DOI:
10.1039/C4RA08471C
(Paper)
RSC Adv., 2014,
4, 48836-48847
Heterogeneous catalysis of transesterification of jatropha curcas oil over calcium–cerium bimetallic oxide catalyst
Received
11th August 2014
, Accepted 4th September 2014
First published on 5th September 2014
Abstract
A series of heterogeneous basic catalysts (mixed oxides of Ca and Ce) with different molar ratios were synthesized via a conventional co-precipitation process using a highly alkaline carbonate salt. Moreover, investigation was conducted batchwise for the transesterification of crude Jatropha curcas oil (JCO) with methanol at 65 °C and 1 atm pressure. The bimetallic oxides possess high thermal stability, since X-ray diffraction (XRD) proved that the crystalline phases present in mixed oxide catalysts were preserved well as pure oxide even up to 900 °C. The co-precipitation synthesis method provided better interaction between vacancies created by the substitution of calcium (Ca) and cerium (Ce) at pH 11. Moreover, the combination of Ca and Ce reduced the temperature maxima and increased the basicity of catalysts, which exhibited better catalytic activity compared with bulk catalysts (CaO and CeO2). The influences of the Ca/Ce atomic ratio in the mixed oxide catalyst, methanol–oil molar ratio, catalyst amount and reaction time on the fatty acid methyl ester (FAME) content were studied. The suitable molar ratio of Ca-to-Ce was 1, and with the optimum conditions of 4 wt% catalyst dosage and 15% methanol–oil molar ratio, a fatty acid methyl ester (FAME) content of 95% was achieved over CaO–CeO2 catalyst at 65 °C. Moreover, the CaO–CeO2 catalyst shows substantial chemical stability and could be reused at least 4 times without major loss in its catalytic activity.
Introduction
The growing energy demands in response to industrialization and urbanization has led to the depletion of world's petroleum reserves and environmental issues. Therefore, it is a great challenge for researchers to identify alternative sources to petroleum-based fuels. At present, the conversion of biomass into renewable transportation fuels is receiving more and more attention.1,2 Currently, alternative methods, such as production of hydrogen from cellulose,3 fermentation of sugar to bioethanol4 and transesterification of vegetable oil or fats into biodiesel1,2,5 are used.
Biodiesel is a clean renewable fuel and it can be used in any compression ignition engine without modification.6 Recently, more than 95% of biodiesel production feedstocks is obtained from edible vegetable oils and the properties of biodiesel produced from these candidates are highly suitable for use as a diesel fuel replacement.1,7
The potential of non-edible oils has not been considered as an alternative feedstock for biodiesel production. Many researchers are interested in non-edible oil sources (non-food vegetable oils, animal fats and waste oils) as a renewable and sustainable solution.8,9 Jatropha curcas oil (JCO) is considered to be a promising feedstock, which is a low-cost and non-food-based feedstock. Hence, biodiesel production cost could be effectively reduced to 60–70% by using this low-cost raw material.9,10
Transesterification is performed in the presence of a suitable catalyst in order to obtain reasonable conversion of triglyceride to biodiesel (Fig. 1). Homogeneous catalysts (acidic: H2SO4 and H3PO4 or basic: NaOH and KOH) are commonly applied in the transesterification of jatropha oil.9,11 The main obstacles to homogeneous catalysts are that they are difficult to recover and lead to downstream waste treatment, increasing the biodiesel production cost.12 Heterogeneous catalysts have been studied for producing biodiesel from jatropha oil. Soares Dias and co-workers13 reported that biodiesel production from soybean oil using a cerium-modified solid basic magnesium alumina (Mg/Al) catalyst showed a FAME yield >90%. Moreover, Margandan et al.14 investigated the catalytic activity of a cerium oxide (CeO2)impregnated alkali zeolite (NaZSM-5) catalyst in the transesterification of jatropha oil with methanol. The result exhibited that the CeO2/NaZSM-5 catalyst showed high activity (90% biodiesel yield); however, it required a high reaction temperature of 100 °C to achieve the high conversion.
 |
| | Fig. 1 Biodiesel (fatty acid methyl esters) production by triglycerides transesterification in the presence of catalyst. | |
Calcium-based catalysts are solid basic catalysts widely investigated in transesterification because they are cheap and have superior performance at a very low reaction temperature. Zhu et al.15 used calcium oxide (CaO) to catalyze the transesterification reaction of jatropha oil with methanol to yield 93% biodiesel. However, the leaching of Ca2+ ion species into the reaction media contaminated the biodiesel product and reduced the service lifetime of the catalyst.
The activity and stability of CaO can be improved by mixing with other metal oxides such as MgO6 La2O38,17 and CeO2.16,17 Among the mixed metal oxides, the CaO-mixed CeO2 catalyst revealed good catalytic activity in the biodiesel industry. The CaO–CeO2 catalyst has been reported as producing biodiesel from edible-grade refined palm16 and soybean17 oils. Thitsartarn and Kawi16 reported a CaO–CeO2 catalyst synthesized by sol–gel with a co-precipitation method. The sol–gel process included precipitation of metal ions with an ammonia aqueous solution at a constant pH to form a white colour gel-like solution. However, the sol–gel process is more time-consuming and needs complicated preparation steps. The CaO–CeO2 catalyst was tested for methanolysis of palm oil, which showed good performance (>10 reaction cycles) during the transesterification reaction. Nevertheless, there were some homogeneous species leaching out from the catalyst into the biodiesel product. High concentrations of leached Ca (102 ppm) and Ce (57 ppm) species were observed in the biodiesel product.
To the best of our knowledge, no report on the catalytic performance of CaO–CeO2 mixed oxides for the transesterification of non-edible crude Jatropha curcas oil (JCO) has been investigated. A series of CaO–CeO2 mixed oxide catalysts have been currently prepared by a simple co-precipitation method to improve the interaction of catalyst components in the bimetallic system. This catalyst was used as an active and stable catalyst for the production of a clean and green alternative fuel. The physicochemical properties of mixed oxide catalysts were characterized and discussed. The effect of Ca-to-Ce molar ratio on the catalytic activity and the leaching behaviour of CaO–CeO2 mixed metal oxide catalysts for the transesterification of crude JCO were determined. Finally, screening of the reaction conditions (i.e. methanol/oil molar ratio, catalyst concentration and reaction time) and reusability of the catalyst for the transesterification reaction has also been performed.
Results and discussion
Crude JCO characteristics
The physicochemical properties of crude JCO are shown in Table 1.18,19 The acid value (AV) of crude JCO was 13.6 mg KOH g−1, whereas it had 6.8% (as oleic) of free fatty acid (FFA). The density and saponification value of crude JCO were 0.92 g cm−3 and 188.40 mg KOH g−1, respectively. Therefore, the average molecular weight of crude JCO was calculated as 962.8 g mol−1.
Table 1 Physicochemical properties and characteristics of crude JCO
| Properties (unit) |
Valuesa |
Valuesb |
Valuesc |
| Adapted from ref. 18. Adapted from ref. 19. Analyzed using Malaysia Palm Oil Board (MPOB) standard methods. Analyzed using Association of Official Analytical Chemists (AOAC) standard methods. |
| Flash point (°C) |
214 |
235 |
— |
| Pour point (°C) |
6 |
8 |
— |
| Cloud point (°C) |
11 |
2 |
— |
| Viscosity at 40 °C (cSt) |
36.92 |
54.8 |
— |
| Specific gravity at 29 °C (g cm−3) |
0.792 |
0.914 |
— |
| Density at 15 °C (g cm−3) |
0.92 |
|
0.92 ± 0.15 |
| Water content (%, w/w) |
— |
0.052 |
0.09 ± 0.01 |
| Acid value (mg KOH g−1) |
38.2 |
0.92–10.0 |
13.60 ± 0.75 |
| Free fatty acid (as oleic, %, w/w) |
— |
9.0–12.0 |
6.80 ± 0.91 |
| Saponification number (mg KOH g−1) |
195.0 |
186.5–193.3 |
188.40 ± 3.10 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Fatty acid composition (%) |
| Palmitic acid (C16:0) |
4.2 |
13.8 |
20.2 ± 0.56d |
| Palmitoleic acid (C16:1) |
— |
— |
1.10 ± 0.05d |
| Stearic acid (C18:0) |
6.9 |
6.8 |
7.20 ± 0.91d |
| Oleic acid (C18:1) (n–9) |
43.1 |
41.7 |
39.8 ± 0.64d |
| Linoleic acid (C18:2) |
34.3 |
35.6 |
31.2 ± 0.41d |
| Linolenic acid (C18:3) |
— |
— |
0.30 ± 0.03d |
| Arachidic acid (C20:0) |
— |
— |
0.20 ± 0.02d |
TG/DTA studies
Fig. 2 shows the TG/DTA curves of the catalyst precursor before calcination. The DTA peaks closely correspond to the weight changes observed on the TGA curves. The total mass loss from 50 to 750 °C was found to be 18.1%. All curves indicated that decomposition mainly occurred via two distinct stages and was complete at about 800 °C. The weight loss found from TGA measurements agrees fairly well with that expected for the decomposition of hydroxycarbonates (M2CO3(OH)2) to different oxides of calcium and cerium. According to the following thermal decomposition equation (eqn (1)), the first stage of mass loss from 50 to 150 °C, associated with the structural water loss, was 6.9%.| | |
CeO2·2H2O →CeO2 + 2H2O (vapor)
| (1) |
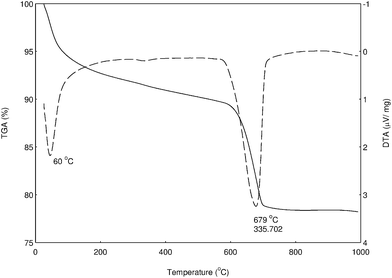 |
| | Fig. 2 TG/DTA spectrum of CaO–CeO2 mixed metal oxide. | |
Ce(OH)4 is a hydrous oxide, represented by CeO2·xH2O, which dehydrates progressively. Therefore, the decomposition of the precursor is a form of the dehydration process of the hydrated CeO2. It was suggested that the precipitate consisted of a mixture of phases such as CeO2·2H2O and CeO2;20 moreover, the weight loss below 100 °C could be due to absorbed moisture. The final weight loss (11.2%) at 550–750 °C signified the decomposition of M2CO3(OH)2.20
Structure and crystallography
Fig. 3(a) and (b) show the powder XRD patterns of the CeO2, CaO and CaO–CeO2 catalysts produced with different preparation variables (pH and Ca/Ce molar ratio). The pure CaO gave well-defined and narrow crystalline diffraction peaks at 2θ values of 32.1°, 37.2°, 53.9°, 64.0° and 67.2° (JCPDS File no. 00-037-1497), corresponding to the presence of a cubic crystal structure associated with reflections from the (110), (200), (220), (311), (222) and (400) planes, respectively. The fluorite-type cubic structure of pure CeO2 showed the characteristic reflections at 2θ values of 28.5°, 33.1°, 47.5°, 56.4°, 59.2°, 69.6°, 76.9° and 79.3° from the (111), (200), (220), (311), (222), (440), (331) and (420) planes, respectively. CaO–CeO2 mixed oxide catalysts (Fig. 3) caused the formation of cubic CaO and fluorite-type cubic CeO2 phases in a binary metal system. The XRD analysis of these co-precipitated CaO–CeO2 catalysts revealed characteristic peaks of their separate metal oxide crystalline phases without the presence of any new formation of mixed oxide phases (homogeneous CaO–CeO2 mixed solid solutions) detectable in the precipitate particles. This was mainly due to the different ionic radii of the metal ions.7 In Fig. 3(a), the increase in synthesis pH (9–12) resulted in an increase and a decrease in the peak intensities of CaO and CeO2, respectively. On the other hand, the addition of CaO to the CeO2 matrix resulted in an insignificant change in the XRD patterns (Fig. 3(b)) of CeO2. The peak intensity of the CaO phase was found to be low, whereas a strong reflection associated with the fluorite-type cubic structure of CeO2 was observed in the XRD profile. With an increase in the Ca/Ce molar ratio, the XRD peaks associated with the cubic CaO phase become more intense, and concomitantly those of CeO2 decrease significantly. This can be attributed to the higher X-ray scattering factor of Ca2+ compared to the Ce4+ ions.21
 |
| | Fig. 3 X-ray diffraction patterns of (a) CeO2 (i), CaCe4.00–pH9 (ii), CaCe4.00–pH10 (iii), CaCe4.00-pH11 (iv), CaCe4.00-pH12 (v) and CaO(VI) catalysts; (b) CeO2 (i), CaCe0.25 (ii), CaCe0.67 (iii), CaCe1.00 (iv), CaCe4.00 (v), CaCe19.0 (vi) and CaO (vii) catalysts. ♣ is characteristic peak of CaO, and Δ is characteristic peak of CeO2. | |
Moreover, the dependence of CeO2 mean crystallite sizes as measured by XRD on the calcium concentration from 0.25 to 19.0 Ca/Ce molar ratio is presented in Table 3. The pure oxides exhibited the metal CaO and CeO2 clusters with 66.3 and 48.6 nm mean crystallite size, respectively, which decreased to become 54.2 and 39.2 nm for the binary oxide CaCe0.25. Nevertheless, the decrease seemed to reach a threshold from a Ca/Ce molar ratio of 0.67. Above a Ca/Ce molar ratio of 0.67, the decrease in CaO and CeO2 crystallite sizes became less pronounced and there was an increase in crystallite sizes to 75.8 and 49.7 nm for CaCe19.0. Initially, the addition of the calcium component into the CeO2 crystalline structure at low concentration was first located between the CeO2 grain boundaries and thus disturbed the normal growth of the CeO2 crystallites at high concentration.20 This indicated that the CeO2 mean crystallite size increased linearly with the loading of calcium (Fig. 4) from low to high concentration. This result demonstrated that the volume of the CeO2 cell increased because of the Ca2+ effective ionic radius (1.12 Å), which is larger than that of Ce4+ (0.97 Å).20 The results indicated that the crystal sizes of the catalysts were in agreement with the line width of the peak, in which the FWHM decreased with the increase in the crystallite size. It was also suggested that the high dispersion of CaO on the composite gave rise to a low crystallite size for CaCe0.25. However, agglomeration and the sintering effect of particle size to form bulk particles resulting from overloading of secondary metal and a high calcination temperature could occur above a Ca/Ce molar ratio of 0.67.2,3,5,7,8
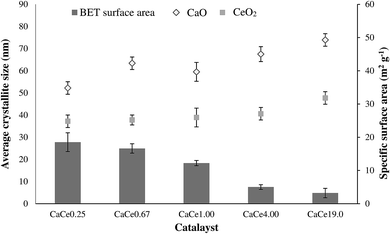 |
| | Fig. 4 Crystallite sizes and BET surface area of CaO–CeO2 mixed metal oxides. | |
Catalyst composition and surface area
Tables 2 and 3 demonstrate the efficiency of CaO–CeO2 catalysts synthesized with different preparation variables (pH and Ca/Ce molar ratio) by evaluating the catalyst compositions. As mentioned above, only the CaO–CeO2 (CaCe4.00-11 and CaCe4.00-12) catalysts (Table 2) synthesized at pH 11 and 12 indicated a high content of CaO in the composites with Ca/Ce molar ratios of 4.39 and 3.53, respectively. Moreover, Ca/Ce atomic ratios were found in the precipitated solids to be close to the theoretical ratios at pH 11–12. This phenomenon was due to the fact that the Ca2+ ion is favourably precipitated at relatively high pH values (pH 11); however, the Ce4+ ion is readily solidified or more soluble at a high pH value of pH 12.7,8 From Table 3, the Ca/Ce molar ratio of the catalyst decreased when the CeO2 content increased. In addition, the BET surface areas of CaO–CeO2 mixed oxide catalysts with different Ca/Ce molar ratios are presented in Table 3. The bulk CeO2 and CaO exhibited high and low surface areas of 52.6 and 9.2 m2g−1. The surface area of the CaO–CeO2 mixed oxide catalysts was in agreement with XRD analysis, which showed that larger crystallite sizes gave lower surface areas (Fig. 4).17,20 The surface areas increased with the addition of CeO2, suggesting that CeO2 was incorporated with the CaO and both were well dispersed on the catalyst surfaces as shown in (Fig. 7(ii)). Moreover, it is well known that an increase in the surface area of a catalyst might be favourable for improving the catalytic performance. Conversely, the low FAME yield (40%) observed with CaCe0.25 might be related to the change in active sites on the catalyst surface due to the addition of low CeO2 content. The surface area of the catalysts (CaCe0.67 to CaCe19.0) decreased when calcium loading increased (Table 3). The overloading of the CaO particles on the surface or into the porous structure of CeO2 that cause saturation or filling of the pores in the catalyst composition also contributed to the reduction in CaO–CeO2 catalyst surface area.8 Furthermore, the decrease in surface area may also be due to the high calcination temperature (900 °C), which led to a sintering effect on fine crystals and promoted cluster agglomeration.8
Table 2 Effect of the precipitation condition (pH) on the FAME yield of crude JCO with CaCe4.00-9, CaCe4.00-10, CaCe4.00-11 and CaCe4.00-12 catalysts
| Catalysts |
Ca/Ce molar ratiob |
FAME yielda (%) |
| Theoretical |
Precipitate |
| Transesterification condition: catalyst dosage, 4%; n(methanol):n(JCO) = 12:1; reaction time, 6 h; reaction temperature, 65 °C. Estimated by XRF analysis. |
| CaCe4.00-9 |
4.00 |
0.52 ± 0.95 |
10.33 ± 1.22 |
| CaCe4.00-10 |
4.00 |
0.41 ± 0.05 |
12.61 ± 0.99 |
| CaCe4.00-11 |
4.00 |
4.39 ± 0.75 |
86.53 ± 5.23 |
| CaCe4.00-12 |
4.00 |
3.53 ± 1.24 |
80.21 ± 2.33 |
Table 3 Elemental composition and physicochemical properties of CaO, CeO2 and CaO–CeO2 mixed metal oxides with various Ca/Ce ratios
| Catalyst |
Ca/Ce molar ratioa |
Crystallite sizeb (nm) |
SBETc (m2g−1) |
| Theoretical |
Experimental |
CaO |
CeO2 |
| Estimated by XRF spectroscopy. Determined from XRD patterns using Scherrer's equation. BET surface area. |
| CaO |
— |
— |
66.3 ± 3.21 |
— |
9.2 ± 1.61 |
| CeO2 |
— |
— |
— |
48.6 ± 2.77 |
52.6 ± 1.77 |
| CaO–CeO2 |
0.25 |
0.28 ± 0.05 |
54.2 ± 2.82 |
39.2 ± 2.54 |
20.5 ± 2.13 |
| |
0.67 |
0.78 ± 0.15 |
65.4 ± 2.64 |
39.4 ± 4.29 |
17.6 ± 1.22 |
| |
1.00 |
1.12 ± 0.27 |
62.5 ± 4.24 |
41.9 ± 2.91 |
12.7 ± 3.41 |
| |
4.00 |
4.39 ± 0.75 |
69.9 ± 3.39 |
42.6 ± 2.39 |
5.5 ± 1.23 |
| |
19.0 |
16.1 ± 2.54 |
75.8 ± 2.89 |
49.7 ± 3.26 |
4.2 ± 1.02 |
Surface functional groups
FTIR spectra of CaO, CeO2 and CaO–CeO2 catalysts are shown in Fig. 5. All spectra present a large absorption band located at around 500 cm−1, which is attributed to the Ca–O, Ce–O and Ca–O–Ce stretching vibration.20 Moreover, the band located at around 1799 cm−1 corresponded to the H–O–H bending vibration, indicating the presence of moisture absorbed after the calcination process.20 In the previous studies, the bands found at around 711 and 873 cm−1 have been attributed to the CO2 asymmetric stretching vibration and CO32− bending vibration, respectively.21 Both bands were also related to the presence of atmospheric CO2 on metallic cations20 and the formation of “carbonate-like” species on the particles' surface21 during the synthesis. Furthermore, another high intensity band was seen at around 1407 cm−1, which was the characteristic vibration mode of isolated CeO2.22
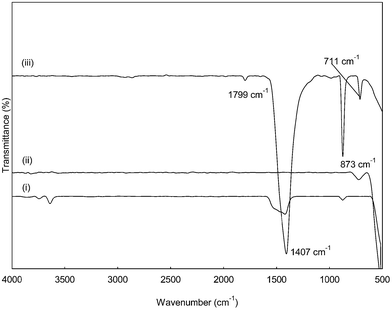 |
| | Fig. 5 FTIR spectrum of CaO (i), CeO2 (ii) and CaO–CeO2 (iii) catalysts. | |
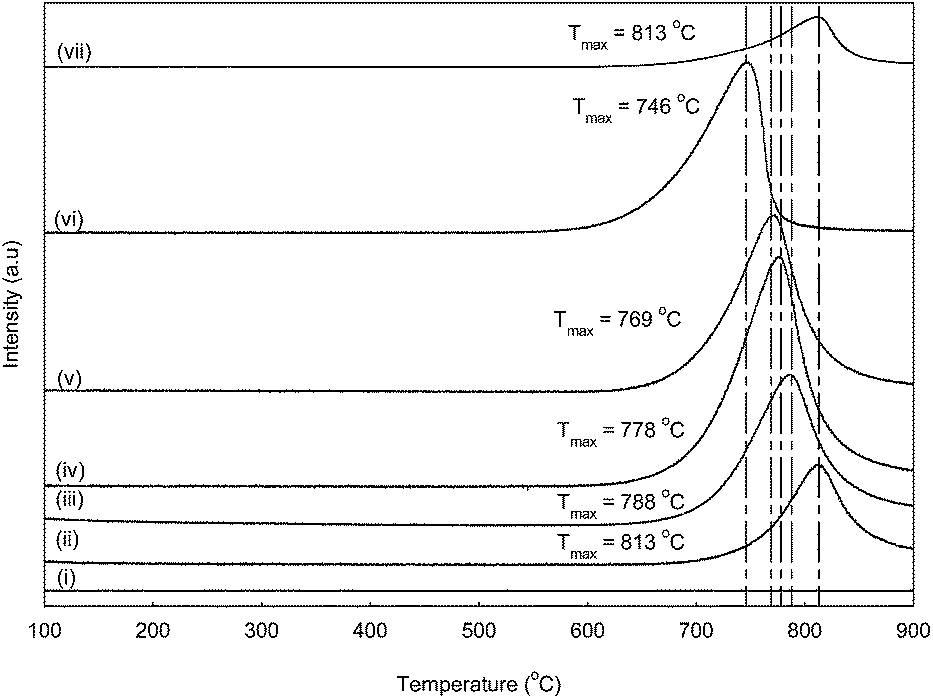
Basicity
The total basicity of the CaO–CeO2 catalysts was evaluated using temperature programmed desorption of CO2. The TPD-CO2 profile of the CaO–CeO2 catalysts is shown in Table 4 and Fig. 6. All mixed oxide catalysts exhibited a high amount of CO2 desorption peaks with peak maxima at 813, 788, 778, 769 and 746 °C, corresponding to basic sites of high strength. The strong basic sites of CaO–CeO2 catalysts showed the existence of oxygen in Ca–O, Ce–O2 ion pairs and isolated O2− anions, which can be expected to possess Lewis base character.8,14 The active sites of the oxide surface may well form an interaction with the proton of methanol and contribute to the breaking of OH bonds, hence causing the formation of an active methanol ion to initiate the transesterification reaction. Basically, the basicity of a CaO–CeO2 bimetallic oxide catalyst was found to be higher than bulk CaO and CeO2. The order of basicity was found to be as follows: CaCe1.00 > CaCe4.00 > CaCe19.0 > CaCe0.67 > CaCe0.25 > CaO > CeO2. The improved basicity of the bimetallic oxide was due to the synergetic effect between the metallic ions of CaO and CeO2. However, the CO2 desorption peak moved toward lower temperatures for bimetallic oxide catalysts with the introduction of more Ca2+ (Ca/Ce molar ratio of 0.25–19.0) to the binary oxide, which indicated that the presence of secondary metal phases (CaO) in CeO2 slightly reduces the basic strength of the catalyst.
Table 4 The basicity of CaO, CeO2 and CaO–CeO2 mixed metal oxides with various Ca/Ce ratios
| Catalysts |
basicity (×10−3 mol g−1) |
Temperature range (°C) |
Peak temperature (°C) |
| CaO |
2.63 ± 0.63 |
658–879 |
813 |
| CaCe0.25 |
2.81 ± 0.12 |
685–860 |
813 |
| CaCe0.67 |
3.57 ± 0.43 |
690–864 |
788 |
| CaCe1.00 |
8.20 ± 0.89 |
672–891 |
778 |
| CaCe4.00 |
4.19 ± 0.21 |
666–850 |
769 |
| CaCe19.0 |
3.73 ± 0.54 |
648–779 |
746 |
| CeO2 |
0 |
— |
— |
 |
| | Fig. 6 CO2-Temperature programmed desorption profiles of CeO2 (i), CaCe0.25 (ii), CaCe0.67 (iii), CaCe1.00 (iv), CaCe4.00 (v), CaCe19.0 (vi) and CaO (vii) catalysts. | |
Morphology
The morphologies of the catalysts were observed by SEM (Fig. 7). A compact and evenly distributed globular granule was found in pure CaO (Fig. 7(i)), whereas pure CeO2 (Fig. 7(viii)) particles were mostly fluorite structure with a large size distribution. All CaO–CeO2 mixed oxide catalysts (Fig. 7(ii) to (vii)) were in the form of agglomerates of irregular-shaped crystalline particles. The particle size increases substantially with an increase in the Ca/Ce molar ratio, indicating an obvious agglomerate of crystalline nature. The mixed metal oxide catalyst showing agglomeration was in agreement with the morphology that has been reported previously for a CeO2–CaO nanocomposite oxide.23 Numerous macropores were found to be present on the surface of the particles (Fig. 7(ii)), which is due to the good dispersion of CaO on the surface and into the pores of CeO2; however, with a decrease in calcium content the catalysts become decreasingly porous. In addition, the SEM image indicated that the catalyst morphology was closely related to the high calcination temperature (900 °C). CaCe1.00 (Fig. 7(iii)) shows homogeneous agglomeration with smaller particles, which is associated with better catalytic performance. Nevertheless, mixed oxide catalysts, CaCe4.00 and CaCe19.0 (Fig. 7(vi) and (vii)), have rough and disproportionate agglomerates. The large aggregating and sintering effect of the catalyst's particles is also considered as one of the reasons why the catalyst (CaCe4.00 and CaCe19.0) has a lower surface area.
 |
| | Fig. 7 SEM micrographs of CaO (i), CaCe0.25 (ii), CaCe0.67 (iii), CaCe1.00 (iv), CaCe4.00 (v), CaCe19.0 (vi), and CeO2 (vii) catalysts. | |
Optimization of process parameters
Effect of the precipitation condition (pH) on the FAME yield. Investigating the effect of a range of CaO–CeO2 mixed oxide (CaCe4.00) catalysts prepared via variable pH (9–12) conditions at the precursor stage upon the molar ratio of each metal component in these catalysts, the concentrations of metal components present in the catalyst composite were dependent on the precipitation pH and this subsequently influenced the activity of the final calcined catalysts. Table 2 shows the optimum pH preparation conditions were identified with respect to the catalytic activity for transesterification of crude JCO to biodiesel. The CaCe4.00-9 and CaCe4.00-10 catalysts exhibited poor catalytic performance due to a low precipitate Ca/Ce molar ratio. The result demonstrated that CeO2 in the catalyst composite is more abundant and it exhibited low basicity, which is not sufficient for performing a complete transesterification reaction.7,15 The CaCe4.00-11 catalyst showed a significant improvement in the FAME yield to 86.53%, due to an increase in the precipitate Ca/Ce molar ratio close to the theoretical molar ratio. The CaCe4.00-11 catalyst with a Ca/Ce molar ratio of 4.39 indicated a high content of Ca and Ce in the bimetallic oxide, which provided more active basic sites for the transesterification process. With a further increase in preparation pH, the FAME yield from the CaCe4.00-12 catalyst was reduced.
Effect of the Ca/Ce molar ratio of catalyst on the FAME yield correlated to the basicity of mixed oxide catalysts. CaO–CeO2 mixed oxide catalysts with different Ca/Ce molar ratios were screened for transesterification activity on crude JCO, which is indicated in Fig. 8. The transesterification was evaluated with a crude JCO-to-methanol ratio of 1:12 at 65 °C with 4 wt% catalyst (with respect to the weight of oil). The results showed that the Ca/Ce molar ratio greatly affected the biodiesel yield. The FAME yield of the CaCe0.67, CaCe1.00, CaCe4.00 and CaCe19.0 catalysts were higher than the CaCe0.25 catalyst. The catalyst with high CeO2 content exhibited low production of FAME. When the Ca/Ce molar ratio increased from 0.25 to 0.67, the FAME yield increased remarkably from 40.61% to 80.59%. A further increase in Ca/Ce molar ratio to CaCe1.00 resulted in the maximum yield of FAME. Unfortunately, a significant fall in catalytic activity was observed beyond a Ca/Ce molar ratio of 4 and 19. Previous studies showed that transesterification activity is dependent on the amount of basic sites in the catalyst.7,8 The results of CO2-TPD supported the findings as shown in Fig. 6 and summarized in Table 4. The increase in FAME yield from 40% to 87% with the increase in Ca content from CaCe0.25 to CaCe1.00 is due to the increase in basicity from 2.81 to 8.20 × 10−3 mol g−1. The relationship between basicity and activity of the catalyst is demonstrated in Fig. 8. The improved basicity of the CaO–CeO2 mixed oxide catalyst was due to the synergetic effect between the metallic ions of CaO and CeO2. Nonetheless, a tremendous amount of catalyst components was observed to leach out from catalysts into the reaction media during the reaction.
 |
| | Fig. 8 Catalytic performance of CaO–CeO2 catalysts with different Ca/Ce molar ratios: (a) using heterogeneous catalyst, (b) using homogeneous species present in methanol solution and (c) basicity of CaO–CeO2 catalysts. Transesterification conditions: oil = 10 g, n(methanol)–n(oil) = 12:1, catalyst dosage = 4 wt%, reaction time = 6 h, reaction temperature = 65 °C. | |
The dissolved homogeneous species are known to be involved in the catalysis of the reaction.17 The reaction parameters, i.e. catalyst amount, 4 wt%; (methanol)/(oil) ratio, 12:1; reaction temperature, 65 °C and reaction time, 6 h, were tested. As illustrated in Fig. 8, bulk CaO exhibited both the highest FAME yield of 90% and the highest contribution of homogeneous catalysis (FAME yield of 32%). These results indicated that pure CaO can easily dissolve into the polar phase (methanol) and form calcium methoxide species as a homogeneous catalyst. Hence, the biodiesel yield was due to the superior mass transfer for this homogeneous catalyst. For mixed metal oxide catalysts, when the Ca/Ce molar ratio increased to 0.25, a concomitant remarkable decrease in homogeneous contribution to 0.4% FAME yield was also observed. Furthermore, the low FAME yield was demonstrated from reactions catalyzed by methanol solutions, containing the active homogeneous species leaching out from CaCe0.67, CaCe1.00 and CaCe4.00 catalysts and the high FAME yield was obtained from CaCe19.0, respectively. The results indicated that the homogeneous species leaching out from solid catalysts into the methanol solution are actively involved in the reaction.
Therefore, the good catalytic performance of CaCe0.67, CaCe1.00, CaCe4.00 and CaCe19.0 catalysts was mainly attributed to the solid basic sites of the catalyst with a very low homogeneous contribution. However, the reasonable Ca/Ce molar ratio for transesterification of crude JCO to FAME is CaCe1.00. By correlation of this tendency with the increased and decreased peak intensity of CaO and CeO2 phases shown in XRD patterns, it can be inferred that an interaction between the surface CaO and the CeO2 most likely exists. Soares Dias et al.13 reported that over 90% of methyl ester yield was obtained with catalyzed soybean oil with a Ce/Mg Al catalyst. The use of a tri-metal catalyst system increased the production cost of biodiesel. On the other hand, the leaching of Ce2+, Mg2+ and Al3+ ions into the product still remained unknown.
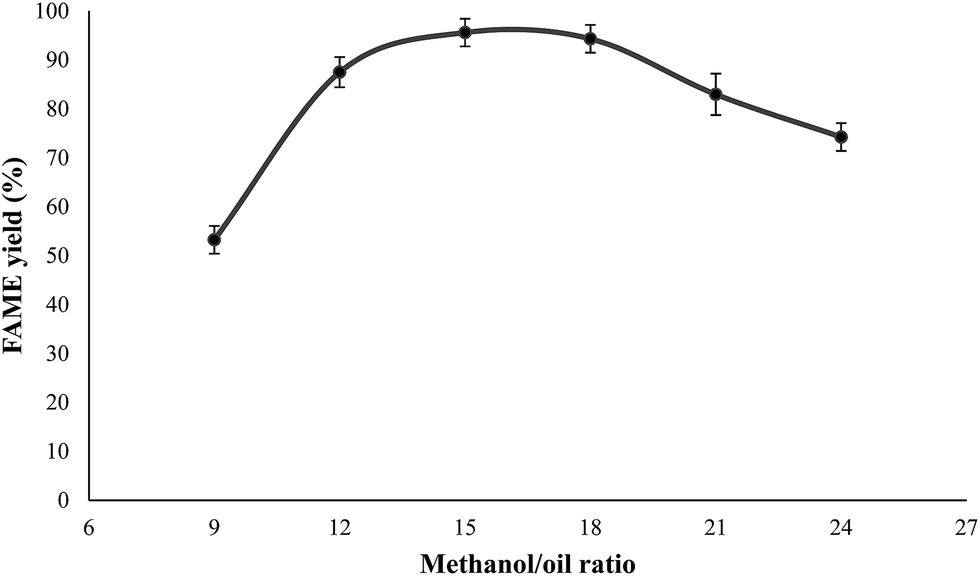
Effect of molar ratio of methanol/oil on FAME yield. The molar ratio of methanol to crude JCO was varied from 9:1 to 24:1 (Fig. 9). The yield of FAME from transesterification of crude JCO increased with the methanol/oil molar ratio up to 15:1, which achieved the maximum ester yield of 95.59%. With a further increase in methanol/oil molar ratio to 18:1, the change in the yield was insignificant. However, the FAME yield decreased considerably to 82.94% and 74.22% at methanol molar ratios of 21:1 and 24:1, respectively. The results showed that a higher methanol/oil molar ratio is required to get better conversion. Nonetheless, an excess of polar OH groups from methanol will cause emulsification that interferes in the separation of as-synthesized glycerine as more energy is required to recover it. Moreover, it can increase the dissolution of oil, intermediates and biodiesel product in a high volume of methanol, which results in wastage of the raw reactants; therefore, 15:1 is the appropriate methanol/oil molar ratio for this reaction. Previously, it was found that transesterification of palm oil required a higher methanol-to-oil molar ratio, which is 20:1, to achieve a high FAME yield with the CaO–CeO2 catalyst prepared by Thitsartarn and Kawi.16
 |
| | Fig. 9 Effect of methanol–oil ratio on the FAME yield of crude JCO. Reaction conditions: oil = 10 g, catalyst dosage = 4 wt%, reaction time = 6 h, reaction temperature = 65 °C. | |
Effect of the catalyst dosage on FAME yield. Fig. 10 shows the influence of the catalyst amount on biodiesel yield. The catalyst amount was varied in the range of 1–6 wt% (with respect to the oil weight). The FAME yield (24%) was initially quite low at the low catalyst amount of 1 wt%. The yield appeared to increase with an increase in catalyst amount due to an increase in the number of active sites. The maximum FAME yield of 95.57% was obtained at 4 wt% catalyst dosage; however, a slight reduction in yield was found at catalyst concentrations above 4 wt%. This effect was attributed to poor diffusion between the methanol–oil–catalyst systems in this case.4,8,11 Kim et al.17 stated that a FAME yield of 91% was recorded with 8 wt% of CaO–La2O3 supported on CeO2 catalysts. Nevertheless, it should be noted that Kim et al.17 used supported transition metal oxides, i.e. La2O3 and CeO2, as catalysts in relatively high concentrations, which increased the biodiesel production cost.
 |
| | Fig. 10 Effect of catalyst loading on the FAME yield of crude JCO. Reaction conditions: oil = 10 g, n(methanol)–n(oil) = 15:1, reaction time = 6 h, reaction temperature = 65 °C. | |
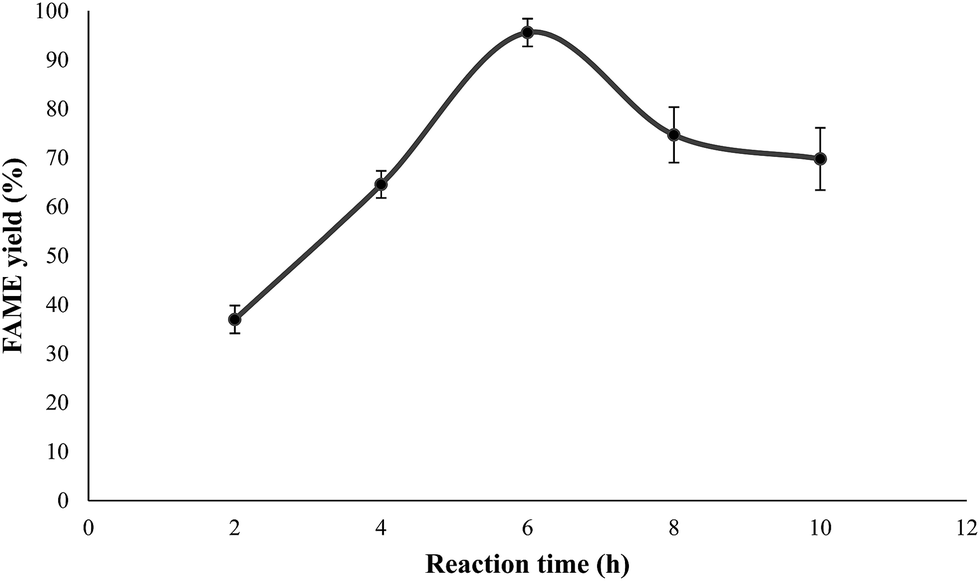
Effect of the reaction time on FAME yield. The dependence of the FAME yield on the reaction time was studied. As demonstrated in Fig. 11, the effect of reaction time on the FAME yield was investigated in the range 2–10 h. Initially, a FAME yield of 36.99% was reached after a short reaction time (2 h). This was due to the limitations of solid mass transfer, which caused poor mixing and dispersion of the solid reactant; however, the conversion for the catalyst increased gradually after 2 h of reaction time. A near-equilibrium FAME yield was found to be around 95% at 6 h reaction time. With a further prolongation of reaction time beyond 6 h, the FAME yield was dramatically reduced to 70% due to the reverse transesterification process.7
 |
| | Fig. 11 Effect of reaction time on the FAME yield of crude JCO. Reaction conditions: oil = 10 g, n(methanol)–n(oil) = 15:1, catalyst dosage = 4 wt%, reaction temperature = 65 °C. | |
 |
| | Fig. 12 Gas chromatography of (a) standard references of fatty acid methyl esters (1000 ppm) and (b) transesterification of jatropha-derived biodiesel (FAME). | |
Quality of biodiesel. As shown in Fig. 12, the FAME compositions of crude JCO contained C16 : 0 (20.16%), C18 : 0 (7.22%), C16 : 1 (1.32%), C18 : 1 (39.77%), C18 : 2 (31.53%) and C20 : 0 (< 0.01%), in which the fatty acids are suitable for biodiesel production.8
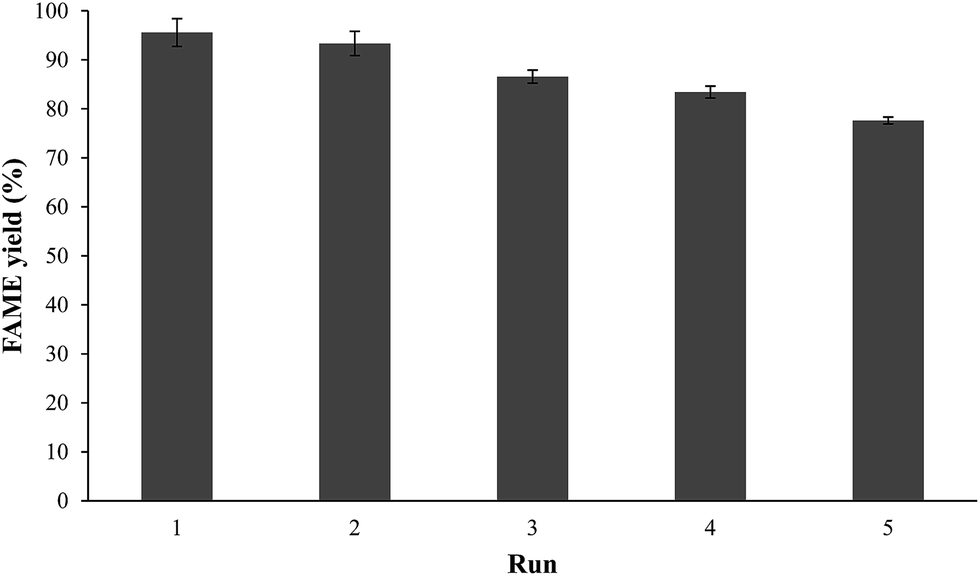
Catalyst stability
Fig. 13 presents the reusability study of CaCe1.00 for transesterification of crude JCO under the best reaction conditions (10 g of oil, methanol–oil molar ratio of 15:1, catalyst amount of 4 wt% and reaction temperature of 65 °C). After each cycle of 6 h of reaction, the catalyst was separated and washed several times with methanol and n-heptane. The resultant dried solid particles were calcined at 900 °C for 3 h and immediately reused in a new batch transesterification process. The FAME yield decreased slowly from 97.57% to 83.41% when this process was repeated 1–4 times. On the other hand, a significant loss of catalytic performance was observed in the 5th run, which indicated a significant reduction of the number of active sites on the catalyst surface after numerous washing and re-calcination processes.16,17 The two possible reasons behind catalyst deactivation are surface poisoning and structural collapse.5,7,8 The surface poisoning might be due to the surface-bound glycerides, i.e. triglycerides (TG), diglycerides (DG) and monoglycerides (MG) on the catalyst, which cannot be removed with the less polar solvent.7 These coated materials inhibited the active sites of the catalyst. Furthermore, the repeated calcination processes will cause a morphology change and reduce the interaction between CaO and CeO2.16,17,20
 |
| | Fig. 13 Recyclability study of catalyst. Reaction conditions: oil = 10 g, catalyst dosage = 4 wt%, (methanol)–(oil) = 15:1, reaction time = 6 h. | |
EDS and AAS analysis were carried out to investigate the leaching active species (Ca and Ce) for the CaCe1.00 mixed oxide catalyst (fresh and fifth-run catalysts) (Table 5). EDS analysis of fresh, 1st, 3rd, and 5th run CaCe1.00 catalysts exhibited a gradual decrease in Ca/Ce molar ratio from 1.33 to 0.81, indicating less leaching of active metal into the reaction medium. Hence, it is implied that the presence of interaction between active phases of the CaO–CeO2 binary metal oxide could stabilize active phases of the catalyst in order to reduce its leaching into the reaction medium. Moreover, the FAME from all reusability cycles was tested using AAS to evaluate the concentration of leached Ca2+ and Ce4+ ions. The results revealed an insignificant loss of active metal ions in the biodiesel product, with concentrations of Ca in the range of 7.23–3.93 ppm and 5.51–3.21 ppm of Ce content, respectively. However, these minor leached metal ions complied with the standard EN 14214 specification, indicating the produced biodiesel was suitable to be used as vehicle fuel. The concentration of Ca and Ce species decreased significantly in biodiesel with subsequent reaction cycles, and reached about 3–4 ppm after the 5th run. This decreased amount of the leached species was attributed to the good interaction between Ca and Ce, which was due to the vacancies created by substitution of Ce4+ by Ca2+ ions via electron transfer.16 Since the content of dissolved Ca and Ce species in the reaction medium was very low (3–4 ppm), the FAME yield shown from the 5th run onward was mainly contributed by the heterogeneous CaCe1.00 catalyst and not by the homogeneous catalytic species dissolved in the reaction mixture. Therefore, the results of reusability and regeneration studies definitely indicated that the CaCe1.00 mixed oxide catalyst is very stable and durable during the transesterification reaction, which is better than that reported by Thitsartarn and Kawi.16
Table 5 Durability studies of CaO–CeO2 catalyst
| Number of runa |
Ca/Ce molar ratiob |
Biodieselc |
| Theoretical |
Experimental |
Ca (ppm) |
Ce (ppm) |
| Transesterification conditions: reaction temperature, 65 °C; reaction time, 6 h; catalyst dosage, 4 wt.%; and methanol–oil ratio of 15:1. Determined by EDS analysis. Concentration of calcium in biodiesel determined by AAS analysis. |
| Fresh |
1.00 |
1.33 ± 0.57 |
1.44 ± 0.04 |
— |
| 1st run |
— |
0.88 ± 0.06 |
7.23 ± 1.11 |
5.51 ± 1.02 |
| 3rd run |
— |
0.87 ± 0.17 |
4.22 ± 0.23 |
3.01 ± 0.12 |
| 5th run |
— |
0.81 ± 0.08 |
3.93 ± 0.79 |
3.21 ± 0.27 |
Experimental
Materials
Commercial crude JCO was obtained from Bionas Sdn Bhd, Malaysia and used without further purification. The reagents were cerium(III) nitrate hexahydrate (Ce(NO3)2·6H2O) (Sigma-Aldrich, 99.9%), calcium(II) nitrate tetrahydrate (Ca(NO3)2·4H2O) (R&M Chemicals, 99.9%), sodium hydroxide (NaOH) (Merck, 99.0%), sodium carbonate anhydrous (Na2CO3) (Bendosen, 99.0%) and commercial CaO (Sigma Aldrich, 99.0%). Anhydrous methanol (Merck, 99.7%) and dichloromethane (Merck, 99.7%) were purchased from Fisher Scientific. Analytical reagent grades were used throughout the experiment. The properties of crude JCO were identified from the data obtained using Malaysia Palm Oil Board (MPOB) standard methods. Therefore, the average molecular weight (M) of crude JCO was calculated by the following equation (eqn (2)):8| |
 | (2) |
where AV is the acid value and SV is the saponification value of crude JCO.
Catalyst preparation
CaO–CeO2 mixed oxides were prepared according to the co-precipitation method following calcination of the precursors. In a typical synthesis, a mixture of Ca(NO3)2·4H2O and Ce(NO3)2·6H2O were dissolved in a calculated quantity of deionized water. The two precursors were mixed homogeneously and allowed to precipitate using a highly basic solution of NaOH (0.04 mol) and Na2CO3 (0.01 mol) at a constant pH. The resulting suspension was stirred at 65 °C for 24 h. The solid product was recovered by filtration, followed by washing with deionized water and drying in an oven at 110 °C overnight. The dried solid was calcined at 900 °C for 6 h with ramping time at 5 °C min−1. The CaO–CeO2 mixed oxide catalysts were referred to as CaCex-y in the text, where x represents the Ca/Ce molar ratio and y is the pH value of the preparation method.
Catalyst characterization
Thermogravimetric. Thermogravimetric and differential thermal analysis (TG/DTA) was performed on a Mettler Toledo thermogravimetric analyzer. These tests were performed under a continuous nitrogen flow (100 ml min−1) over a temperature range of 35–1000 °C at a heating rate of 10 °C min−1.
Structure and crystallography. The powder X-ray diffraction (XRD) analysis was performed with a XRD6000 powder X-ray diffractometer (Shimadzu Corporation, Japan) at ambient temperature. The Cu Kα radiation was generated by a Philips glass diffraction X-ray tube (broad focus 2.7 kW type), with a step size of 0.04° in the 2θ range from 10 to 80°, and it was used to generate diffraction patterns from the powder crystalline samples. The data was processed with the X'Pert HighScore Plus software. The peaks were identified using the Powder Diffraction File (PDF) database created by the International Centre for Diffraction Data (ICDD). Then, the crystallite size of the powder catalysts was calculated with Debye-Scherrer's equation.8
Elemental analysis. The real molar ratio of the catalyst components (Ca/Ce) was determined by energy-dispersive X-ray fluorescence (XRF) spectrometry using a Bruker AXS. The measurements were performed using calibration curves based on the XRF measurements for the prepared mixtures from silica (Degussa). The metal concentration in the examined samples was determined by the amount of emitted X-ray radiation related to the values in the calibration curves.
Surface functional groups. Infrared spectra (IR) were recorded on a Perkin Elmer (PC) Spectrum 100 FTIR spectrometer using attenuated total reflection-Fourier transform-infrared (ATR-FTIR) to identify the surface functional groups of the catalysts over the wavenumber range of 650–4000 cm−1 with 4 cm−1 resolution. All measurements were conducted at room temperature.
Specific surface area. The total surface area of the catalysts was measured using the Brunauer–Emmett–Teller (BET) method, corresponding to the multi-point N2 adsorption–desorption isotherms at −196 °C. The analysis was conducted using a Micromeritics ASAP-2020. Prior to measurements, all catalysts were degassed for 8 h at 200 °C.
Basicity. The basicity was evaluated using a temperature programmed desorption method with CO2 as probe molecule (CO2-TPD). These experiments were carried out using a Thermo Finnigan TPD/R/O 1100 series apparatus equipped with a thermal conductivity detector (TCD). In a typical experiment, approximately 0.1 g catalyst was pre-treated in an N2 gas flow (30 ml min−1) at 500 °C for 1 h. Subsequently, the catalyst was brought to room temperature and saturated with CO2 gas. Desorption of carbon dioxide was performed after flushing using carrier gas over a temperature range of 30–900 °C at a heating rate of 10 °C min−1.
Morphology. The surface structure and elemental composition of the catalysts were observed with a Hitachi S-3400 scanning electron microscope coupled with an energy-dispersive X-ray detector (SEM-EDX) at room temperature. The catalysts were coated with gold using a sputter coater and the accelerating voltage was 20 kV. The elemental composition was analyzed using an EDS detector mounted on the microscope.
Catalytic activity and biodiesel analysis. The transesterification reactions were carried out by mixing crude JCO, catalyst and methanol in a 250 ml two-necked reaction flask. The reactor was magnetically stirred and equipped with a condenser, a thermometer, and a heating mantle. Unless otherwise noted, catalytic activity tests were performed in the presence of 4 wt% catalyst, a methanol–oil ratio of 12:1, and a reaction temperature of 65 °C for 6 h. At the end of the experiment, the catalyst was separated from the mixture by centrifugation and the mixture was then loaded into a rotary evaporator to remove excess methanol. After the methanol evaporation, the liquid phase (biodiesel and glycerol) was dissolved in hexane and then washed with hot distilled water several times for refinement. The moisture of the washed biodiesel was subsequently removed using anhydrous magnesium sulphate. Finally, the liquid phase was kept in a separating funnel to separate the lower glycerol layer from the upper FAME layer. The glycerol could be separated because it was insoluble in the esters and had a much higher density.The quantification and characterization of biodiesel (FAME) were carried out using a Shimadzu GC-14C gas chromatograph system equipped with a flame ionization detector (FID), a split/splitless injector and a polar BP-20 capillary column (30 m × 0.5 mm × 0.25 μm) with helium as the carrier gas and a flow rate of 1.5 ml min−1. The ester content was quantified according to EN 141037 in the presence of methyl heptadecanoate (C18H36O2) as an internal standard. The analysis of biodiesel for each sample was carried out by dissolving 1 g biodiesel sample into 10 ml n-hexane and injecting 0.1 ml of this solution for each injection. The amount of FAME was calculated and expressed as a mass fraction in percentage terms using the following equation (eqn (3)):
| |
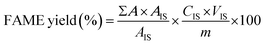 | (3) |
where Σ
A is the total peak area of JCO fatty acid methyl esters with carbon numbers C16–20 with degree of unsaturation of 0–2;
AIS is the internal standard peak area;
CIS is the concentration of the internal standard solution in mg ml
−1;
VIS is the volume of the internal standard solution used in ml;
m is the mass of the sample in mg. Each experiment was conducted in triplicate and the data reported as mean ± standard deviation.
Catalyst stability. To evaluate the stability of the CaO–CeO2 catalysts, a leachate test was conducted by mixing the catalyst (CaO–CeO2 catalysts with different Ca/Ce molar ratios) with methanol, under the same conditions as those used in the transesterification process without the presence of oil. After that, the catalyst was separated, and the remaining methanol solution was reacted with the necessary volume of fresh crude JCO. The leachate test was performed to evaluate the contribution of homogeneous catalysis originating from the leaching of active sites (Ca2+).In addition, the catalyst was recycled to study its reusability. After completion of each run, the used catalyst was separated from the reaction mixture and washed several times with methanol and n-heptane solvents simultaneously to remove the surface-bound glycerides, i.e. triglycerides (TG), diglycerides (DG) and monoglycerides (MG), from the catalyst. The resultant solid particles were treated at 900 °C for 3 h and used for further recycling studies.
Conclusions
The CaO–CeO2 mixed oxide catalysts prepared by a highly alkaline co-precipitation method were successfully used for transesterification of crude JCO to biodiesel. The activity and stability of CaO–CeO2 mixed oxide catalysts were greatly influenced by the synthesis pH and Ca/Ce molar ratio. Among these CaO–CeO2 catalysts, the CaCe1.00 (which comprised a Ca/Ce molar ratio of 1 and was calcined at 900 °C) catalyst has superior catalytic performance for transesterification as it showed the highest BET surface area and total basicity. Under the optimized conditions at 65 °C, a 4% catalyst dose with a 15:1 molar ratio of methanol to jatropha oil, the catalyst exhibited 95.57% biodiesel yield. The CaCe1.00 catalyst also revealed a low leaching of homogeneous catalytic species (i.e. Ca and Ce) into the reaction media. Lower amounts of Ca2+ and Ce4+ ions, around 3–4 ppm, showed that the dissolved metal species from calcium-based mixed oxides into the reaction medium are insignificant and lower than the standard limit according to biodiesel quality standards. In addition, the catalyst could be reused 4 times with good performance (>90% FAME yield). The bimetallic oxide system improved the heterogeneous catalytic stability remarkably, due to the defects induced by substitution of Ca ions for Ce ions on the surface. The result clearly suggests that the CaCe1.00 catalyst is very stable and durable during the transesterification reaction and contamination of the catalyst component in the biodiesel product is no longer a problem for the long-term use of this catalyst.
References
- M. K. Lam, K. T. Lee and A. R. Mohamed, Biotechnol. Adv., 2010, 28(4), 500 CrossRef CAS PubMed.
- J. C. Juan, D. A. Kartika, T. Y. Wu and Y. H. Taufiq-Yap, Bioresour. Technol., 2010, 102, 452 CrossRef PubMed.
- Y. H. Taufiq-Yap, S. Sivasangar and A. Salmiaton, Energy, 2012, 47(1), 158 CrossRef CAS PubMed.
- H. T. Tan, K. T. Lee and A. R. Mohamed, Bioresour. Technol., 2010, 101, 5719 CrossRef CAS PubMed.
- A. Islam, Y. H. Taufiq-Yap, C. M. Chu, E. S. Chan and P. Ravindra, Process Saf. Environ. Prot., 2013, 91, 131 CrossRef CAS PubMed.
- F. Muhammad, R. Anita and S. Duvvuri, J. Cleaner Prod., 2013, 59, 131 CrossRef PubMed.
- H. V. Lee, R. Yunus, J. C. Juan and Y. H. Taufiq-Yap, Fuel Process. Technol., 2011, 92, 2420 CrossRef CAS PubMed.
- Y. H. Taufiq-Yap, S. H. Teo, U. Rashid, A. Islam and M. Z. Hussien, Energy Convers. Manage., 2014 DOI:10.1016/j.enconman.2013.12.075.
- M. M. Gui, K. T. Lee and S. Bhatia, Energy, 2008, 33, 1646 CrossRef CAS PubMed.
- K. E. Abebe, K. Yohannes and Z. Rolando, Energy, 2011, 36, 2693 CrossRef PubMed.
- P. D. Patil, V. Gnaneswar and S. G. Deng, Ind. Eng. Chem. Res., 2009, 48, 10850 CrossRef CAS.
- S. Tamalampudi, M. R. Talukder, S. Hama, T. Numata, A. Kondo and H. Fukuda, Biochem. Eng. J., 2008, 39, 185 CrossRef CAS PubMed.
- A. P. Soares Dias, J. Bernardo, P. Felizardo and M. J. Neiva Correia, Energy, 2012, 41, 344 CrossRef CAS PubMed.
- B. Margandan, V. Mari and N. G. Andrews, Bull. Korean Chem. Soc., 2013, 34, 3059 CrossRef.
- H. Zhu, Z. Wu, Y. Chen, P. Zhang, S. Duan, X. Liu and Z. Mao, Chin. J. Catal., 2006, 27, 391 CrossRef CAS.
- W. Thitsartarn and S. Kawi, Green Chem., 2011, 13, 3423 RSC.
- M. H. Kim, D. M. Craig, S. L. Yan, O. Steven Salley and K. Y. Simon Ng, Green Chem., 2011, 13, 334 RSC.
- S. A. Raja, D. S. Robinson smart and C. L. Robert Lee, Res. J. Chem. Sci., 2011, 1(1), 81 Search PubMed.
- Bionas biofuels: the next generation sustainable energy, Properties of jatropha oil, 2014, http://www.biofuelbionas.com/fuel.html.
- T. Laurianne, M. T. Ta, D. Thierry, K. Konstantin, H. Valérie, S. Cyriaque, A. Caroline, P. N. Ivan, P. Alain, V. Olivier and J. P. Blondeau, Mater. Res. Bull., 2010, 45, 527 CrossRef PubMed.
- A. R. West, Solid State Chemistry and Its Applications, Wiley India Pvt. Ltd, 2007 Search PubMed.
- J. K. Jasmine and A. N. Samson, J. Ceram. Process. Res., 2011, 12(1), 74 Search PubMed.
- S. Samantary, D. K. Pradhan, G. Hota and B. G. Mishra, J. Chem. Eng., 2012, 193–194, 1 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.