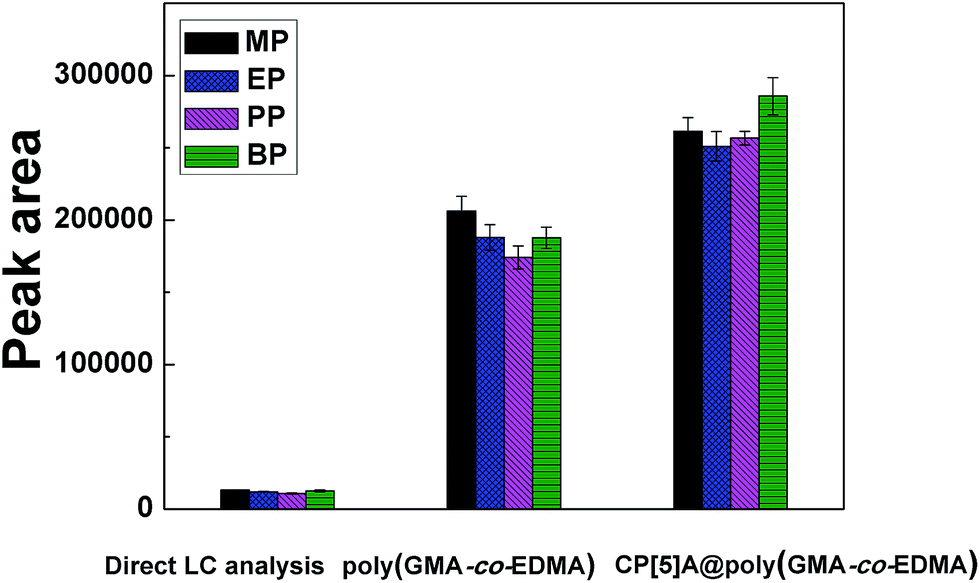 | ||
| Fig. 7 Comparison of the analytical performance of MP, EP, PP, and BP enrichment by direct HPLC analysis, poly(GMA-co-EDMA) and CP[5]A@poly(GMA-co-EDMA) monolithic columns under the optimized conditions. | ||
The peak areas with CP[5]A@poly(GMA-co-EDMA) column were significantly higher than that with the other two methods, which may be due to various kinds of interactions including electrostatic affinity, hydrophobic interactions, π–π interactions, and hydrogen bonding between the target analytes and the carboxyl groups (and/or the cavity) of CP[5]A. Nevertheless, the adsorbed parabens on the poly(GMA-co-EDMA) column were only based on hydrophobic interactions in the extraction process. This result also indicated that the functionalized monolithic material had high extraction efficiency for parabens.
Determination of parabens in red wine samples
To evaluate the reliability of the present method for extraction and preconcentration of parabens from real samples, different brands of red wines and spiked real samples were selected for polymer monolith SPME-HPLC analysis (Level 1 0.5 μg mL−1 and Level 2 1.0 μg mL−1). Results were presented in Table 3. Fig. 8 exhibited typical chromatograms obtained from spiked samples. Since most parabens in samples are present in concentrations close to or below the detection limits of analytical equipment, the direct determination is a significant challenge. Table 3 showed that some of the parabens can be detected after polymer monolith SPME and acceptable relative recoveries (70.1–106.1%) of the added parabens could be obtained.| Sample | MP | EP | PP | BP | |
|---|---|---|---|---|---|
| S 1 | Found (μg mL−1) | <LOD | <LOD | <LOD | <LOD |
| Recovery (%, Level 1) | 101.3 ± 0.9 | 83.3 ± 1.5 | 78.9 ± 1.3 | 71.3 ± 1.8 | |
| Recovery (%, Level 2) | 75.6 ± 1.1 | 98.8 ± 0.7 | 96.1 ± 1.9 | 73.8 ± 2.7 | |
| S 2 | Found (μg mL−1) | <LOD | <LOD | 0.029 | <LOD |
| Recovery (%, Level 1) | 102.4 ± 1.3 | 96.2 ± 0.7 | 73.7 ± 1.7 | 88.1 ± 1.6 | |
| Recovery (%, Level 2) | 97.0 ± 0.9 | 93.8 ± 2.7 | 88.9 ± 2.0 | 95.8 ± 1.4 | |
| S 3 | Found (μg mL−1) | <LOD | <LOD | <LOD | <LOD |
| Recovery (%, Level 1) | 105.9 ± 1.4 | 90.3 ± 0.9 | 93.2 ± 1.2 | 88.7 ± 0.3 | |
| Recovery (%, Level 2) | 76.2 ± 0.8 | 97.8 ± 1.1 | 75.5 ± 1.0 | 90.6 ± 0.2 | |
| S 4 | Found (μg mL−1) | 0.023 | <LOD | <LOD | 0.030 |
| Recovery (%, Level 1) | 103.8 ± 2.2 | 86.4 ± 1.5 | 97.2 ± 0.5 | 83.4 ± 0.9 | |
| Recovery (%, Level 2) | 98.9 ± 0.8 | 105.7 ± 2.1 | 91.9 ± 0.7 | 81.0 ± 0.5 | |
| S 5 | Found (μg mL−1) | 0.011 | <LOD | 0.044 | <LOD |
| Recovery (%, Level 1) | 83.1 ± 1.2 | 102.3 ± 1.1 | 89.0 ± 2.0 | 86.3 ± 2.3 | |
| Recovery (%, Level 2) | 75.0 ± 0.8 | 98.8 ± 0.9 | 72.2 ± 0.8 | 94.7 ± 1.5 | |
| S 6 | Found (μg mL−1) | <LOD | <LOD | 0.031 | 0.027 |
| Recovery (%, Level 1) | 96.0 ± 2.0 | 105.8 ± 2.7 | 79.8 ± 1.7 | 98.8 ± 0.6 | |
| Recovery (%, Level 2) | 86.5 ± 0.8 | 72.8 ± 0.7 | 83.4 ± 1.7 | 100.7 ± 2.9 | |
| S 7 | Found (μg mL−1) | <LOD | <LOD | 0.027 | <LOD |
| Recovery (%, Level 1) | 98.3 ± 2.1 | 92.5 ± 0.4 | 98.5 ± 1.7 | 70.9 ± 1.9 | |
| Recovery (%, Level 2) | 95.2 ± 2.4 | 97.3 ± 1.0 | 83.6 ± 2.0 | 79.4 ± 0.1 | |
| S 8 | Found (μg mL−1) | 0.020 | 0.041 | <LOD | <LOD |
| Recovery (%, Level 1) | 96.5 ± 2.6 | 71.1 ± 0.2 | 75.1 ± 0.5 | 101.3 ± 1.1 | |
| Recovery (%, Level 2) | 70.1 ± 1.8 | 82.5 ± 2.0 | 79.7 ± 1.3 | 76.7 ± 0.7 | |
| S 9 | Found (μg mL−1) | 0.017 | <LOD | 0.088 | 0.060 |
| Recovery (%, Level 1) | 84.2 ± 2.3 | 94.1 ± 1.2 | 71.8 ± 0.4 | 77.3 ± 0.3 | |
| Recovery (%, Level 2) | 89.6 ± 1.0 | 94.7 ± 1.5 | 91.2 ± 0.5 | 75.8 ± 0.3 | |
| S 10 | Found (μg mL−1) | 0.038 | <LOD | 0.039 | <LOD |
| Recovery (%, Level 1) | 106.1 ± 2.1 | 104.5 ± 1.7 | 92.6 ± 0.6 | 78.8 ± 1.7 | |
| Recovery (%, Level 2) | 99.1 ± 1.5 | 80.1 ± 0.9 | 94.3 ± 1.8 | 103.1 ± 0.5 |
 | ||
| Fig. 8 Chromatograms of red wine samples obtained by the preconcentration procedures. (a) Blank sample, (b) sample spiked with Level 1 (all 0.5 μg mL−1), and (c) sample spiked with Level 2 (all 1.0 μg mL−1). | ||
Conclusions
CP[5]A@poly(GMA-co-EDMA) monolithic column was successfully synthesized and characterized by SEM, XPS, TGA, and FTIR. Based on the novel functional polymer materials, polymer monolith SPME-HPLC method was developed and applied for the extraction and determination of parabens, MP, EP, PP, and BP, in red wine samples. Optimal extractive conditions were obtained by optimizing experimental parameters. Compared with traditional poly(GMA-co-EDMA) monolith, the functional monolith displayed good enrichment capacity of parabens via the electrostatic, hydrophobic, hydrogen bonding, and π–π interactions. Due to the satisfactory precision and accuracy, the present polymer monolith SPME-HPLC method proved itself to be sensitive, accurate, and has promising applications for parabens determinations in food samples.Acknowledgements
The project was supported by National Natural Science Foundation of China (21205047, 51473061), Jilin Provincial Science & Technology Department (201105102), and the Innovation Program of the State Key Laboratory of Supramolecular Structure and Materials for financial support. We also thank the Key Laboratory of Pesticide and Chemical Biology, Ministry of Education, Central China Normal University (201301A01) for financial support.Notes and references
- F. Han, Y. Z. He and C. Z. Yu, Talanta, 2008, 74, 1371 CrossRef CAS PubMed.
- E. Tahmasebi, Y. Yamini, A. Mehdinia and F. Rouhi, J. Sep. Sci., 2012, 35, 2256 CrossRef CAS PubMed.
- L. P. Melo and M. E. C. Queiroz, J. Sep. Sci., 2010, 33, 1849 CrossRef CAS PubMed.
- D. N. Huang, C. Fu, Z. B. Li and C. H. Deng, J. Sep. Sci., 2012, 35, 1667 CrossRef CAS PubMed.
- S. Ouane, M. Kallel, H. Trabelsi, F. Safta and K. Bouzouita, J. Pharm. Biomed. Anal., 1998, 17, 361 CrossRef.
- J. Regueiro, E. Becerril, C. Garcia-Jares and M. Llompart, J. Chromatogr. A, 2009, 1216, 4693 CrossRef CAS PubMed.
- A. M. Casas Ferreira, M. Moder and M. E. Fernandez Laespada, Anal. Bioanal. Chem., 2011, 399, 945 CrossRef CAS PubMed.
- I. Maijo, F. Borrull, C. Aguilar and M. Calull, Electrophoresis, 2013, 34, 363 CrossRef CAS PubMed.
- S. L. He, Y. F. Zhao, Z. W. Zhu, H. W. Liu, M. X. Li, Y. H. Shao and Q. K. Zhuang, Talanta, 2006, 69, 166 CrossRef CAS PubMed.
- M. S. Noorashikin, M. Raoov, S. Mohamad and M. R. Abas, Int. J. Mol. Sci., 2013, 14, 24531 CrossRef CAS PubMed.
- J. Ballesta Claver, M. C. Valencia and L. F. Capitan-Vallvey, Talanta, 2009, 79, 499 CrossRef CAS PubMed.
- M. A. Farajzadeh, D. Djozan and R. F. Bakhtiyari, Talanta, 2010, 81, 1360 CrossRef CAS PubMed.
- M. Zhang, F. Wei, Y. F. Zhang, J. Nie and Y. Q. Feng, J. Chromatogr. A, 2006, 1102, 294 CrossRef CAS PubMed.
- X. Wang, Q. Ma, M. Li, C. L. Chang, Y. Bai, Y. Q. Feng and H. W. Liu, J. Chromatogr. A, 2013, 1317, 121 CrossRef CAS PubMed.
- C. Li, Y.-W. Yang, Z. Liang, G. Wu and H. Gao, Polym. Chem., 2013, 4, 4366 RSC.
- Z. Liang, X. Wu, Y.-W. Yang, C. Li, G. Wu and H. Gao, Polym. Chem., 2013, 4, 3514 RSC.
- X. Du, N. Song, Y.-W. Yang, G. Wu, J. Ma and H. Gao, Polym. Chem., 2014, 5, 5300 RSC.
- W.-X. Gu, Y.-W. Yang, J. Wen, H. Lu and H. Gao, Polym. Chem., 2014, 5, 6344 RSC.
- Q.-L. Li, W. X. Gu, H. Gao and Y.-W. Yang, Chem. Commun., 2014 10.1039/c4cc03036b.
- Q.-L. Li, L. Wang, X.-L. Qiu, Y.-L. Sun, P.-X. Wang, Y. Liu, F. Li, A.-D. Qi, H. Gao and Y.-W. Yang, Polym. Chem., 2014, 5, 3389 RSC.
- J. L. Zhang, G. Chen, M. M. Tian, R. G. Li, X. J. Quan and Q. Jia, Talanta, 2013, 115, 801 CrossRef CAS PubMed.
- K. Liu, H. D. Tolley, J. S. Lawson and M. L. Lee, J. Chromatogr. A, 2013, 1321, 80 CrossRef CAS PubMed.
- M. L. Chen, X. M. Fu, J. Q. Liu, T. T. Ye, S. Y. Hou, Y. Q. Huang, B. F. Yuan, Y. Wu and Y. Q. Feng, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 905, 67 CrossRef CAS PubMed.
- F. Maya and F. Svec, J. Chromatogr. A, 2013, 1317, 32 CrossRef CAS PubMed.
- A. Vaast, L. Novakova, G. Desmet, B. de Haan, R. Swart and S. Eeltink, J. Chromatogr. A, 2013, 1304, 177 CrossRef CAS PubMed.
- F. Maya and F. Svec, Polymer, 2014, 55, 340 CrossRef CAS PubMed.
- Y. Y. Li, H. D. Tolley and M. L. Lee, Anal. Chem., 2009, 81, 9416 CrossRef CAS PubMed.
- Z. Lin, H. Huang, X. B. Sun, Y. Lin, L. Zhang and G. N. Chen, J. Chromatogr. A, 2012, 1246, 90 CrossRef CAS PubMed.
- Y. Fan, M. Zhang, S. L. Da and Y. Q. Feng, Analyst, 2005, 130, 1065 RSC.
- Y. Q. Lv, D. Y. Fu, T. W. Tan and M. Y. Wang, J. Mol. Catal. B: Enzym., 2010, 62, 149 CrossRef CAS PubMed.
- C. P. Bisjak, R. Bakry, C. W. Huck and G. K. Bonn, Chromatographia, 2005, 62, s31 CAS.
- S. S. Tong, X. Zhou, C. H. Zhou, Y. Y. Li, W. J. Li, W. H. Zhou and Q. Jia, Analyst, 2013, 138, 1549 RSC.
- Y.-W. Yang, Y. L. Sun and N. Song, Acc. Chem. Res., 2014, 47, 1950 CrossRef CAS PubMed.
- S. Y. Dong, B. Zheng, F. Wang and F. H. Huang, Acc. Chem. Res., 2014, 47, 1982 CrossRef CAS PubMed.
- D. S. Guo and Y. Liu, Acc. Chem. Res., 2014, 47, 1925 CrossRef CAS PubMed.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052 CrossRef CAS PubMed.
- H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106 CrossRef CAS PubMed.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941 CrossRef CAS PubMed.
- M. Saaid, B. Saad, I. A. Rahman, A. S. M. Ali and M. I. Saleh, Talanta, 2010, 80, 1183 CrossRef CAS PubMed.
- Q. F. Zhang, H. Y. Cheung, X. C. Shangguan and G. D. Zheng, J. Sep. Sci., 2012, 35, 3347 CrossRef CAS PubMed.
- O. I. Kalchenko, S. O. Cherenok, A. V. Solovyov and V. I. Kalchenko, Chromatographia, 2009, 70, 717 CAS.
- X. L. Huang, Y. B. Tan, Y. X. Wang, H. Yang, J. Cao and Y. J. Che, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5999 CrossRef CAS.
- L. L. Tan, Y. M. Zhang, B. Li, K. Wang, S. X. A. Zhang, Y. C. Tao and Y.-W. Yang, New J. Chem., 2014, 38, 845 RSC.
- T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022 CrossRef CAS PubMed.
- L. L. Tan and Y.-W. Yang, J. Inclusion Phenom. Macrocyclic Chem., 2014 DOI:10.1007/s10847-014-0441-3.
- N. Song and Y.-W. Yang, Sci. China: Chem., 2014, 57, 1185 CrossRef CAS.
- M. Xue, Y. Yang, X. D. Chi, Z. B. Zhang and F. H. Huang, Acc. Chem. Res., 2012, 45, 1294 CrossRef CAS PubMed.
- K. Wang, Y.-W. Yang and S. X. A. Zhang, Chem. J. Chin. Univ., 2012, 33, 1 CAS.
- P. J. Cragg and K. Sharma, Chem. Soc. Rev., 2012, 41, 597 RSC.
- Y. L. Sun, Y.-W. Yang, D. X. Chen, G. Wang, Y. Zhou, C. Y. Wang and J. F. Stoddart, Small, 2013, 9, 3224 CAS.
- Q. P. Duan, Y. Cao, Y. Li, X. Y. Hu, T. X. Xiao, C. Lin, Y. Pan and L. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10542 CrossRef CAS PubMed.
- G. C. Yu, Y. J. Ma, C. Y. Han, Y. Yao, G. P. Tang, Z. W. Mao, C. Y. Gao and F. H. Huang, J. Am. Chem. Soc., 2013, 135, 10310 CrossRef CAS PubMed.
- Z. Y. Li, Y. Y. Zhang, C. W. Zhang, L. J. Chen, C. Wang, H. W. Tan, Y. H. Yu, X. P. Li and H. B. Yang, J. Am. Chem. Soc., 2014, 136, 8577 CrossRef CAS PubMed.
- L. Chen, W. Si, L. Zhang, G. F. Tang, Z. T. Li and J. L. Hou, J. Am. Chem. Soc., 2013, 135, 2152 CrossRef CAS PubMed.
- D. D. Zheng, D. Y. Fu, Y. Q. Wu, Y. L. Sun, L. L. Tan, T. Zhou, S. Q. Ma, X. Zha and Y.-W. Yang, Chem. Commun., 2014, 50, 3201 RSC.
- J. Z. Fan, Y. Chen, D. R. Cao, Y.-W. Yang, X. S. Jia and C. J. Li, RSC. Adv., 2014, 4, 4330 RSC.
- X. S. Hou, C. F. Ke, C. Y. Cheng, N. Song, A. K. Blackburn, A. A. Sarjeant, Y. Y. Botros, Y.-W. Yang and J. F. Stoddart, Chem. Commun., 2014, 50, 6196 RSC.
- H. C. Zhang and Y. L. Zhao, Chem.–Eur. J., 2013, 19, 16862 CrossRef CAS PubMed.
- H. Li, D. X. Chen, Y. L. Sun, Y. B. Zheng, L. L. Tan, P. S. Weiss and Y.-W. Yang, J. Am. Chem. Soc., 2013, 135, 1570 CrossRef CAS PubMed.
- M. M. Tian, D. X. Chen, Y. L. Sun, Y.-W. Yang and Q. Jia, RSC. Adv., 2013, 3, 22111 RSC.
- S. Y. Zhou, N. Song, S. X. Liu, D. X. Chen, Q. Jia and Y.-W. Yang, Microchim. Acta, 2014, 181, 1551 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |