
A highly efficient, enantioselective and recyclable mesoporous silica-based Mn(II) catalyst for asymmetric oxidation of thioanisole
Sohaila Alavia,
Hassan Hosseini-Monfared*a and
Pavlo Aleshkevychb
aDepartment of Chemistry, University of Zanjan 45195-313, Zanjan, Islamic Republic of Iran. E-mail: monfared@znu.ac.ir
bInstitute of Physics, Polish Academy of Sciences (PAN), Al. Lotnikow 32/46, PL-02-668 Warsaw, Poland. E-mail: pavloa@ifpan.edu.pl
First published on 19th September 2014
Abstract
The development of environmentally benign reactions is an important goal in synthetic organic chemistry and chemical engineering. However, catalytic enantioselective oxidations using transition-metal complexes are limited when the oxidant is hydrogen peroxide. A hydrazone based asymmetric ligand (H2L) was used to prepare a heterogeneous catalyst by the immobilization of its manganese complex, [{Mn(H2O)2Cl2}2(H2Btar)] (1), on mesoporous support SBA-15 via the post grafting method (H2L = 2,3-O-4-hydroxybenzhydrazidebenzylidene-D-tartrate). The heterogeneous asymmetric catalyst 1-SBA15 was characterized by elemental analysis; small angle X-ray diffraction (SAX), scanning electron microscopy (SEM), nitrogen sorption measurement, Fourier transform infrared spectroscopy and EPR spectroscopy. The asymmetric oxidation of thioanisole (PhSMe) was achieved with hydrogen peroxide in the presence of 1-SBA15 in excellent conversion and enantioselectivity (>99% ee). Compared to a homogeneous catalyst, the heterogenized catalyst is more stable and can be recycled four times without any significant loss of activity. Immobilization of complex 1 onto SBA-15 increased the selectivity toward sulfone in the oxidation.
Introduction
Due to the great importance of the chiral compounds for the total synthesis of natural products, pharmaceuticals and agricultural agents,1 the synthesis of enantiopure epoxide and sulfoxide building blocks has attracted special attention. Enantiopure epoxides are highly valuable chiral molecules, useful for the synthesis of various biologically active molecules.2 Chiral sulfoxides are an important class of compounds as chiral auxiliaries in asymmetric carbon–carbon bond forming reactions,3,4 as bioactive ingredients in the pharmaceutical industry5 and constitute chiral synthons in organic synthesis for the preparation of biologically active compounds.6 Therefore, design of simple and efficient chiral catalysts for an enantioselective epoxidation/or sulfoxidation7,8 is one of the core research activities in asymmetric catalysis8 and requires the preparation of readily accessible chiral metal complexes.9Enantioselective transformations have been extensively studied using homogeneous asymmetric catalysts for many years. However, these catalysts do not applied significantly in the industrial synthesis of fine chemicals. A central reason is that homogeneous asymmetric catalyst design requires relatively bulky ligands and catalyst re-use through recovery and recycle often causes problems. One mechanism to overcome this problem is to immobilize the asymmetric catalyst onto a support and the resulting heterogeneous asymmetric catalyst can, in principle, be readily re-used.10 Heterogeneous chiral catalysts which are friendly to the environment have the inherent advantages of the easier handling, separation and recovery from the reaction mixture over the homogeneous catalysts.11 Current research on preparation of heterogeneous chiral catalysts is very active and needed in pharmaceutical, food, and agricultural industries.12 The Jacobsen catalyst (Mn(III) salen complexes) is now widely used as enantioselective epoxidation catalyst in laboratory and industrial scale.13 Various approaches have been used for heterogenization of the chiral Mn(III) salen catalysts (anchoring the catalysts on a solid support,11,14–20 onto activated carbons,21 dendrimer,22 mesoporous silica17–19,23 and encapsulation in zeolite using ship-in-a-bottle methodology24) where the Mn(III) complexes were supported via axial/apical coordination or through covalent bonding of the ligand to the support. All of these approaches are interesting; however, the complex immobilization onto supports often requires complicated synthetic manipulations/or the structure modification of the homogeneous catalyst, which ultimately results in lower activity and enantioselectivity. In general, the heterogeneous Mn(III) salen catalysts led to the lower activities and/or enantioselectivities. Although manganese complexes of many chiral ligands (in particular, the Jacobsen catalyst) with variety of donor atoms are known today and used comprehensively for asymmetric catalytic reactions, there exists no report on the use of a chiral heterogeneous Mn(II)–tartrate catalyst. Very recently we have reported25 manganese(II) complexes containing chiral and achiral dicarboxylate ligands. In this context we are attracted to the chiral Mn(II)–tartrate catalyst synthesized by immobilization on mesoporous silica.
The incorporation of chiral catalyst on mesoporous silica materials for asymmetric catalysis has attracted a great deal of interest due to its tunable pore dimension, well-defined pore arrangement, relatively large specific surface area and pore volume.26 The large specific surface area and pore volume can enhance the loading amounts of the metal catalyst and increase its catalytic efficiency, while the tunable pore dimension and well-defined pore arrangement can avoid the aggregation of catalytic active species and maintain excellent stereocontrol performance. More importantly, these mesoporous silica-supported chiral catalysts do not swell or dissolve in common organic solvents, they are easy and reliable to be reused via simple precipitation or nanofiltration methods. Furthermore, they also exhibit superior thermal and mechanical stability in catalytic process, showing a potential application in industry.
Herein, we report the results of the catalytic potential of a Mn(II) complex by an easy to synthesis and cheap benzhydrazide-D-tartrate ligand immobilized onto the mesoporous silicate SBA-15, [{Mn(H2O)2Cl2}2(HBtar)]/SBA15 (H2Btar = 2,3-(−)-O-4-hydroxybenzhydrazidebenzylidene-D-tartrate), in asymmetric oxidation of thioanisole and alkenes with H2O2 as the oxidant. Katsuki and Sharpless reported in 1980, a highly enantioselective epoxidation of allylic alcohols using a titanium/tartrate/t-butyl hydroperoxide system.27 Kagan28 and Modena29 described enantioselective sulfoxidation with alkyl hydroperoxides using titanium tartrate catalysts. The selective oxidation of sulfides to sulfoxides has attracted much attention since 1980s and there are excellent reviews on this topic.30–35
Results and discussion
Enantiomerically pure (−)-diethyl-D-tartrate was synthesized by treatment of D-(−)-tartaric acid with ethanol and then its dihydroxyl groups protected by benzaldehyde in the presence of p-toluenesulfonic acid to produce diethyl-2,3-benzylidene-D-tartrate in high yield. Ligand H2Btar was obtained by the condensation reaction of diethyl-2,3-benzylidene-D-tartrate with 4-hydroxybenzhydrazide in good yield and purity (Scheme 1). Ligand H2Btar is potentially bischelating and could coordinate to two different metal centers by its symmetric two bidentate units. | ||
| Scheme 1 (A) The ligand H2Btar synthesis and (B) covalent modification of SBA-15 silica surface by silanization reaction with 3-(chloropropyl)-triethoxysilane and conversion to organofunctionalized SBA-15 silica with H2Btar followed by the formation of supported [{Mn(H2O)2Cl2}2(HBtar)] complex (1-SBA15): (a) toluene, reflux, 6 h; (b) H2Btar, ethanol, reflux, 3 h; (c) MnCl2·4H2O, ethanol, reflux, 3 h. | ||
The infrared spectra of the SBA-15, HBtar/SBA-15 and 1-SBA15 in the range 400–4000 cm−1 are shown in Fig. 1. The ν(OH) stretching vibrations observed in the 3600–3400 cm−1 region are attributed to the hydrogen-bonded silanol groups.36 The band at 1088 cm−1 is assigned to the asymmetric νas(Si–O–Si) vibrations and the band at 798 cm−1 is assigned to symmetric vibrations of (Si–O) SBA-15, while the band at 960 cm−1 is attributed to ν(Si–OH) vibrations. The bands due to 1-SBA15 are weak and masked by the Si–OH bands due to the low concentration of the former in 1-SBA15.
 | ||
| Fig. 1 FT-IR spectra of (a) SBA-15, (b) HBtar/SBA-15 and (c) 1-SBA15. | ||
The powder XRD patterns of pristine SBA-15 show a very intense peak assigned to reflection at (100) and two additional peaks with low intensities at (110) and (200) reflections, which can be indexed for a hexagonal unit cell (Fig. 2). The strongest peak for d100 = 9.4 nm appears in the region 2θ = 0.979°. It is observed that the intensities of peaks (110) and (200) decrease marginally with a little shift toward lower 2θ values after the attachment of complex [{Mn(H2O)2Cl2}2(H2Btar)] to functionalized SBA-15; suggesting that the structure of mesoporous SBA-15 does not collapse by immobilization.37 Some loss in the intensities of the peaks was observed after the attachment of complex revealing that silylation has indeed occurred inside the mesopores of SBA-15.38
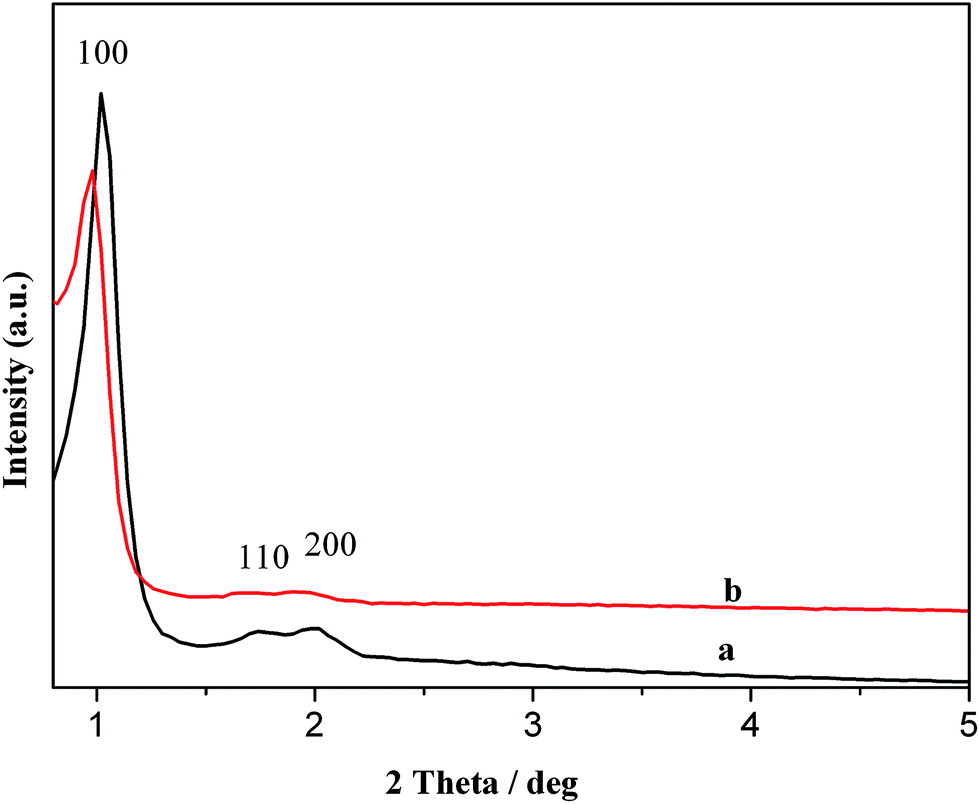 | ||
| Fig. 2 Small angle XRD patterns of (a) SBA-15 and (b) 1-SBA15. | ||
The process for preparing 1-SBA15 was monitored by N2 adsorption–desorption analysis and the structural parameters for these materials are presented in Table 1. N2 adsorption–desorption isotherm of 1-SBA15 sample shows Type IV isotherm, According to IUPAC classification, type IV isotherms relate to capillary condensation steps, characteristic of the mesoporous materials (20–500 Å),39–41 with completely reversible nature, uniformly sized mesopores. It is well known that the introduction of homogenous catalysts or metals on porous supports shows a decrease in its specific surface area and pore volume (Fig.3). The surface area of the used catalyst 1-SBA15 is almost in the same order of magnitude with that of the fresh catalyst.
| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore diameter (nm) |
|---|---|---|---|
| SBA15 | 730.38 | 0.8761 | 4.798 |
| SBA15-Cl | 535.68 | 0.7108 | 5.307 |
| Fresh 1-SBA15 | 533.71 | 0.7030 | 5.268 |
| Used 1-SBA15 | 543.05 | 0.6312 | 4.649 |
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherm of SBA-15, SBA15-Cl, fresh 1-SBA15 and used 1-SBA15 after catalysis. | ||
SEM images of SBA-15 and 1-SBA15 show the primary particles of SBA-15 present a curved cylinders form that remained unaffected on immobilization of [{Mn(H2O)2Cl2}2(H2Btar)] (1) complex thus confirming the retention of the physical structure of the siliceous material (Fig. 4). The curved cylinder-particles have a diameter of about 500–700 nm and length up to 1–2 μm, which is typical morphology for SBA-15 mesoporous materials.42,43 Furthermore, the complex immobilized SBA-15 (1-SBA15) is stable and preserved its morphology at the end of the catalytic oxidation reaction (see the next section), Fig. 4e and f.
 | ||
| Fig. 4 SEM images of (a and b) SBA-15 and (c and d) 1-SBA15 and (e and f) recovered 1-SBA15 after catalysis. | ||
The electronic spectra of [{Mn(H2O)2Cl2}2(H2Btar)] (1) and 1-SBA15 are shown in Fig. 4. Solid UV-Vis spectrum support the successful immobilization of the complex as the characteristic π → π* and n → π* bands at 244 and 279 nm of homogeneous complex 1 that they are present as broad bands for the immobilized complex (Fig. 5). For complex 1, no d–d bands are detected and this finding along with pale yellow color of the complex suggest the existence of manganese as Mn(II).
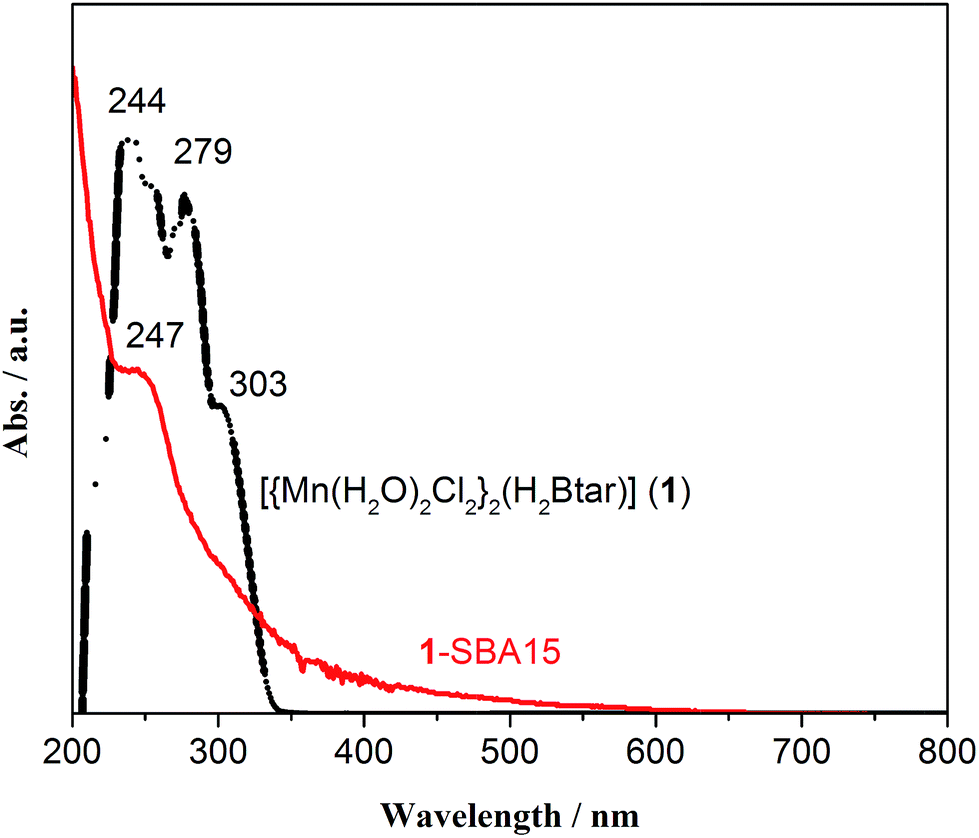 | ||
| Fig. 5 UV-Vis spectra of [{Mn(H2O)2Cl2}2(H2Btar)] (1) in CH3OH (crème color) and 1-SBA15 recorded after dispersion in Nujol. | ||
EPR studies
Fig. 6 shows the EPR spectra recorded for the powders of complex 1 and 1-SBA15 at liquid helium temperature. Since they were powders, the paramagnetic centers are here randomly orientated with respect to the magnetic field and the spectrum represents the envelope of all resonance lines summed over all possible orientations. As it is seen from Fig. 6, the spectrum of 1-SBA15 exhibits intensive sextet centered at g = 2.0. Additionally, a much less intensive resonance can be distinguished in low magnetic field region (Hres ∼ 1500 Oe). Such spectrum is proved to be associated with Mn2+ ions (3d5 electronic configuration) by many reports in the literature.44–46 It was shown for example in the review of Griscom44 and reference therein that in case of small axial distortion as to compare with Zeeman splitting (|D| < gμBH), the spectrum will giving rise to the predominant absorption at g-factor close to 2. In this case the observed sextet is the hyperfine splitting structure due to nuclear spin I = 5/2 of 55Mn ion within doublet characterized by electronic quantum numbers ms = +1/2, −1/2.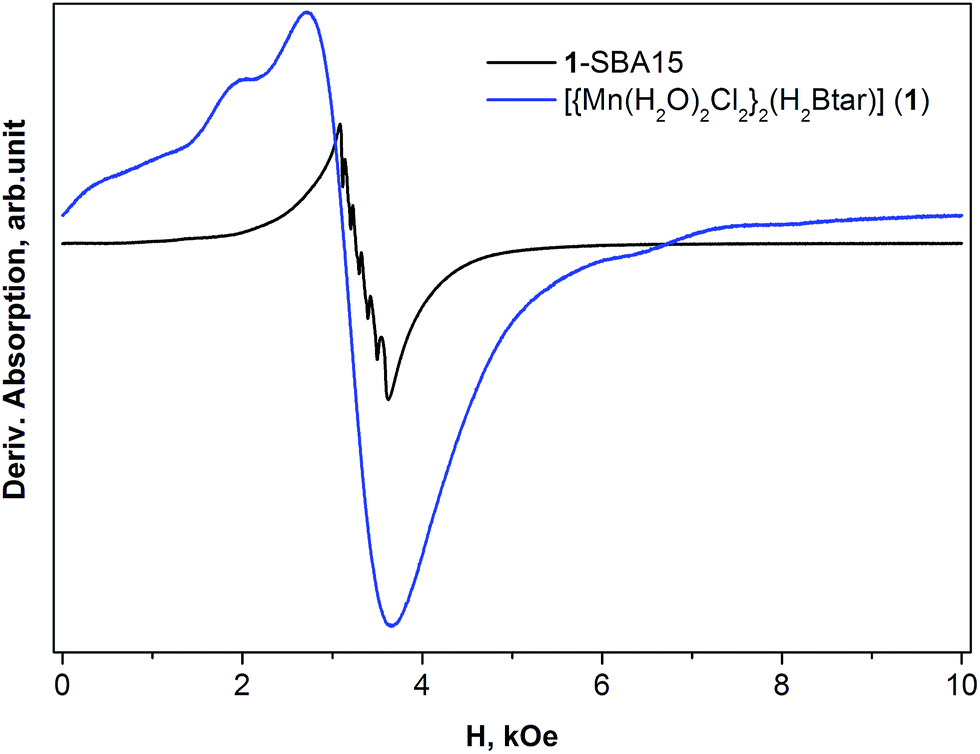 | ||
| Fig. 6 The EPR spectra for powders of complexes [{Mn(H2O)2Cl2}2(H2Btar)] (1) and 1-SBA15, recorded at T = 5 K. | ||
The spectrum of complex 1 is much intensive than that of 1-SBA15 and shows poorly resolved traces of fine structure located below (Hres = 400, 2000 Oe) and above (H = 6300 Oe) the main resonance line at H = 3100 Oe. This spectrum is also associated with Mn2+ ions. The difference between spectra of 1 and 1-SBA15 could be explained by different local environment of Mn in these complexes. Considering that the number of Mn ions per gram is the same in both 1-SBA15 and 1 samples the much weaker resonance in 1-SBA15 could be explained by fact that only part of Mn ions is in 2+ electronic state, contributing to the resonance absorption with the rest of Mn ions are in 3+ electronic state which is “silent” in the X-band EPR spectroscopy.
Catalytic activity
The oxidation of thioanisole with H2O2 catalyzed by 1-SBA15 was carried out in acetonitrile for 2–60 min by using 0.3 mol% of the catalyst, 1.0 mmol of substrate and 2.0 mmol of H2O2. The reactions were monitored by GC until completion. The results are summarized in Table 2. Besides sulfoxide, sulfone was also formed. The formation of sulfones is observed in various reactions as by-products.31 Efforts were first directed to optimize the solvent of the reaction in the catalysis with thioanisole as substrate. As is obvious from entries 1–4 in Table 2, in comparison with acetonitrile, both the substrate conversion and sulfoxide selectivity decrease when the reaction was carried out in chloroform, ethanol and methanol. Therefore, acetonitrile was used as solvent for all further studies.
|
|||||||
|---|---|---|---|---|---|---|---|
| No. | Catalyst/solvent | H2O2/sulfide molar ratio | Conv.b (%) | Sulfoxide selectivityb (%) | Sulfoxide ee (Confign)c | Temp. (°C) | TONd |
| a Reaction conditions: catalyst 1-SBA15 10 mg (contain 2.9 μmol Mn) or homogeneous 1 1.0 mg (contain 2.19 μmol), methyl phenyl sulfide (thioanisole) 1 mmol, solvent 3 mL, aqueous 30% H2O2 2.0 mmol, temperature 25 °C, reaction time 2 min.b Conversions and yields are based on the starting substrate.c ee, % enantiomeric excess (enantioselectivity) = (|R − S|/|R + S|) × 100. Enantioselectivity was determined by chiral GC-FID (ID-CYDEX-B column). The absolute configuration of the major isomer was determined by comparing the GC data with those observed for R-(+)-limonene.d TON: turnover number = number of moles of product formed per mole of the catalyst.e reaction time 60 min. | |||||||
| Solvent effect | |||||||
| 1 | 1-SBA15/CH3Cl | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
2 | 20 | 98 (R) | 25 | 14 |
| 2 | 1-SBA15/EtOH | 2:1 |
59 | 34 | 99 (R) | 25 | 407 |
| 3 | 1-SBA15/MeOH | 2:1 |
69 | 35 | 100 (R) | 25 | 476 |
| 4 | 1-SBA15/CH3CN | 2:1 |
70 | 36 | 98 (R) | 25 | 483 |
|
|||||||
| Temperature effect | |||||||
| 5 | 1-SBA15/CH3CN | 2:1 |
67 | 24 | 99 (R) | 40 | 462 |
| 6 | 1-SBA15/CH3CN | 2:1 |
69 | 40 | 98.5 (R) | 60 | 476 |
| 7 | 1-SBA15/CH3CN | 2:1 |
70 | 19 | 51 (R) | 80 | 483 |
|
|||||||
| H2O2 amount effect | |||||||
| 8 | 1-SBA15/CH3CN | 1:1 |
46 | 29 | 100 (R) | 25 | 317 |
| 9 | 1-SBA15/CH3CN | 3:1 |
52 | 33 | 27 (R) | 25 | 359 |
|
|||||||
| Control experiments | |||||||
| 10 | 1-SBA15/CH3CN | no H2O2 | 7 | 100 | 88 (R) | 25 | 48 |
| 11 | No Cat./CH3CN | 2:1 |
0 (26)e | 0 (40) | — | 25 | — |
| 12 | SBA15/CH3CN | 2:1 |
30 | 50 | — | 25 | — |
| 13 | SBA15-HBtar/CH3CN | 2:1 |
30 | 53 | 100 (R) | 25 | — |
| 14 | 1/CH3CN | 2:1 |
63 | 58 | 100 (R) | 25 | 575 |

| Substrate | Catalyst/solvent | H2O2/sub. molar ratio | Conv.b % | Product/selectivity % | Sulfoxide ee (Confign)c | Temp. (°C) | TON |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1-SBA15 10 mg (contain 2.9 μmol Mn), substrate 1 mmol, acetonitrile 3 mL, time 5 h.b Conversions and yields are based on the starting substrate.c ee, % enantiomeric excess (enantioselectivity) = (|R − S|/|R + S|) × 100. Enantioselectivity was determined by chiral GC-FID (ID-CYDEX-B column). The absolute configuration of the major isomer was determined by comparing the GC data with those observed for R-(+)-limonene.d 1-SBA15 20 mg, n-octane 0.1 g. | |||||||
 |
1-SBA15/CH3CN | 2:1 |
39 | RSO2/54 RSO/46 | 100 (R) | 25 | 269 |
 |
1/CH3CN | 2:1 |
73 | RSO2/3 RSO/97 | 100 (R) | 25 | 503 |
 |
1-SBA15d/CH3CN | 3:1 |
55 | Epoxide/>99 | — | 80 | 379 |
The reaction temperature shows a significant effect on the sulfoxide yield. A temperature increase from 25 °C led to decrease in the yield of sulfoxide (Table 2, entries 5–7). Hydrogen peroxide has to be used in a controlled manner to overcome the over-oxidation reaction to sulfone. This formation may suggest the existence of a kinetic resolution process during the course of the reaction. A reduction or increase in H2O2 content resulted also to the lower conversion and sulfoxide selectivity (Table 2, entries 8 and 9).
Control experiments proved the essential presence of the catalyst 1-SBA15 and H2O2 in catalytic oxidation of thioanisole (Table 2, entries 10 and 11). The catalyst support SBA-15 by itself is able to oxidize thioanisole up to 30% and the presence of the immobilized ligand H2Btar does not add extra activity (Table 2, entries 12 and 13). Under the optimized condition homogeneous catalyst [{Mn(H2O)2Cl2}2(H2Btar)] (1) showed the conversion and sulfoxide selectivity of 63 and 58%, respectively (Table 2, entry 14). Comparison of the immobilized and free complex 1 activity (Table 2, entries 14 and 4) proves that the incorporation of the complex mainly inside the mesoporous channels of SBA-15 increases its activity to the expense of sulfoxide selectivity. Reduction, however, in enantioselectivity is only 2%. Although immobilization can sometimes enhance the activity of homogeneous catalysts, a common problem encountered in asymmetric catalysis is a decrease of enantioselectivity upon heterogenisation.47 Similar to our findings, all the heterogenised catalysts developed for epoxidation of unfunctionalised alkenes exhibit considerably lower48–51 or no higher52,25 enantioseletivity than their homogeneous counterparts.
Extension of the above oxidation reaction to other thiol also gave high ees at room and 80 °C temperatures (Table 3). The oxidation rate of 4-chlorobenzenethiol is lower than that of thioanisole. The activity and selectivity of homogeneous [{Mn(H2O)2Cl2}2(H2Btar)] (1) were also higher than those of immobilized complex 1. The significant catalytic activity of 1-SBA15 is obviously seen in the successful oxidation of cyclohexene (conversion 55%), Table 3.
The heterogeneity of 1-SBA15 was confirmed by the reusability of 1-SBA15 in the oxidation of thioanisole by H2O2. After the oxidation reaction was complete, the catalyst could be recovered simply by filtration followed by washing with CH3CN. The absence of Mn in the reaction solution through leaching of 1 was also studied by measuring manganese. Investigation of the filtrate after separation of the catalyst by atomic absorption method at wavelength 279.5 nm corresponding to trace amounts, showed only 0.003% manganese. This finding ensured that leaching of the active supported Mn(II)-components occur in trace amounts. Thus, the obtained catalytic results derive mainly from the heterogeneous catalyst. Table 4 gives the results obtained for reusing 1-SBA15 up to four times. Notably, this catalyst could be reused at least four times with a total turnover number of 2408 without a decrease in enantioselectivity. In contrast, a reported Mn–MCM-41–salen catalyst suffered from a loss of enantioselectivity of 40% ee when reused for the first time.55 Li and co-workers53 reported the immobilisation of a variant of the Sharpless epoxidation catalyst on MCM-41 and silica. This was achieved by reacting tartaric acid with 3-aminopropyltriethoxysilane using a number of steps and the triethoxysilane moiety used to attach the modified tartramide to the silica surface. The final step was addition of the titanium centre in the form of titanium tetraisopropoxide to give the catalyst. The turnover numbers were 10–14 (15 for the homogeneous reaction) and the ees 78–86 (83% for the homogeneous reaction). Unfortunately no catalyst reuse data and the catalyst stability were reported. Various homogeneous and heterogeneous mono- and dinuclear Mn-catalysts have been used for the oxidation of cyclohexene to cyclohexene oxide54–57 with H2O2 and t-BuOOH. However, none of the reported catalysts shows such a high activity or/and selectivity as complex 1. In addition to the chemical simplicity and easy synthesis of complex 1 and 1-SBA15, they have also the ability to limit the H2O2 disproportionation reaction to a great extent relative to most Mn(II)/Mn(III) compounds, the H2O2/acetic acid/[LMn(μ-O)3MnL][PF6]2 system (L = trimethyl-1,4,7-triazacyclononane) which needs 4.6 to 13.9 equiv. of H2O2(ref. 58) and [Mn(μ-terph) (H2O)2]n ([H2O2]/[substrate] = 10) (terph = terephthalate).26 Despite their power as a synthetic tool and abundant use in academic research, oxidation reactions as a whole comprise as little as 3% of the reactions used on a preparative scale in the pharmaceutical industry.58 Perhaps the greatest factor influencing the hesitation to employ oxidation reactions on a large scale is the safety of these processes. Using less amount of H2O2 improves oxidation process safety in large scale syntheses.
| Catalyst | Conv.b (%) | Selectivityb (%) sulfoxide/sulfone | ee (Confign)c |
|---|---|---|---|
| a Reaction conditions: 1-SBA15 10 mg (contain 2.9 μmol Mn), methyl phenyl sulfide (thioanisole) 1.0 mmol, CH3CN 3 mL, aqueous 30% H2O2 2.0 mmol, temperature 25 °C, reaction time 2 min.b Conversions and yields are based on the starting substrate.c ee (% enantiomeric excess), see Table 2 for detail. | |||
| Fresh | 70 | 36:64 |
98% R |
| Reused-1 | 70 | 37:63 |
100% R |
| Reused-2 | 69 | 36:64 |
100% R |
| Reused-3 | 70 | 35:65 |
100% R |
| Reused-4 | 70 | 36:64 |
100% R |
Conclusions
The highly efficient syntheses of [{Mn(H2O)2Cl2}2(H2Btar)] (1) and 1-SBA15 have been developed. The catalytic oxidation of thioanisole indicates that the asymmetric oxidation using homogeneous and SBA-15 immobilized chiral manganese complex as catalysts and hydrogen peroxide as oxidant is possible, with good results. The asymmetric sulfoxidation and epoxidation of alkenes were achieved successfully. The use of chiral heterogeneous catalysts under ambient conditions offers several advantages compared with their homogeneous counter-parts, such as ease of recovery and recycling and enhanced stability. Immobilization of complex 1 onto SBA-15 increased the selectivity toward sulfone in the oxidation.Experimental
D-(−)-tartaric acid, tetraethyl orthosilicate (TEOS), poly(ethylene oxide), poly(propylene oxide), poly(ethylene oxide), Pluronic P123 [EO20–PO70–EO20], 3-chloropropyl triethoxysilane (CPTES), benzaldehyde, 4-hydroxy-benzoic acid hydrazide and all other reagents were obtained from commercial sources (Merck or Fluka) and were used as received without further purification. Aqueous 30% hydrogen peroxide was used and its exact concentration was determined before use by titration with standard KMnO4. Mesoporous SBA-15 and chlorine-functionalized SBA-15 material, SBA15-Cl were synthesized according to the procedure described already.59The reaction products of oxidation were determined and analyzed by HP Agilent 6890 gas chromatograph equipped with a HP-5 capillary column (phenyl methyl siloxane 30 m × 320 μm × 0.25 μm) with flame-ionization detector. The enantiomeric excess (ee) was determined by chiral GC (HP 6890 GC) using a SGE-CYDEX-B capillary column (25 m × 0.22 mm ID × 0.25 μm). Temperatures: column 50 °C, step 10 °C min−1, final 150 °C, injector 250 °C, detector 250 °C; mobile phase nitrogen, rate 0.7 mL min−1. Retention time for sulfoxide: Rt (S)-enantiomer: 17.9 min; Rt (R)-enantiomer: 19.1 min. Sulfone: Rt: 26.5 min.
1H NMR spectra were recorded by use of a Bruker 250 MHz spectrometer. UV-Vis spectra of solution were recorded on a Shimadzu 160 spectrometer. Chemical analyses of the catalysts were performed by atomic absorption (Varian spectrometer AAS-110) upon chemical attack of the material using concentrated HNO3. Microanalyses were carried out using a Heraeus CHN–O– Rapid analyzer. IR spectra were recorded using Perkin-Elmer 597 and Nicolet 510P spectrophotometers. Powder X-ray diffraction patterns were collected at the Bruker, D8 ADVANCE, Germany, wavelength 1.5406 Å (Cu Kα), voltage: 40 kV, current, 40 mA. The size and morphology of solid compounds were recorded by using a Hitachi F4160 scanning electron microscope (SEM) operated at an accelerating voltage of 10 KV and the textural properties determined from N2 adsorption isotherms measured on a Belsorp mini II (Japan) instrument. The electron paramagnetic resonance (EPR) measurements were carried out using Bruker EMX spectrometer working at fixed frequency 9.25 GHz (X-band) with Oxford Instruments helium-flow cryostat operating in the temperature range from 3.8 K to 300 K. A 100 kHz magnetic field modulation and phase sensitive detection were used to record the derivative of the absorbed microwave power.
Synthesis of (−)-diethyl-D-tartrate
A 50 mL round-bottomed flask equipped with a condenser was charged with 0.15 g (1 mmol) of D-(S,S)-(−)-tartaric acid, 7.5 mL of 96% ethanol, 4.9 mL of chloroform and 0.123 mL of HCl (1 M). The stirred mixture was heated at reflux for 60 h so that no more water separated. A dark yellow colored oily product was obtained. Yield: 99.6% (0.205 g). IR (KBr, cm−1): 3467 (s, vb), 2983 (s), 1747 (vs.). 1H NMR (250 MHz, CDCl3): δ = 1.27, 1.30, (2 t, 6H, J = 7.1, 2 CH2CH3), 4.25, 4.52 (2 q, 4H, J = 7.1, 2 CH2CH3), broad 4.90 (s, 2H, 2 OHCH), 6.5 (s, 2H, OH). 13C NMR (62.90 MHz, CDCl3) δ: 13.9, 14.1 (2C, CH3), 62.5, 62.6 (2C, CH2), 72.1 (2C, CH–OH), 171.6, 171.7 (2C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
Synthesis of diethyl-2,3-O-benzylidene-D-tartrate
This compound was prepared following a similar procedure reported in the literature.60 A 50 mL, round-bottomed flask was charged with (−)-diethyl-D-tartrate (206 mg, 1 mmol), benzaldehyde (0.106 g, 1 mmol), cyclohexane (10 mL) and p-toluenesulfonic acid monohydrate (5.6 mg, 0.029 mmol). The homogeneous mixture was refluxed for 16 h. The solution was allowed to cool to room temperature and then was concentrated by rotary evaporation. The residual yellow oil was dissolved in 10 mL of diethyl ether, transferred to a separatory funnel and washed with saturated aqueous potassium bicarbonate (5 mL) and water (10 mL). The ethereal layer was dried over magnesium sulfate and filtered. The solution was concentrated by rotary evaporation, followed by removal of solvent. The solid product was collected by suction filtration through a sintered-glass funnel to give diethyl 2,3-O-benzylidene-D-tartrate as yellow crystals. Yield: 87% (0.256 g). Mp 45 °C. IR (KBr, cm−1): 3368 (vb, w), 3237 (w), 2982 (s), 2931 (m), 1754 (vs.), 1599 (s). 1H NMR (250 MHz, CDCl3) δ: 1.32, 1.35 (2 t, 6H, J = 7.1, 2 CH2CH3), 4.23, 4.28 (2 q, 4H, J = 7.1, 2 CH2CH3), 4.83, 4.95 (2 d, 2H, J = 4.0, 2 CHO), 6.16 (s, 1H, CHPh), 7.40, 7.58 (2 m, 5H, C6H5).Synthesis of 2,3-O-4-hydroxybenzhydrazidebenzylidene-D-tartrate (H2Btar)
A mixture of 2.56 g (8.7 mmol) diethyl-2,3-O-benzylidene-D-tartrate, 2.65 g (17.4 mmol) 4-hydroxybenzhydrazide and 0.02 g (0.105 mmol) p-toluenesulfonic acid monohydrate in benzene (15 mL) was refluxed for 5 days. After evaporation of the solvent, it was again refluxed for 24 h with absolute ethanol. The product 2,3-O-4-hydroxybenzhydrazidebenzylidene-D-tartrate (H2Btar) was separated as a white solid. Yield: 46.7% (2.06 g). IR (KBr, cm−1): 3460 (w), 3316 (m), 3269 (m), 3196 (s), 3043 (m), 1641 (vs.), 1615 (vs.). 1H NMR (250 MHz, DMSO) δ: 4.39 (s, Ar–OH), 6.77, 6.85 (2 d, 2H, J = 8.0, 2 O–CH–CO), 7.41–7.82 (m, 13H, 2C6H4, C6H5), 8.41 (s, 1H, CHPh), 9.51, 9.96, 10.15, 11.66 (s, 4H, 4 NH). 13C-NMR (62.90 MHz, DMSO) δ: 115.5, 115.3 (2C, 2 O–CH–CO), 124.4 (1C, CHPh), 127.40, 129.3, 130.2 (18C, 2C6H4, C6H5), 134.9 (2C, 2 Ar–CO), 166.4, 160.4 (2C, 2 CO).Synthesis of complex [{Mn(H2O)2Cl2}2(H2Btar)] (1)
H2Btar (0.020 g, 0.04 mmol) and manganese(II) chloride tetrahydrate (0.016 g, 0.08 mmol) were dissolved in absolute ethanol and the resulting solution allowed to reflux for 3 h. The excess of solvent was removed under vacuum and a dark yellow colored product resulted. Yield: 68% (0.0237 g). IR (KBr, cm−1): 3456 (br, s), 3289 (br, vs.), 3202 (br, vs.), 3043 (m), 2924 (w), 2851 (w), 1647 (s, CO), 1609 (s, CO), 1569 (s), 1507 (vs., CC). Anal. for C25H30Cl4Mn2N4O12 ([{Mn(H2O)2Cl2}2(H2Btar)], MW = 830.21): calc. C 36.17, H 3.64, Mn 13.23%; found C 36.00, H 3.14, Mn 13.25%
Immobilization of [{Mn(H2O)2Cl2}2(H2Btar)] over SBA-15, 1-SBA15
Following a reported procedure,61 to a suspension of freshly dried chlorine-functionalized SBA-15 material, SBA15-Cl (1 g), in absolute ethanol (40 mL), a solution of H2Btar (0.1 g) in absolute ethanol (10 mL) was added and the resulting solution was refluxed for 3 h. The white colored solid was separated, Soxhlet extracted with dichloromethane to remove the unreacted starting material adsorbed on the external surface of SBA15-Cl and vacuum dried for 24 h. The ligand gets attached to the SBA-15 through the spacer by the nucleophilic displacement of chlorine of SBA15-Cl by the basic hydroxo group of the ligand H2Btar. Then to a suspension of the resulting solid (1 g) in absolute ethanol (40 mL) a solution of MnCl2·4H2O (0.8 g) in absolute ethanol (25 mL) was added. This was refluxed for 3 h. The white solid, 1-SBA15, was separated by filtration, was dried and Soxhlet extracted with absolute ethanol to remove any unreacted manganese from the surface. Analyses for SBA15-Cl: C, 2.97; H, 0.75%. Anal. found for 1-SBA15: C 8.64, H 1.42, N, 1.61, Mn 3.24%.Acknowledgements
The authors are grateful to the University of Zanjan, Iran National Science Foundation (INSF 92036382) and Polish Academy of Sciences (PAN) for financial support of this study.Notes and references
- I. Ojima, N. Clos and C. Bastos, Tetrahedron, 1989, 45, 6901 CrossRef CAS.
- A. Li, J. Liu, S. Q. Pham and Z. Li, Chem. Commun., 2013, 49, 11572 RSC.
- I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651 CrossRef CAS PubMed.
- M. C. Carreno, M. Ribagorda and G. H. Posner, Angew. Chem., Int. Ed., 2002, 41, 2753 CrossRef CAS.
- H. Cotton, T. Elebring, M. Larsson, L. Li, H. Sorensen and S. von Unge, Tetrahedron: Asymmetry, 2000, 11, 3819 CrossRef CAS.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19 CrossRef CAS.
- T. Ohkuma, M. Kitamura and R. Noyori, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, Weinheim, 2nd edn, 2000, pp. 1–110 Search PubMed.
- H.-U. Blaser, B. Pugin and F. Spindler, J. Mol. Catal. A: Chem., 2005, 231, 1 CrossRef CAS PubMed.
- I. Ojima, N. Clos and C. Bastos, Tetrahedron, 1989, 45, 6901 CrossRef CAS.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
- H.-U. Blaser and B. Pugin, in Chiral Reactions in Heterogeneous Catalysis, ed. G. Jannes and V. Dubois, Plenum Press, New York, 1995, p. 33 Search PubMed.
- C. Brown, Chirality in Drug Design and Synthesis, Academic Press, New York, 1990 Search PubMed.
- E. N. Jacobsen and M. H. Wu, in. Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, H. Yamamoto and A. Pfaltz, Springer-Verlag, Berlin, 1999, vol. 2, p. 649 Search PubMed.
- X. Huang, X. Fu, Z. Jia, Q. Miao and G. Wang, Catal. Sci. Technol., 2013, 3, 415 CAS.
- J. Huang, X. Fu, G. Wang, Q. Miao and G. Wang, Dalton Trans., 2012, 41, 10661 RSC.
- K. Yu, L.-L. Lou, F. Ding, S. Wang, Z. Wang and S. Liu, Catal. Commun., 2006, 7, 170 CrossRef CAS PubMed.
- K. Yu, Z. Gu, R. Ji, L. L. Lou, F. Ding, C. Zhang and S. Liu, J. Catal., 2007, 2, 312 CrossRef PubMed.
- K. Yu, Z. Gu, R. Ji, L. L. Lou and S. Liu, Tetrahedron, 2009, 65, 305 CrossRef CAS PubMed.
- Z. Xu, X. Ma, Y. Ma, Q. Wang and J. Zhou, Catal. Commun., 2009, 10, 1261 CrossRef CAS PubMed.
- C. Freire, C. Pereira and S. Rebelo, Catalysis, 2012, 24, 116 CAS.
- F. Maia, R. Silva, B. Jarrais, A. R. Silva, C. Freire, M. F. R. Pereira and J. L. Figueiredo, J. Colloid Interface Sci., 2008, 328, 314 CrossRef CAS PubMed.
- M. Peng, Y. Chen, R. Tan, W. Zheng and D. Yin, RSC Adv., 2013, 3, 20684 RSC.
- S. Xiang, Y. Zhang, Q. Xin and C. Li, Chem. Commun., 2002, 2696 RSC.
- S. B. Ogunwumi and T. Bein, Chem. Commun., 1997, 901 RSC.
- H. Hosseini-Monfared, A. Mohajeri, A. Morsali and C. Janiak, Monatsh. Chem., 2009, 140, 1437 CrossRef CAS PubMed.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732 CrossRef CAS PubMed.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS.
- P. Pitchen, E. Dunach, M. N. Deshmukh and H. B. Kagan, J. Am. Chem. Soc., 1984, 106, 8188 CrossRef CAS.
- F. Di Furia, G. Modena and R. Seraglia, Synthesis, 1984, 4, 325 CrossRef.
- K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315 CrossRef CAS PubMed.
- H. B. Kagan, in Organosulfur Chemistry in Asymmetric Synthesis, ed. T. Toru, C. Bolm, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, p. 1 Search PubMed.
- M. C. Carreno, G. Hernandez-Torres, M. Rbagorda and A. Urbano, Chem. Commun., 2009, 6129 RSC.
- G. E. O'Mahony, P. Kelly, S. E. Lawrence and A. R. Maguire, ARKIVOC, 2011, i, 1 CrossRef.
- T. Katsuki, in Pharmaceutical Process Chemistry, ed. T. Shiori, I. Kunisuke and T. Konoike, WILEY-VCH Verlag GmbH & Co., Weinheim, Germany, 2011, p. 59 Search PubMed.
- H. Srour, P. L. Maux, S. Chevance and G. Simonneaux, Coord. Chem. Rev., 2013, 257, 3030 CrossRef CAS PubMed.
- M. D. Alba, Z. Luan and J. Klinowski, J. Phys. Chem., 1996, 100, 2178 CrossRef CAS.
- K. Pathak, I. Ahmad, S. H. R. Abdi, R. I. Kureshy, N. Khan and R. V. Jasra, J. Mol. Catal. A: Chem., 2008, 280, 106 CrossRef CAS PubMed.
- T. Joseph, S. S. Deshpande, S. B. Halligudi, A. Vinu, S. Ernst and M. Hartman, J. Mol. Catal. A: Chem., 2003, 206, 13 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56 CrossRef CAS.
- T. Horikawa, D. D. Do and D. Nicholson, Adv. Colloid Interface Sci., 2011, 169, 40 CrossRef CAS PubMed.
- Z. Y. Wang, F. X. Zhang, Y. L. Yang, B. Xue, J. Cui and N. Guan, J. Chem. Mater., 2007, 19, 3286 CrossRef CAS.
- L. N. Sun, H. J. Zhang, J. B. Yu, S. Y. Yu, C. Y. Peng, S. Dang, X. M. Guo and J. Feng, Langmuir, 2008, 24, 5500 CrossRef CAS PubMed.
- J. Griscom, J. Non-Cryst. Solids, 1980, 40, 211 CrossRef.
- L. Gacema, A. Artemenko, D. Ouadjaout, J. P. Chaminade, A. Garcia, M. Pollet and O. Viraphon, Solid State Sci., 2009, 11, 1854 CrossRef PubMed.
- A. Fedotovs, Dz. Berzins, O. Kiselova, A. Sarakovskis and U. Rogulis, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 23, 012018 CrossRef.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994, ch. 8, p. 346 Search PubMed.
- B. B. De, B. B. Lohray, S. Sivaram and P. K. Dhal, Tetrahedron: Asymmetry, 1995, 6, 2105 CrossRef CAS.
- F. Minutolo, D. Pini, A. Petri and P. Salvadori, Tetrahedron: Asymmetry, 1996, 7, 2293 CrossRef CAS.
- M. J. Sabater, A. Corma, A. Domenech, V. Fornés and H. García, Chem. Commun., 1997, 1285 RSC.
- P. Piaggio, P. McMorn, C. Langham, D. Bethell, P. C. Bulman-Page, F. E. Hancock and G. J. Hutchings, New J. Chem., 1998, 1167 RSC.
- I. F. J. Vankelecom, D. Tas, R. F. Parton, V. V. de Vyver and P. A. Jacobs, Angew. Chem., Int. Ed. Engl., 1996, 35, 1346 CrossRef CAS.
- S. Xiang, Y. Zhang, Q. Xin and C. Li, Angew. Chem., Int. Ed., 2002, 41, 821 CrossRef CAS.
- D. Veghini, M. Bosch, F. Fischer and C. Falco, Catal. Commun., 2008, 10, 347 CrossRef CAS PubMed.
- N. Nakayama, S. Tsuchiya and S. Ogawa, J. Mol. Catal. A: Chem., 2007, 277, 61 CrossRef CAS PubMed.
- V. Mirkhani, M. Moghadam, S. Tangestaninejad and B. Bahramian, Appl. Catal., A, 2006, 313, 122 CrossRef CAS PubMed.
- M. S. Saeedi, S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and A. R. Khosropour, Polyhedron, 2013, 49, 158 CrossRef CAS PubMed.
- R. W. Dugger, J. A. Ragan and D. H. Brown Ripin, Org. Process Res. Dev., 2005, 9, 253 CrossRef CAS.
- S. Alavi, H. Hosseini-Monfared and M. Siczek, J. Mol. Catal. A: Chem., 2013, 377, 16 CrossRef CAS PubMed.
- B. Steuer, V. Wehner, A. Lieberknecht and V. Jäger, Org. Synth., 1997, 74, 1 CrossRef CAS . (Coll. Vol IX, p. 39).
- T. Joseph and S. B. Halligudi, J. Mol. Catal. A: Chem., 2005, 229, 2417 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |