DOI:
10.1039/C4RA07533A
(Paper)
RSC Adv., 2014,
4, 49386-49394
Tunable 3D hierarchical graphene–BiOI nanoarchitectures: their in situ preparation, and highly improved photocatalytic performance and photoelectrochemical properties under visible light irradiation†
Received
24th July 2014
, Accepted 16th September 2014
First published on 17th September 2014
Abstract
Novel 3D hierarchical graphene–BiOI (GR–BiOI) nanoarchitectures have been successfully fabricated via an in situ self-assembly approach for the first time. More attractively, the hierarchical nanoarchitectures can be adjusted by simply controlling the amount of graphene oxide, which determines the improved level of photocatalytic performance. Photochemical measurements reveal that the as-obtained 5% GR–BiOI composite exhibits the most significantly enhanced photocatalytic activities for the degradation of Rhodamine B (RhB) and photocurrent (PC) generation under visible light irradiation (λ > 420 nm). This remarkably improved photocatalytic performance of GR–BiOI could be attributed to the well-established interfacial interaction between graphene and BiOI, which can greatly facilitate the separation and easy transfer of photogenerated electrons and holes to generate abundant ˙O2− and ˙OH active species with powerful oxidability. This was verified by the photoluminescence (PL) spectra, electrochemical impedance spectra (EIS), active species trapping, and ˙O2− and ˙OH quantification experiments. Our work provides a new strategy for the construction of hierarchical nanoarchitectures of high-performance composite photocatalysts and paves an alternative way to the design and synthesis of graphene-based composites for special applications.
1. Introduction
Over the next few decades, semiconductor photocatalysis is expected to prove its promise as a green technology for environmental remediation and energy conversion.1–3 As a new type of photocatalytic material, bismuth compounds, including BiVO4,4 Bi2MoO6,5 Bi2WO6,6 Bi2SiO5,7 BiPO4,8 BiOX (X = Cl, Br, I),9,10 and other new Bi-based photocatalysts11–15 have attracted considerable attention for their promising practical industrial applications, due to their high activity, high stability, and nontoxicity. Generally, Bi-based oxide semiconductors possess the hybridized valence band (VB) of O 2p and Bi 6s, which can effectively narrow the band gap and make them harvest more light with long wavelengths.16
BiOX (X = Cl, Br, I), with the simplest Sillén structure featuring its active (Bi2O2)2+ layers and intergrown single halogen layers, exhibits excellent photo-oxidation ability and an interesting structure–property relationship.9,10 In particular, BiOI with a narrow band gap (Eg = 1.72–1.92 eV), responding to most visible light, is a very valuable photocatalyst for utilizing solar energy.17 Nevertheless, there are also some drawbacks in the use of individual BiOI, e.g., high recombination of photoinduced electron–hole pairs. Therefore, some modifications have been made to increase the lifetime of the photogenerated charge carriers of BiOI and to obtain a more satisfying photocatalytic performance. These methods mainly include BiOI self doping,18 and BiOI-based heterojunctions, such as TiO2/BiOI,19 ZnO/BiOI,20 BiPO4/BiOI,21 Bi2O3/BiOI,22 BiOCl/BiOI,23 and BiOBr/BiOI.24,25 On the other hand, a two-dimensional (2D) hybridized semiconductor graphene (GR) with a conjugative π structure material has received ever-increasing attention for its fascinating structural and electronic properties.26 Owing to its excellent conductivity, GR has been proven to be effective for reducing the recombination of photoinduced charge carriers and enhancing the photocatalytic activity. Graphene-based composites, including TiO2/GR,27 ZnO/GR,28 ZnWO4/GR,29 BiOCl/GR,30 and BiOBr/GR,31,32 all exhibit obvious enhancements of photocatalytic activity compared to bare samples. Nevertheless, most of the above GR-based composites were synthesized by the solution-mixing method, which provides much less intimate interfacial contact than the approaches of in situ crystallization and ex situ hybridization.33
Currently, fabrication of a high-performance photocatalyst system via a facile and efficient route has become the focus of new photocatalytic material design. In particular, it is difficult to achieve the controllable synthesis of 3D hierarchical nanoarchitectures with tunability. Conventional methods suffer from high temperature and pressure, long reaction times, the need for special capping agents (like surfactants), and high cost. Thus, a simple and efficient approach to fabricate a high-performance photocatalyst system with 3D hierarchical nanoarchitectures is challenging but highly desirable.
Herein, we demonstrate for the first time a facile, fast, and efficient ethylene glycol (EG)-assisted approach for preparing hierarchical GR–BiOI nanoarchitectures. It is interesting to find that the assembly mode of BiOI flowers on GR varies with changing the GR content. The photochemical properties of GR–BiOI composites were demonstrated by the photodecomposition of rhodamine B (RhB) and by photocurrent (PC) measurements under visible light irradiation (λ > 420 nm). In particular, the possible mechanisms for the remarkably improved photocatalytic performance were systematically investigated by photoluminescence (PL) emission spectra, electrochemical impedance spectra (EIS), active species trapping, and ˙O2− and ˙OH quantification experiments.
2. Experimental section
2.1 Synthesis
All chemicals were analytical reagent grade and were used without further purification. Graphene oxide (GO) was synthesized from natural graphite powders by a modified Hummers' method.34
GR–BiOI composites were synthesized by an in situ crystallization method, as illustrated in Scheme 1. Different amounts of as-prepared GO and 1 mmol of Bi(NO3)3·5H2O were ultrasonically dispersed into 20 mL ethylene glycol. 1 mmol KI was dissolved in 20 mL deionized water, and then added dropwise into the above GO and Bi(NO3)3·5H2O suspension under strong magnetic stirring. Subsequently, the suspension was stirred for 10 min at room temperature. Afterward, the suspension was transferred into a 100 mL Teflon-lined stainless autoclave and heated at 120 °C for 10 min to reduce GO. After cooling, the products were filtered, washed with deionized water and ethanol several times, and dried at 60 °C for 12 h. The mass fractions of GO 1%, 3%, 5%, and 7% were donated as 1%, 3%, 5%, and 7% GR–BiOI, respectively. For reference, pure BiOI was prepared under the same conditions without adding GO.
 |
| | Scheme 1 Schematic illustration of the preparation of the GR–BiOI nanocomposites. | |
2.2 Characterization
A D8 Advance X-ray diffractometer (Bruker AXS, Germany) with Cu Kα radiation was employed to analyse the crystal structures of the obtained samples. The FTIR spectra of the as-obtained samples were recorded on a PerkinElmer Spectrum 100 FTIR spectrophotometer in the 4000–400 cm−1 range. The morphology and microstructure of the products were investigated by a S-4800 scanning electron microscope (SEM). Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained using a JEM-2100 electron microscopy (JEOL, Japan). Specific surface areas of the samples were characterized by the nitrogen adsorption BET method with a Micromeritics 3020 instrument. UV-vis diffuse reflectance spectra (DRS) were recorded from a PerkinElmer Lambda 35 UV-vis spectrometer. The spectra were collected at 200–1000 nm, referenced to BaSO4. Room temperature emission spectra were measured on a Hitachi F-4600 fluorescence spectrophotometer with a 150 W xenon lamp as the excitation lamp.
2.3 Photocatalytic activity experiment
The photocatalytic activity of GR–BiOI composites were evaluated by photodegradation of the organic dyes (rhodamine B) RhB under visible light with a xenon lamp coupled with 420 nm filters (λ > 420 nm, 1000 W). 0.05 g of as-prepared GR–BiOI or pure samples was dispersed into 50 mL of 3 × 10−5 mol L−1 RhB solution. First, the reaction mixture was stirred in the dark for 2 h to ensure the complete adsorption–desorption equilibrium of the organic dye by the catalyst. Afterward, about 3 mL of the suspension was taken at certain intervals under irradiation, and then separated through centrifugation. The concentration of RhB was determined by recording the absorption spectra of the residual dye by UV-vis spectroscopy based on the absorbance at 553 nm.
2.4 Active species trapping and ˙O2− and ˙OH− quantification experiments
For detecting the active species in the photocatalytic system, 1 mM ethylenediaminetetraacetic acid disodium salt (EDTA-2Na), 1 mM benzoquinone (BQ), and 1.0 mM iso-propanol (IPA) were added as the active species quenchers of the holes (h+), ˙O2−, and ˙OH, respectively.35,36 The method was similar to the former photocatalytic activity experiment. In order to determine the amount of ˙O2− generated from the potocatalytic system, 0.025 mM nitroblue tetrazolium (NBT), which exhibits an absorption maximum at 259 nm and which can react with ˙O2−, was utilized.37 The production of ˙O2− was quantitatively analyzed through recording the concentration of NBT with an UV-vis spectrophotometer. Moreover, to estimate the relative concentration of hydroxyl radicals (˙OH radicals), the terephthalic acid (TA) fluorescence method was utilized, because it can react readily with ˙OH to produce a highly fluorescent 2-hydroxyterephthalic acid.36 The production of ˙OH generated from BiOI and GR–BiOI was quantitatively analyzed by detecting the concentration of 2-hydroxyterephthalic acid on a Hitachi F-4600 fluorescence spectrophotometer at 425 nm with the excitation wavelength of 315 nm. The NBT transformation and TA-PL methods were also similar to the former photocatalytic activity test and the above active species trapping, but with NBT and TA replacing the RhB.
2.5 Photoelectrochemical measurements
The photoelectrochemical tests were carried out in a three-electrode system with a 0.1 M Na2SO4 electrolyte solution by using an electrochemical system (CHI-660B, China). Platinum wire and saturated calomel electrodes (SCE) were used as the counter electrode and reference electrode, respectively. GR–BiOI and pure BiOI film electrodes on ITO served as the working electrode. The intensity of light was 1 mW cm−2. The photocurrent (PC) generation and electrochemical impedance spectra (EIS) with visible light on and off were measured at 0.0 V. A 5 mV sinusoidal ac perturbation was applied to the electrode.
3. Results and discussion
3.1 Morphological structure
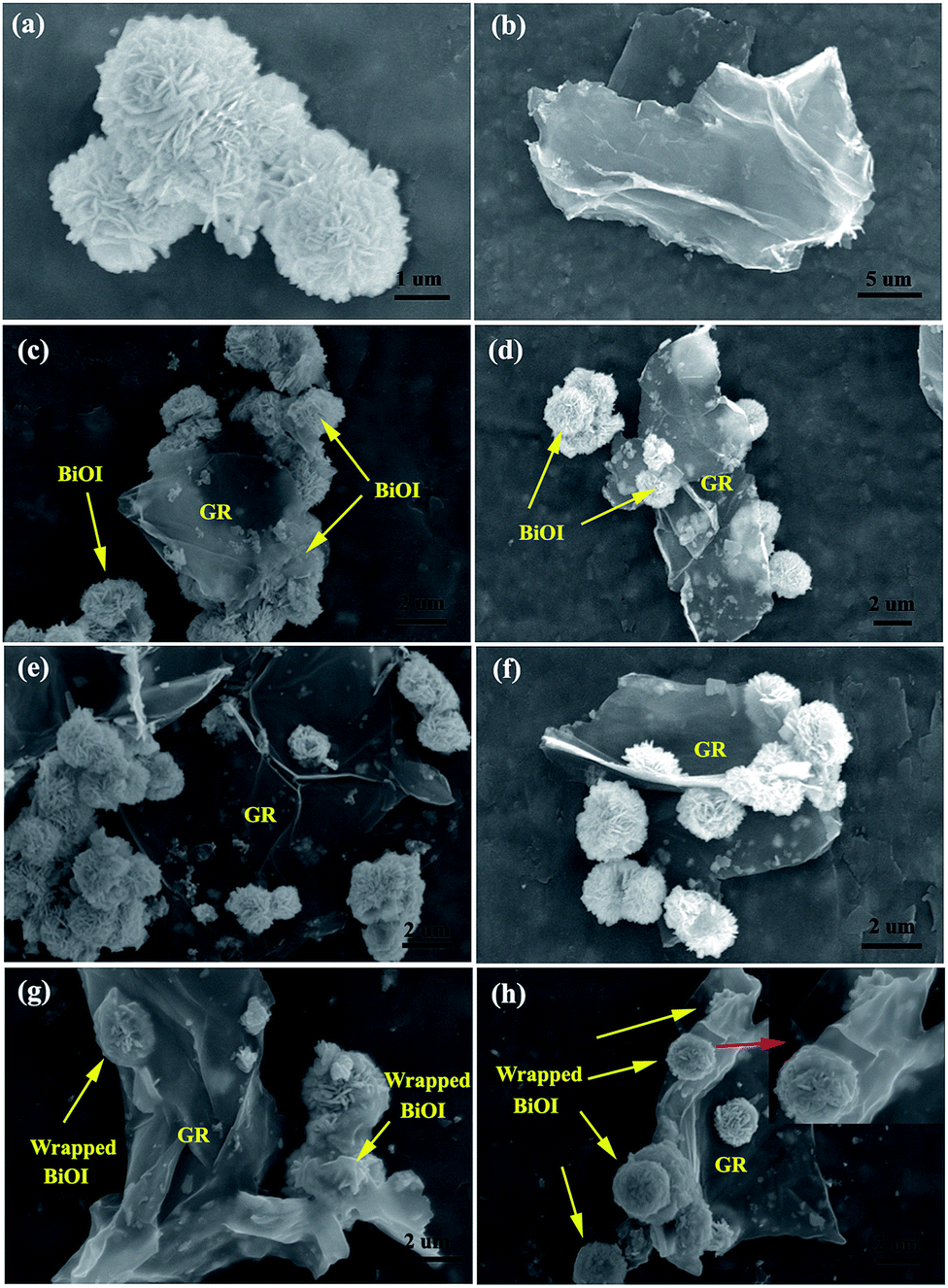
The morphologies and microstructures of BiOI, GR, and GR–BiOI hybrid composites were obtained by scanning electron microscopy (SEM). Fig. 1a and b show the SEM images of pure BiOI and GR, respectively. It is very important to notice that the as-prepared BiOI exhibits a 3D hierarchical flower-like morphology, which was assembled by numerous BiOI nanosheets. The diameters of the entire BiOI micro-flowers are about 2 μm. After graphene was introduced into the BiOI self-assembly system, the assembly mode of GR and BiOI were found to vary with their proportions. Fig. 1c and d present the 1% and 3% GR–BiOI composites, respectively, where it can be seen that only partial BiOI microspheres are equipped on the surface of the GR, as GR was low in content. When the content of GR increased to 5%, the flower-like BiOI products were all uniformly assembled on the surface of GR, as shown in Fig. 1e and f. As a result, perfect 3D self-assembled hierarchical graphene–BiOI nanoarchitectures were obtained. Moreover, another interesting phenomenon observed was that graphene-wrapped BiOI could also be obtained when the GR content reached 7%. As shown in Fig. 1g and h, most of BiOI micro-flowers were well-encapsulated in the GR sheets. It can hence be inferred that in situ synthesis is conducive to intimate interfacial contact between flower-like BiOI and GR.
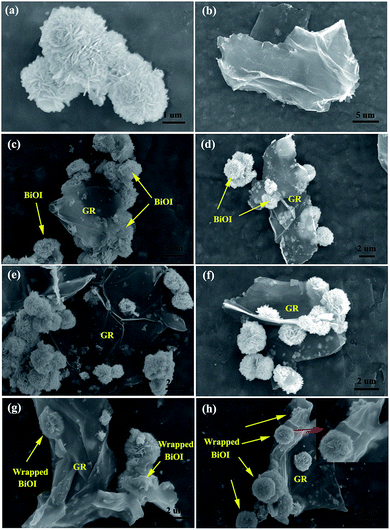 |
| | Fig. 1 SEM images of as-prepared (a) pure BiOI, (b) graphene, and GR–BiOI composites with GR amounts of (c) 1%, (d) 3%, (e and f) 5% and (g and h) 7%. | |
The pure BiOI, GR, and GR–BiOI composites were further characterized by TEM and HRTEM. As presented in Fig. 2a, the as-prepared BiOI micro-flowers are constructed by a large number of nanoplates, and the average thicknesses of these nanoplates are about 20–40 nm. Fig. 2c confirms the 3D hierarchical nanoarchitectures of the 5% GR–BiOI composites, in which BiOI micro-flowers are firmly assembled on the surface of GR. The contact interface between BiOI and GR can also be clearly seen. Compared with GO–BiOI (Fig. S1†), clearly the hydrothermal process did not alter the morphology of the nanocomposites. Fig. 2d is the TEM image of 7% GR–BiOI. It is clear to see that the BiOI micro-flower is completely wrapped by GR, which indicated that the self-assembly of the BiOI micro-flower took place in the space between the graphene layers with high graphene content. In Fig. 2e and f, the high-resolution transmission electron microscopy (HRTEM) image shows the characteristic lattice fringes of BiOI nanoplates in the composites. In order to obtain a clearer lattice fringe, fast Fourier transform (FFT) images (Fig. 2g and i) and reduced FFT images (Fig. 2h and j) were obtained. The interplanar instances resolved from the lattice fringe are 0.301 and 0.281 nm (as seen in Fig. 2h and j, respectively), which is consistent with the spacing of the corresponding (102) and (110) planes of the tetragonal phase of BiOI. This was also confirmed by the following XRD results.
 |
| | Fig. 2 TEM images of (a) pure BiOI, (b) graphene, (c) 5% GR–BiOI (d) 7% GR–BiOI, (e and f) HRTEM images, (g and i) FFT (fast Fourier transition) patterns, and (h and j) inverse FFT (fast Fourier transition) patterns of the lattice fringe of BiOI. | |
3.2 Crystal structure and phase analysis
Fig. 3 shows the crystal structure of BiOI along the a–c and a–b planes. Tetragonal phase BiOI possesses a Sillén structure with a typically layered configuration, which is composed of [Bi2O2]2+ layers with I− ions between them. Its space group is P4/nmm(129) with the cell parameters a = 4.0900 Å and c = 7.2100 Å. The corresponding (102) and (110) crystal faces are also highlighted.
 |
| | Fig. 3 Crystal structure of BiOI along the a–c and a–b planes (110 and 102 planes are highlighted). | |
The crystal structure and crystallization phases of the products were characterized by X-ray powder diffraction (XRD). Fig. 4a presents the XRD patterns of BiOI, graphene oxide (GO), and GR–BiOI composites. The pure BiOI displays a series of neat diffraction peaks with high purity and crystallinity, which can be indexed into the tetragonal phase of BiOI (JCPDS 10-0445). Similar with those in other graphene composites, the characteristic diffraction peaks of GO and GR were all invisible, due to the destruction of the regular stacking of GO by the reduction process under hydrothermal conditions and the low amount and intensity of graphene in the composite.35–37
 |
| | Fig. 4 (a) XRD patterns and (b) IR spectra of pure BiOI and GR–BiOI composites. | |
3.3 Chemical composition
Fig. 4b shows a comparison of the FTIR spectra of pure BiOI, GO, and GR–BiOI composites. The characteristic peaks of GR are verified by the bands at 1731 cm−1, 1727 cm−1, 1397 cm−1, 1227 cm−1, and 1063 cm−1, which refer to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of COOH groups, skeletal vibrations of unoxidized graphitic domains, the O–H deformation vibration of the C–OH groups, the epoxy C–OH stretching vibration, and C–O–C stretching vibrations, providing evidence for the presence of different types of oxygen-containing functional groups, including –COOH, –C–OH, –CO, and –C–O–C, on the graphite oxide.38 For pure BiOI, the peaks at 3440 cm−1 and 1630 cm−1 originate from the absorption of water or O–H groups.39 The IR peak observed at 1385 cm−1 results from the OH absorption of hydrogen-related defects.40 In the case of the GR–BiOI composites, the characteristic peaks of GO become weaker in intensity or even disappear, compared with that of the pure GO, demonstrating that the oxygenous functional groups attached to the basal GO layers have been removed after the hydrothermal treatment process. The peak intensity at 1385 cm−1 in GR–BiOI composites is also dramatically decreased, indicating that the defect sites of BiOI may be occupied by graphene. In addition, a new absorption band at 1582 cm−1, attributed to the skeletal vibration of the aromatic –CC– bonds of the graphene network, has been observed and it gradually increases in intensity with increasing the GR loading content.41 Compared with pure GO and BiOI, the 7% GR–BiOI shows a much broader peak around 1227 cm−1. This enhanced C–OH stretching interaction may indicate that the graphene and BiOI are chemically bonded.34
O stretching vibration of COOH groups, skeletal vibrations of unoxidized graphitic domains, the O–H deformation vibration of the C–OH groups, the epoxy C–OH stretching vibration, and C–O–C stretching vibrations, providing evidence for the presence of different types of oxygen-containing functional groups, including –COOH, –C–OH, –CO, and –C–O–C, on the graphite oxide.38 For pure BiOI, the peaks at 3440 cm−1 and 1630 cm−1 originate from the absorption of water or O–H groups.39 The IR peak observed at 1385 cm−1 results from the OH absorption of hydrogen-related defects.40 In the case of the GR–BiOI composites, the characteristic peaks of GO become weaker in intensity or even disappear, compared with that of the pure GO, demonstrating that the oxygenous functional groups attached to the basal GO layers have been removed after the hydrothermal treatment process. The peak intensity at 1385 cm−1 in GR–BiOI composites is also dramatically decreased, indicating that the defect sites of BiOI may be occupied by graphene. In addition, a new absorption band at 1582 cm−1, attributed to the skeletal vibration of the aromatic –CC– bonds of the graphene network, has been observed and it gradually increases in intensity with increasing the GR loading content.41 Compared with pure GO and BiOI, the 7% GR–BiOI shows a much broader peak around 1227 cm−1. This enhanced C–OH stretching interaction may indicate that the graphene and BiOI are chemically bonded.34
3.4 Optical property
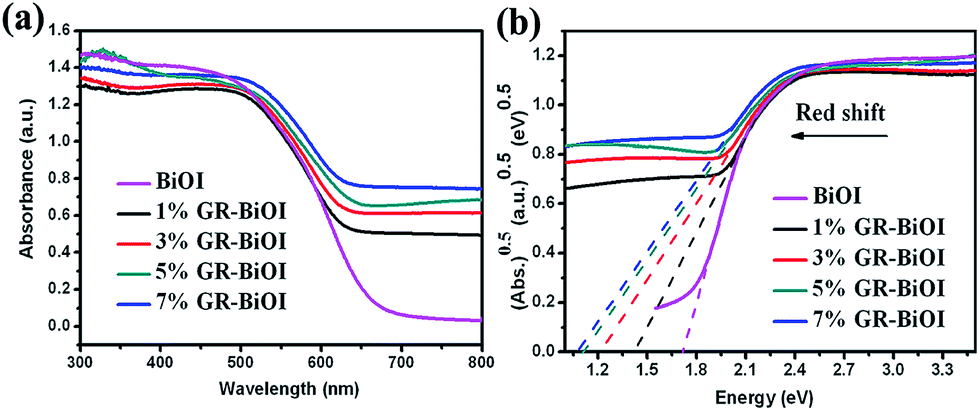
The ultraviolet-visible diffuse reflectance spectra of pure BiOI and GR–BiOI composites are shown in Fig. 5a. Compared to pure BiOI with an absorption edge of 690 nm, all the GR–BiOI composites exhibit an obvious enhancement in their visible-light absorption. In semiconductors, different optical transitions can result in different types of band gap. The band gap (Eg) can be determined by the Kubelka–Munk function:9,42,43where α, h, ν, Eg, and A are the absorption coefficient, Planck constant, light frequency, band gap energy, and a constant, respectively. Among these, n is determined by the type of optical transition of a semiconductor (n = 1 for direct transition and n = 4 for indirect transition). For BiOI, the n value is 4. The indirect band gaps estimated from the Kubelka–Munk function (Fig. 5b) are calculated to be 1.72, 1.42, 1.22, 1.11, and 1.07 for BiOI, 1%, 3%, 5%, and 7% GR–BiOI, respectively. The optical results indicate that the incorporation of GR into BiOI effectively narrows the band gap from the valence band to the conduction band.
 |
| | Fig. 5 (a) UV-vis diffuse reflectance spectra and (b) the band gap energies of pure BiOI, 1%, 3%, 5%, and 7% GR–BiOI composites. | |
3.5 Photocatalytic activity under visible light irradiation
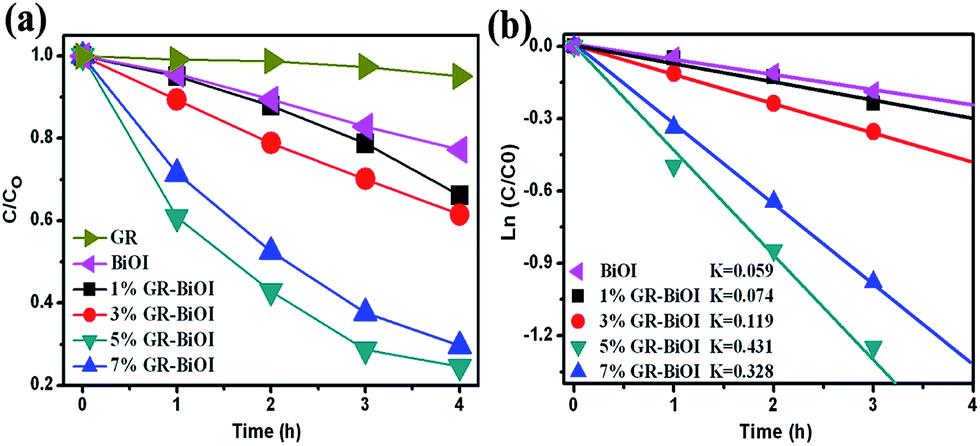
The photocatalytic performances of the as-prepared pure BiOI and GR–BiOI composite photocatalysts were evaluated by degradation of RhB molecules in aqueous solution under visible light irradiation (λ > 420 nm), as shown in Fig. 6. The photodegradation curves with error bars are also presented in Fig. S2.† As displayed in Fig. 6a, the pure BiOI shows relatively poor activity, with only 20% RhB degraded under visible light for 4 h. Comparatively, the degradation efficiencies were 34%, 39%, 76%, and 70% for 1% GR–BiOI, 3% GR–BiOI, 5% GR–BiOI, and 7% GR–BiOI, respectively within the same irradiation time. It can be seen that the photocatalytic activity gradually increased with increases in the loading content of graphene. Nevertheless, the photocatalytic activity decreased when the content of graphene reached 7%. As shown in Fig. 6a, the single GR almost has no photodegradation effect on RhB. With the further increase of GR content to 7%, most of BiOI microspheres were covered or wrapped by graphene, as shown in Fig. 1g and h, which leads to the largely reduced visible-light absorption of BiOI. Hence, the amount of photoexcited electron–hole pairs in BiOI in the 7% GR–BiOI composite also decreased. As a result, the 5% GR–BiOI possesses the most improved photocatalytic performance, with a RhB degradation efficiency of approximately 80%.
 |
| | Fig. 6 (a) Photocatalytic degradation curves of RhB over GR, pure BiOI, 1%, 3%, 5%, and 7% GR–BiOI composites under the irradiation of visible light (λ > 420 nm). (b) Apparent rate constants for the photodegradation of RhB. | |
In order to quantitatively understand the reaction kinetics of the photocatalytic degradation process of RhB, the apparent pseudo-first-order model according to the Langmuir–Hinshelwood (L–H) kinetics model was applied, as expressed by the following equation:12,13
where
kapp is the apparent pseudo-first-order rate constant (h
−1),
C0 is the initial RhB concentration (mol L
−1), and
C is the RhB concentration in aqueous solution at time
t (mol L
−1). It can be found that there is a good correlation to the pseudo-first-order model. As shown in
Fig. 6b, the apparent rate constant
k is 0.059 h
−1, 0.074 min
−1, 0.119 h
−1, 0.431 h
−1, and 0.328 h
−1 for the pure BiOI, 1% GR–BiOI, 3% GR–BiOI, 5% GR–BiOI, and 7% GR–BiOI, respectively. Thus, the optimized photocatalytic efficiency was obtained in 5% GR–BiOI, which is about 7.3 times higher than that of pure BiOI. As the specific surface area of a the photocatalyst is closely related to its photocatalytic activity, the BET surface areas of the pristine BiOI, 1% GR–BiOI, 3% GR–BiOI, 5% GR–BiOI, and 7% GR–BiOI composites were measured, and their values were found to be 8.34, 9.82, 13.64, 19.57, and 27.68 m
2 g
−1, respectively. Comparisons between our photocatalysts on the “same surface area” basis were also performed. The apparent rate constants per surface areas were 0.0071, 0.0075, 0.0087, 0.0220, and 0.0119 (h m
2 g
−1)
−1 for BiOI, 1% GR–BiOI, 3% GR–BiOI, 5% GR–BiOI, and 7% GR–BiOI, respectively. The 5% GR–BiOI still possesses the highest photocatalytic efficiency, which is approximately 3.1 times higher than that of pure BiOI.
3.6 Investigation of the photocatalytic mechanism
As photoluminescence (PL) emission spectra mainly result from the recombination of free carriers, therefore, PL spectra can be employed to survey the separation efficiency of the photogenerated electrons and holes in a semiconductor.12,44 Generally, a lower PL irradiance reflects the lower recombination of the charge carriers, which results in higher photocatalytic activity. Fig. 7 depicts the PL spectra of BiOI and GR–BiOI composites at room temperature. From the figure, the highest and lowest emission intensities were observed in pure BiOI and 5% GR–BiOI, respectively. Thus, the lowest recombination of photogenerated electrons and holes occurred in 5% GR–BiOI, which is in good agreement with the results from the photodegradation experiments.
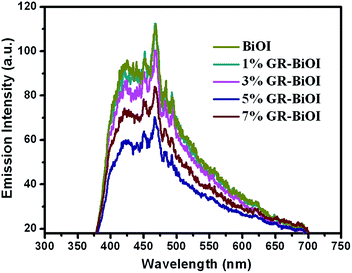 |
| | Fig. 7 PL spectra with pure BiOI and GR–BiOI composites. | |
The photocurrent–time responses, which may directly correlate with the recombination efficiency of the photogenerated carriers, were utilized to investigate the interfacial charge transfer dynamics of BiOI and GR–BiOI.45 As shown in Fig. 8a, photocurrent generation with good reproducibility for the two samples can be observed under visible light irradiation, indicating that the electrodes are stable and that the photoresponses are reversible. The much larger photocurrent density observed in GR–BiOI is 6 times that of pristine BiOI, which is in good agreement with the photodegradation tests. This result reflects the more efficient transfer of the photoinduced charge carriers of GR–BiOI composite, owing to the interfacial interaction between BiOI and graphene.
 |
| | Fig. 8 (a) Comparison of transient photocurrent responses, and (b) EIS Nyquist plots of pure BiOI and 5% GR–BiOI composite with light on/off cycles under visible-light irradiation (λ > 420 nm, [Na2SO4] = 0.1 M). | |
The migration and transfer processes of photogenerated electron–hole pairs in semiconductors can also be elucidated by the electrochemical impedance spectra (EIS) Nyquist plots.13 The smaller arc diameter in Nyquist plots implies a lower resistance of the interfacial charge transfer in the electrode–electrolyte interface region on the surface of electrode. As shown in Fig. 8b, the 5% GR–BiOI exhibits a significantly smaller arc diameter in EIS compared to BiOI, indicating a more effective separation of photogenerated electron–hole pairs and that a fast interface charge transfer occurred in the GR–BiOI composite.
To reveal the photocatalytic mechanism in the degradation process over the GR–BiOI composite, active species trapping experiments were performed. The sacrificial agents, including 1,4-benzoquinone (BQ), disodium ethylenediaminetetraacetate (EDTA), and iso-propanol (IPA), were used as the scavengers of the superoxide radical (˙O2−), hole (h+), and hydroxyl radical (˙OH), respectively.35,36 As shown in Fig. 9, 1 mM of EDTA (a hole scavenger) almost has no effect on the photocatalytic activity of GR–BiOI, indicating that very few of the holes (h+) were involved in the degradation of RhB. Conversely, the degradation rate for RhB significantly decreased with the addition of 1 mM of ˙O2− scavenger BQ, demonstrating the existence of abundant ˙O2− radical species. In addition, a clear decrease in the degradation rate was also observed when 1 mM of tertbutyl alcohol (TBA) was added as a scavenger for the ˙OH radical species, suggesting that the ˙OH pathways also have a crucial role in RhB photo-oxidation. This indicated that, in our photocatalyst systems, the holes are easier to transform into ˙OH radicals than to react with RhB or scavengers. Thus, ˙O2− and ˙OH are the two main active species in the degradation process of RhB over the GR–BiOI composite.
 |
| | Fig. 9 (a) Photodegradation of RhB over 5% GR–BiOI composite in the presence of different scavengers. | |
Accordingly, the possible photocatalytic mechanism of GR–BiOI composite was proposed, as depicted in Fig. 10. Under visible light irradiation, the separation of the electron and hole pairs occurred, and the electrons were injected into the conduction band of BiOI. Due to the strong interfacial interaction between BiOI micro-flowers and graphene, originating from the in situ crystallization, the photogenerated electrons are very easy to transfer from the conduction band of BiOI onto the surface of graphene. As is well known, graphene possesses a two-dimensional conjugated π structure and superior electrical conductivity. Thus, graphene can serve as an acceptor of the photogenerated electrons of BiOI in the GR–BiOI composites. These electrons further react with O2 adsorbed on the surface of graphene to generate abundant ˙O2−, which play a crucial role in the photo-oxidation of RhB. On the other hand, the holes left in the VB of BiOI can oxidize the OH− ions in solution to produce active ˙OH radicals with powerful oxidization. Therefore, the 3D self-assembled hierarchical graphene–BiOI nanoarchitectures could greatly facilitate the separation of photogenerated charge carriers and effectively suppress the recombination of electrons and holes, leaving more ˙O2− and ˙OH radicals as active species for the highly efficient photo-oxidation degradation of RhB. Furthermore, the incorporation of graphene can enhance the surface absorption ability of the photocatalyst system, which is also beneficial for the photocatalytic reaction, as it is essentially a two-step process of surface absorption and photo-oxidation.
 |
| | Fig. 10 Schematic diagram of GR–BiOI composite under visible light irradiation. | |
In order to further confirm the supposed photocatalytic mechanism and verify the contribution of ˙O2− and ˙OH radicals, quantification experiments of the ˙O2− and ˙OH production over GR–BiOI were performed. Fig. 11a presents the absorption spectra centered at 259 nm of NBT (detection agent of ˙O2− production) with 4 h visible light irradiation (λ > 420 nm) during the photocatalytic reaction. It can be found that the characteristic absorption peak at 259 nm of NBT obviously decreased with increases in the irradiation time, indicating that NBT was transformed by the abundant ˙O2− radicals generated from the GR–BiOI photocatalyst system.37 It was further revealed that in the GR–BiOI composite, the photogenerated electrons on BiOI could easily transfer to graphene, leading to even more photogenerated electrons reacting with O2 to produce ˙O2− and taking part in the decomposition of RhB. Moreover, the terephthalic acid photoluminescence probing technique (TA-PL) was also utilized to detect the ˙OH radicals, because TA can react with ˙OH radicals to form highly fluorescent 2-hydroxyterephthalic acid (TAOH). As revealed in Fig. 11b, the emission irradiance of TAOH dramatically increased with prolonging the irradiation time, which demonstrated that the ˙OH species play an important role in the photocatalytic reaction over the GR–BiOI composite. These results further confirmed that the self-assembled hierarchical GR/BiOI nanoarchitecture photocatalyst system can indeed facilitate the separation of photogenerated charge carriers greatly, and can thus generate a large number of ˙O2− and ˙OH species with powerful oxidability for highly efficient photocatalytic activity.
 |
| | Fig. 11 (a) Absorption spectra of NBT and (b) fluorescence spectra of a TAOH solution over 5% GR–BiOI composite with 4 h visible light irradiation (λ > 420 nm). | |
4. Conclusions
In summary, we successfully developed a very simple in situ self-assembly method using ethylene glycol as a chelating agent for the construction of tunable 3D hierarchical graphene–BiOI (GR–BiOI) nanoarchitectures for the first time. It was found that the BiOI micro-flowers can be not only assembled on the surface of graphene, but can also be totally wrapped by graphene sheets. All the GR–BiOI composites were found to exhibit much higher photocatalytic efficiency than pure BiOI under visible light irradiation (λ > 420 nm). The optimum photocatalytic activity, obtained in 5% GR–BiOI with a perfect surface-equipped configuration, is about 7.3 and 6 times higher than those of pure BiOI for the photodecomposition of RhB and for photocurrent generation, respectively. The significantly improved photocatalytic performance was demonstrated to be due to the high separation efficiency of the photogenerated charge carriers originating from the well-established interfacial interaction between graphene and BiOI, as revealed in the PL and EIS tests. The active species trapping and quantification experiments indicated that ˙O2− and ˙OH play crucial roles in the photo-oxidation degradation reaction, which further confirmed the high separation efficiency of the photogenerated electrons and holes in GR–BiOI. This work may serve as a guide for fabricating other high-performance Bi-based composite-photocatalysts with hierarchical nanoarchitectures and for designing graphene-based composites for special applications.
Acknowledgements
This work was supported by the National Natural Science Foundations of China (Grant no. 51302251), the Fundamental Research Funds for the Central Universities (2652013052), and the National High Technology Research and Development Program (863 Program 2012AA06A109) of China.
Notes and references
- L. Shang, T. Bian, B. Zhang, D. H. Zhang, L. Z. Wu, C. H. Tung, Y. D. Yin and T. R. Zhang, Angew. Chem., Int. Ed., 2014, 53, 250–254 CrossRef CAS PubMed.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2011, 112, 1555–1614 CrossRef PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- J. Bi, L. Wu, J. Li, Z. Li, X. Wang and X. Fu, Acta Mater., 2007, 55, 4699–4705 CrossRef CAS PubMed.
- C. Zhang and Y. F. Zhu, Chem. Mater., 2005, 17, 3537–3545 CrossRef CAS.
- R. G. Chen, J. H. Bi, L. Wu, W. J. Wang, Z. H. Li and X. Z. Fu, Inorg. Chem., 2009, 48, 9072–9076 CrossRef CAS PubMed.
- C. S. Pan and Y. F. Zhu, Environ. Sci. Technol., 2010, 44, 5570–5574 CrossRef CAS PubMed.
- X. Zhang, Z. H. Ai, F. L. Jia and L. Z. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CAS.
- J. Henle, P. Simon, A. Frenzel, S. Scholz and S. Kaskel, Chem. Mater., 2007, 19, 366–373 CrossRef CAS.
- X. Xiao, C. L. Xing, G. P. He, X. X. Zuo, J. M. Nan and L. S. Wang, Appl. Catal., B, 2014, 148, 154–163 CrossRef PubMed.
- H. W. Huang, S. B. Wang, N. Tian and Y. H. Zhang, RSC Adv., 2014, 4, 5561–5567 RSC.
- H. W. Huang, Y. He, Z. S. Lin, L. Kang and Y. H. Zhang, J. Phys. Chem. C, 2013, 117, 22986–22994 CAS.
- H. W. Huang, Y. He, R. He, Z. S. Lin, Y. H. Zhang and S. C. Wang, Inorg. Chem., 2014, 53, 8114–8119 CrossRef CAS PubMed.
- H. W. Huang, K. Liu, K. Chen, Y. L. Zhang, Y. H. Zhang and S. C. Wang, J. Phys. Chem. C, 2014, 118, 14379–14387 CAS.
- J. W. Tang, Z. G. Zou and J. H. Ye, J. Phys. Chem. C, 2007, 111, 12779–12785 CAS.
- X. Xiao and W. D. Zhang, J. Mater. Chem., 2010, 20, 5866–5870 RSC.
- X. Zhang and L. Z. Zhang, J. Phys. Chem. C, 2010, 114, 18198–18206 CAS.
- X. Zhang, L. Z. Zhang, T. F. Xie and D. J. Wang, J. Phys. Chem. C, 2009, 113, 7371–7378 CAS.
- J. Jiang, X. Zhang, P. B. Sun and L. Z. Zhang, J. Phys. Chem. C, 2011, 115, 20555–20564 CAS.
- J. Cao, B. Y. Xu, H. L. Lin and S. F. Chen, Chem.–Eur. J., 2013, 228, 482–488 CAS.
- Y. Y. Li, J. S. Wang, H. C. Yao, L. Y. Dang and Z. J. Li, Catal. Commun., 2011, 12, 660–664 CrossRef CAS PubMed.
- T. B. Li, G. Chen, C. Zhou, Z. Y. Shen, R. C. Jin and J. X. Sun, Dalton Trans., 2011, 40, 6751–6758 RSC.
- W. D. Wang, F. Q. Huang, X. P. Lin and J. H. Yang, Catal. Commun., 2008, 9, 8–12 CrossRef CAS PubMed.
- J. Cao, B. Y. Xu, B. D. Luo, H. L. Lin and S. F. Chen, Catal. Commun., 2011, 13, 63–68 CrossRef CAS PubMed.
- Y. Zhang, Y. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- Y. H. Zhang, Z. R. Tang, X. Z. Fu and Y. J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS PubMed.
- Q. Luo, X. Yu, B. Lei, H. Chen, D. Kuang and C. Su, J. Phys. Chem. C, 2012, 116, 8111–8117 CAS.
- X. J. Bai, L. Wang and Y. F. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS.
- F. D. Gao, D. W. Zeng, Q. W. Huang, S. Q. Tian and C. S. Xie, Phys. Chem. Chem. Phys., 2012, 14, 10572–10578 RSC.
- Z. H. Ai, W. K. Ho and S. C. Lee, J. Phys. Chem. C, 2011, 115, 25330–25337 CAS.
- X. M. Zhang, X. F. Chang, M. A. Gondal, B. Zhang, Y. S. Liu and G. B. Jia, Appl. Surf. Sci., 2012, 258, 7826–7832 CrossRef CAS PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- L. Ye, J. Liu, C. Gong, L. Tian, T. Peng and L. Zan, ACS Catal., 2012, 2, 1677–1683 CrossRef CAS.
- X. J. Bai, L. Wang and Y. F. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS.
- L. Q. Ye, J. Y. Liu, Z. Jiang, T. Y. Peng and L. Zan, Appl. Catal., B, 2013, 142, 1–7 Search PubMed.
- T. F. Yeh, J. M. Syu, C. Cheng, T. H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- Z. S. Li, T. Yu, Z. G. Zou and J. H. Ye, Appl. Phys. Lett., 2006, 88, 071917 CrossRef PubMed.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS PubMed.
- H. W. Huang, J. Y. Yao, Z. S. Lin, X. Y. Wang, R. He, W. J. Yao, N. X. Zhai and C. T. Chen, Angew. Chem., Int. Ed., 2011, 50, 9141 CrossRef CAS PubMed.
- H. W. Huang, L. J. Liu, S. F. Jin, W. J. Yao, Y. H. Zhang and C. T. Chen, J. Am. Chem. Soc., 2013, 135, 18319–18322 CrossRef CAS PubMed.
- X. J. Wang, Q. Wang, F. T. Li, W. Y. Yang, Y. Zhao, Y. J. Hao and S. J. Liu, Chem. Eng. J., 2013, 234, 361–371 CrossRef CAS PubMed.
- H. Kim, P. Borse, W. Choi and J. Lee, Angew. Chem., Int. Ed., 2005, 44, 4585–4589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07533a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.