
A nanostructured CeO2 promoted Pd/α-alumina diethyl oxalate catalyst with high activity and stability†
Erlei Jina,
Leilei Hea,
Yulong zhangb,
Anthony R. Richarda and
Maohong Fan*ac
aDepartment of Chemical and Petroleum Engineering, University of Wyoming, Laramie, WY 82071, USA. E-mail: mfan@uwyo.edu
bWestern Research Institute, Laramie, WY 82070, USA
cSchool of Energy Resources, University of Wyoming, Laramie, WY 82071, USA
First published on 24th September 2014
Abstract
A Pd/α-Al2O3 nanocatalyst was synthesized and investigated as a catalyst for CO oxidative coupling to diethyl oxalate and CeO2 was used as a promoter. With the highest activity and stability found so far, great CO conversion and diethyl oxalate selectivity were achieved due to the addition of CeO2.
Ethylene glycol (EG) is a crucial raw chemical with a global demand of around 25 million tons each year, which is mostly produced through traditional petrochemical technology.1,2 However, the cost of this production is relatively high due to the continuously increasing price of natural gas and crude oil, and dwindling sources of petroleum. Furthermore, strong acids or alkalis such as sulphuric acid or sodium hydroxide have to be used during the traditional method, which causes severe corrosion to the equipment and environmental problems.3 Therefore, a green route which is independent of petroleum while achieves a high yield of EG is in demand and of great significance.
Coal is the most abundant energy reserve in the world that some people like because of their needs while others hate due to the various emissions resulting from its combustion.4 To reduce CO2 emission and produce high-value fuels and chemicals from coal, coal gasification and liquefaction technologies have attracted increasing interest during the past few decades.5–8 Coal to ethylene glycol, as a potentially green and economic coal liquefaction technology, has been attracting extensive attention in both academic and business circles in the past decades.9–12 Although it is challenging to achieve high industrial production levels, due primarily to achieving good performance of the catalysts, this technology has been scaled-up to industrial levels of production in China and Europe. Until now, China has leaded the word in this area and has successfully built the world's first annual 200 thousand ton coal to ethylene glycol production plant in 2009.13
Syngas to ethylene glycol process contains several steps. The step of CO oxidative coupling to di-alkyl oxalate is the critical one since di-alkyl oxalate is needed for production of EG with hydrogenation. Two major chemical reactions are involved with the overall CO oxidative coupling step, CO coupling and RONO regeneration, as shown in R1 and R2 respectively. R in both R1 and R2 could be methyl, ethyl or butyl groups. R1 needs catalyst while R2 does not. Esterification between oxalic acid and alcohol has been employed as a traditional way of synthesizing oxalic ester. However, this method has several problems, such as severe pollution, high energy consumption and high capital costs. Therefore, oxidative coupling CO with alkyl nitrite for formation of oxalic ester is gaining increasing interest.3,14–20
| 2CO + 2RONO → (COOR)2 + 2NO | (R1) |
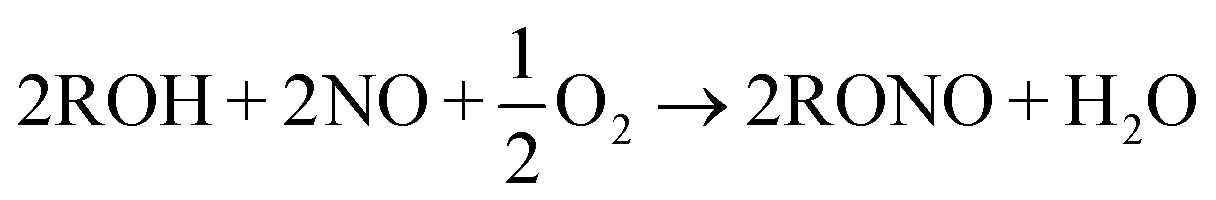 | (R2) |
Various supported palladium catalysts for gas-phase synthesis of dimethyl oxalate (DMO) or diethyl oxalate (DEO) have been investigated, and the results have demonstrated that higher conversion and selectivity are realized on Pd/α-Al2O3 compared to Pd on active carbon or γ-Al2O3.21,22 However, the relatively high Pd loading (around 2 wt%) is always an issue for industrial application of CO oxidative coupling to DMO which greatly increases the cost of production. Therefore, the design of low Pd loaded catalysts with high performance is important to industry. A Pd/α-Al2O3 nanocatalyst with ultra-low Pd loading that exhibits high activity and stability for CO oxidative coupling to DMO was developed recently.23 This catalyst was prepared by a Cu2+ assisted in situ reduction method at room temperature, which significantly increased the dispersion and the specific surface area of active component Pd, and also decreased the ensemble size of Pd nanoparticles dispersed over the Pd/α-Al2O3. To further enhance the activity and stability of Pd/α-Al2O3, several metals such as Fe,24,25 Ni, and Ce were reported as promoters to enhance the dispersion of Pd on the support or decrease the Pd particles size.24–27 CeO2 was reported as a promoter for Pd/α-Al2O3 catalyst.28 However, the activity of the CeO2 promoted catalyst was only evaluated up to 100 min.
Although methyl nitrite has been maturely used, especially in China, for the industrial synthesis of DMO, it is controlled in the US due to its highly flammable, explosive, and toxic properties. Ethyl nitrite, however, is a safe and non-explosive alkyl nitrite that also can be used for CO oxidative coupling reaction.18,20,29–32 Therefore, to find a good catalyst with low Pd loading and high catalytic activity for CO oxidative coupling to DEO is of great significance. Herein, we report a Pd–CeO2 α-Al2O3 nanocatalyst with 0.8% Pd (wt%) loading and 0.2 wt% CeO2 as a catalyst for CO oxidative coupling to DEO. We present the preparation and characterization of two catalysts with and without CeO2 as a promoter. The comparison of catalytic activities between the two catalysts is discussed and the interaction among Pd, ceria and the support leading to the activity differences is also presented.
The textural characteristics of Pd–CeO2/α-Al2O3 catalyst were investigated by transmission electronic microscopy (TEM) (Fig. 1), scanning transmission electronic microscopy (STEM) and scanning electronic microscopy (SEM) (Fig. S5†). TEM images presented in Fig. 1a and c clearly show that the Pd nanoparticles of Pd–CeO2/α-Al2O3 are dispersed on the α-Al2O3 relatively uniformly while the dispersion of Pd nanoparticles of Pd/α-Al2O3 is poor. Moreover, the results in Fig. 1b and d show that the average Pd nanoparticles size of the Pd–CeO2/α-Al2O3 catalyst is 13.2 nm which is smaller than that of the Pd/α-Al2O3 catalyst (17.3 nm). The Pd nanoparticles size distribution of the Pd–CeO2/α-Al2O3 catalyst is narrower than that of the Pd/α-Al2O3 catalyst as well. CeO2 was difficult to detect by TEM which may be due to its low loading concentration. However, the red circles in Fig. S5a† indicate the dispersion of CeO2 on the α-Al2O3 support, which is confirmed by energy dispersive X-ray (EDX) spectra. The dispersion of CeO2 particles was not as good as Pd particles. Small portion of the added CeO2 aggregated into the large particles while most of it which played the promoter role existed in the form of small nanoparticles that hardly detectable with SEM or TEM. In summary of the results from TEM and SEM, it can be concluded that the promoter CeO2 not only promotes the dispersion of Pd on the support, but also decreases the nanoparticle size of Pd.
 | ||
| Fig. 1 TEM (a) and size distribution (b) of catalyst Pd/α-Al2O3; TEM (c) and size distribution (d) of catalyst Pd–CeO2/α-Al2O3. | ||
The two catalysts, Pd/α-Al2O3 and Pd–CeO2/α-Al2O3, were analyzed with X-ray photoelectron spectroscopy (XPS) (Pd 3d) before and after the reaction with CO and EN at 140 °C (Fig. S3†). Although there were small differences between Pd/α-Al2O3 and Pd–CeO2/α-Al2O3, the obtained Pd 3d3/2 and Pd 3d5/2 values for both Pd(0) and Pd(II) were consistent with the published literatures.33–35 In Fig. 2a and c, both the Pd 3d5/2 and Pd 3d3/2 of catalysts Pd/α-Al2O3 and Pd–CeO2/α-Al2O3 are around 335 and 340 eV, respectively, which indicates that the oxidation state of Pd in the catalysts is Pd(0). However, after reaction, two new peaks appeared in both of the catalysts (Fig. S3c and d†), which are assigned to Pd(II),35 indicating that some Pd(0) in the two catalysts was oxidized to Pd(II) by ethyl nitrite to form an intermediate, CH3CH2O–Pd(II)–OCH2CH3.13 The peaks area of the Pd(II) in Fig. S3d† is much bigger than the peaks area in Fig. S3b,† which indicates that more intermediate was generated on the surface of Pd–CeO2/α-Al2O3 catalyst, and therefore Pd–CeO2/α-Al2O3 may have higher catalytic activity with the addition of CeO2. Furthermore, the percentage of the Pd on both catalysts was calculated using the peaks area of the XPS and the Pd–CeO2/α-Al2O3 catalyst showed higher Pd concentration (0.92%) than that of the Pd/α-Al2O3 catalyst (0.81%), which suggests that the promoter CeO2 can also enhance the Pd loading concentration on the support.21 X-ray diffraction (XRD) (Fig. S4†) was also attempted to further confirm the XPS results. However, no detectable CeO2 or Pd peak was found which may be due to their low concentrations and the high dispersion of Pd.28
 | ||
| Fig. 2 Conversion of CO (blue lines) and EN (red lines) of CO oxidative coupling to DEO with different catalysts within 72 h (a) and DEO selectivity of CO oxidative coupling to DEO with different catalysts within 72 h (b). | ||
The catalytic performances of the two catalysts were evaluated under the same conditions. With the addition of CeO2, the conversion of CO and EN was increased from 39% to 65% and 64–92%, respectively (Fig. 2a), which is 50% more conversion for both of the reactants. The STY of DEO with Pd–CeO2/α-Al2O3 was also greatly increased, which is 60% higher than that of Pd/α-Al2O3 at 140 °C (Table 1). Meanwhile, the selectivity of DEO with these two catalysts was almost the same (92–95%). Since there was no catalytic activity found for the catalyst CeO2/α-Al2O3, the CeO2 must play an important role as a promoter and the interaction of CeO2 with Pd was responsible for the high activity and selectivity in CO oxidative coupling to DEO. Most of all, the catalytic activity of catalyst Pd–CeO2/α-Al2O3 can be maintained for at least 72 h (Fig. 2b), which lays a good foundation for a further long-term stability test.
| Catalysts | Pd content (wt%) | Ce content (wt%) | Conversionb (%) | Selectivity (%) | STY (g L−1 h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: 3.5 g of catalyst, 1200 h−1 of gas hourly space velocity (GHSV), reactants volume ratio CO–EN is 1.2. 0.1 MPa, 140 °C.b Conversion of CO. | |||||
| Pd/α-Al2O3 | 0.8 | — | 39 | 95 | 195 |
| Pd–CeO2/α-Al2O3 | 0.8 | 0.15 | 65 | 93 | 318 |
| CeO2/α-Al2O3 | — | 0.2 | — | — | — |
Fig. 3 illustrates the in situ DR-FTIR spectra for the reaction of CO with ethyl nitrite to DEO. The band at 1768 cm−1 is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the DEO product. It is obvious to note that the intensity of the band at 1768 cm−1 in the spectrum of Fig. 3b is stronger than that in the spectrum of Fig. 3a and from the integration results of the two peaks (Fig. S6†), the peak area of Pd–CeO2/α-Al2O3 catalyst is 20% bigger than that of the catalyst without CeO2, which demonstrates the superior catalytic activity of Pd–CeO2/α-Al2O3 relative to Pd/α-Al2O3, consistent with the results of catalytic activity evaluation, TEM, and XPS results.
O stretching vibrations of the DEO product. It is obvious to note that the intensity of the band at 1768 cm−1 in the spectrum of Fig. 3b is stronger than that in the spectrum of Fig. 3a and from the integration results of the two peaks (Fig. S6†), the peak area of Pd–CeO2/α-Al2O3 catalyst is 20% bigger than that of the catalyst without CeO2, which demonstrates the superior catalytic activity of Pd–CeO2/α-Al2O3 relative to Pd/α-Al2O3, consistent with the results of catalytic activity evaluation, TEM, and XPS results.
 | ||
| Fig. 3 In situ FTIR spectra for the CO oxidative coupling to DEO reaction with Pd/α-Al2O3 (a) and Pd–CeO2/α-Al2O3 (b). | ||
The effect of temperature on both EN and CO conversion where Pd–CeO2/Al2O3 was used as the catalyst for CO oxidative coupling to DEO reaction was also evaluated (Fig. S7†). The conversions of EN and CO increased by 20% when the temperature increased from 120 °C to 140 °C, while the selectivity of DEO had little change until the temperature reached to 180 °C. Both the CO conversion and DEO selectivity decreased at 180 °C due to the decomposition of the EN. Therefore, the future plan for this project is optimizing the Pd–CeO2/Al2O3 catalyst to achieve a lower reaction temperature while maintaining high DEO selectivity.
In summary, a low Pd loading Pd/Al2O3 nanocatalyst with 0.8% Pd (wt%) loading and an average Pd size of 13.2 nm was synthesized for CO oxidative coupling to DEO. After the introduction of 0.2 wt% CeO2, Pd–CeO2/Al2O3 catalyst showed remarkably higher catalytic activity compared with the catalyst without CeO2. The CO conversion was increased by 50% (from 39% to 62%) with the DEO selectivity higher than 90% when the CeO2 was used as a promoter and, importantly, the high activity and selectivity could be maintained up to 72 h without visible decrease. TEM results showed clearly that CeO2 not only improved the dispersion of palladium on the surface of the support but also decreased the palladium size as well, thus resulting in the excellent catalytic activity. In consideration of the facile synthesis and low Pd loading of this catalyst as well as the risky factors of methyl nitrite, this highly efficient and stable nanocatalyst may have a promising industrial application, especially in the US, of coal to ethylene glycol.
Acknowledgements
This research was supported by the Department of Energy and Wyoming Clean Coal Program.Notes and references
- W. Kotowski, J. Freiberg, W. Spisak and S. Zamorowskabiernacik, Przem. Chem., 1989, 68, 73–76 CAS.
- D. F. Othmer and M. S. Thakar, Ind. Eng. Chem., 1958, 50, 1235–1244 CrossRef CAS.
- T. J. Zhao, D. Chen, Y. C. Dai, W. K. Yuan and A. Holmen, Ind. Eng. Chem. Res., 2004, 43, 4595–4601 CrossRef CAS.
- E. Jin, Y. Zhang, L. He, H. G. Harris, B. Teng and M. Fan, Appl. Catal., A, 2014, 476, 158–174 CrossRef CAS PubMed.
- F. Zhang, D. Xu, Y. Wang, X. Guo, L. Xu and M. Fan, Appl. Energy, 2014, 130, 1–6 CrossRef CAS PubMed.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS PubMed.
- A. Kumar, D. D. Jones and M. A. Hanna, Energies, 2009, 2, 556–581 CrossRef CAS PubMed.
- M. E. Dry, J. Am. Chem. Soc., 2000, 219, U254–U254 Search PubMed.
- H. Y. Song, R. H. Jin, M. R. Kang and J. Chen, Chin. J. Catal., 2013, 34, 1035–1050 CrossRef CAS.
- Q. L. Chen, W. M. Yang and J. W. Teng, Chin. J. Catal., 2013, 34, 217–224 CAS.
- J. S. Bae, I. S. Hwang, Y. J. Kweon, Y. C. Choi, S. J. Park, H. J. Kim, H. Jung and C. Han, Korean J. Chem. Eng., 2012, 29, 868–875 CrossRef CAS PubMed.
- F. X. Li and L. S. Fan, Energy Environ. Sci., 2008, 1, 248–267 CAS.
- Z. N. Xu, J. Sun, C. S. Lin, X. M. Jiang, Q. S. Chen, S. Y. Peng, M. S. Wang and G. C. Guo, ACS Catal., 2013, 3, 118–122 CrossRef CAS.
- F. D. Meng, G. X. Xu, R. Q. Guo, H. F. Yan and M. Q. Chen, Chem. Eng. Process., 2004, 43, 785–790 CrossRef CAS.
- B. Sadeghi and S. Ghamami, Chem. Eng. Commun., 2013, 200, 178–184 CrossRef CAS.
- C. W. Jiang, Z. W. Zheng, Y. P. Zhu and Z. H. Luo, Chem. Eng. Res. Des., 2012, 90, 915–925 CrossRef CAS PubMed.
- X. C. Gao, Y. J. Zhao, S. P. Wang, Y. L. Yin, B. W. Wang and X. B. Ma, Chem. Eng. Sci., 2011, 66, 3513–3522 CrossRef CAS PubMed.
- Z. H. Gao, C. Q. Hu, Z. H. Li, F. He and G. H. Xu, Chin. J. Catal., 2004, 25, 205–209 CAS.
- Z. H. Gao, Q. Wu, F. He, Z. H. Li and G. H. Xu, Chin. J. Catal., 2002, 23, 95–98 CAS.
- Q. Wu, Z. H. Gao, F. He, Z. H. Li and G. H. Xu, Chin. J. Catal., 2003, 24, 289–293 CAS.
- G. H. Xu, Y. C. Li, Z. H. Li and H. J. Wang, Ind. Eng. Chem. Res., 1995, 34, 2371–2378 CrossRef CAS.
- Q. Lin, X. G. Zhao, W. Bi and W. D. Xiao, Chin. J. Catal., 2006, 27, 911–915 CAS.
- S. Y. Peng, Z. N. Xu, Q. S. Chen, Y. M. Chen, J. Sun, Z. Q. Wang, M. S. Wang and G. C. Guo, Chem. Commun., 2013, 49, 5718–5720 RSC.
- X. Gao, Y. P. Zhu and Z. H. Luo, Chem. Eng. Sci., 2011, 66, 6028–6038 CrossRef CAS PubMed.
- Z. H. Gao, Z. C. Liu, F. He and G. H. Xu, J. Mol. Catal. A: Chem., 2005, 235, 143–149 CrossRef CAS PubMed.
- Q. Lin, Y. Ji, Z. D. Jiang and W. D. Xiao, Ind. Eng. Chem. Res., 2007, 46, 7950–7954 CrossRef CAS.
- Y. Yamamoto, T. Matsuzaki, S. Tanaka, K. Nishihira, K. Ohdan, A. Nakamura and Y. Okamoto, J. Chem. Soc., Faraday Trans., 1997, 93, 3721–3727 RSC.
- X. G. Zhao, Q. Lin and W. D. Xiao, Appl. Catal., A, 2005, 284, 253–257 CrossRef CAS PubMed.
- G. L. Zhuo and X. Z. Jiang, Chin. J. Catal., 2003, 24, 509–512 CAS.
- F. D. Meng, G. H. Xu and Q. R. Guo, J. Mol. Catal. A: Chem., 2003, 201, 283–288 CrossRef CAS.
- Z. H. Li, Y. Song, P. Du, X. B. Ma, B. W. Wang and G. H. Xu, React. Kinet. Catal. Lett., 2001, 73, 135–142 CrossRef CAS.
- F. Meng, G. Xu, B. Wang and X. Ma, J. Nat. Gas Chem., 2002, 11, 57–62 CAS.
- A. Tressaud, S. Khairoun, H. Touhara and N. Watanabe, Z. Anorg. Allg. Chem., 1986, 541, 291–299 CrossRef.
- C. J. Jenks, S. L. Chang, J. W. Anderegg, P. A. Thiel and D. W. Lynch, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 6301–6306 CrossRef CAS.
- W. E. Moddeman, W. C. Bowling, D. C. Carter and D. R. Grove, Surf. Interface Anal., 1988, 11, 317–326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08170f |
| This journal is © The Royal Society of Chemistry 2014 |