
Recent advances in the production of polyols from lignocellulosic biomass and biomass-derived compounds
Xiaoran Liu
ab,
Xicheng Wang
*a,
Shengxi Yao
a,
Yijun Jiang
a,
Jing Guan
a and
Xindong Mu
*a
aKey Laboratory of Biobased Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, China. E-mail: wangxc@qibebt.ac.cn; muxd@qibebt.ac.cn; Fax: +86-532-80662724; Tel: +86-532-80662723
bUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 15th September 2014
Abstract
The conversion of renewable, non-edible and resource-abundant lignocellulose to fuels, chemicals and materials has received significant attention as it holds the possibility of using carbon neutral technologies to combat global changes. Considering the relatively high oxygen content in cellulose, it is more desirable to be transformed into oxygenated chemicals rather than hydrocarbon fuels in view of atom efficiency. Among the oxygen-rich chemicals from biomass, polyols, such as ethylene glycol and propylene glycol, are widely used in polymer synthesis, food industry and manufacturing of pharmaceuticals. Hydrolysis, coupled with hydrogenation and hydrogenolysis serves as an effective approach to transform biomass to polyols. This review summarizes the recent advances in biomass upgrading reactions for the production of polyols with a special emphasis on the formation of glycols.
 Xiaoran Liu | Xiaoran Liu is a PhD candidate under the supervision of Prof. Xindong Mu at Qingdao Institute of Bioenergy and Bioprocess Technology (QIBEBT), Chinese Academy of Sciences. He received his B.S. degree from Qingdao University of Science and Technology in 2012. His current research interests focus on the catalytic conversion of renewable biomass into high value-added chemicals and fuels. |
 Xicheng Wang | Dr Xicheng Wang received his B.S. degree in environmental science from Wuhan University of Technology in 2007. He obtained his PhD degree in chemical engineering from QIBEBT, Chinese Academy of Sciences in 2012 under the tutelage of Prof. Xindong Mu. He continued his academic career as a research associate at QIBEBT. His research interests involve energy and green chemical processes including renewable fuels and oxygenated chemicals from biomass, catalyst design and catalyst preparation. |
 Shengxi Yao | Shengxi Yao is a research assistant in Prof. Xindong Mu's group at QIBEBT, Chinese Academy of Sciences. He obtained his Master's degree from QIBEBT in 2011, and his thesis focused on the catalytic conversion of carbohydrates into diols. Currently, his research interests include synthesis and application of porous solid materials for energy and environmental catalysis. |
 Yijun Jiang | Yijun Jiang is an Associate Professor at QIBEBT. He received his PhD degree in 2007 from Shanghai Institute of Ceramics, Chinese Academy of Sciences. His current research interests are to develop novel bio-based hybrid materials and exploit their applications in the fields of green chemistry and clean energy. His papers have been cited more than 300 times, and some of them were highlighted in Science or published as cover articles. |
 Jing Guan | Dr Jing Guan obtained her PhD from Dalian Institute of Chemical Physics, Chinese Academy of Sciences, in 2010, and is presently a research associate at QIBEBT, Chinese Academy of Sciences. Her research areas include DFT modeling of reaction mechanisms in heterogeneous catalysis, with focus on the metal-catalyzed conversion of biomass-derived oxygenates to produce value-added chemicals such as polyols and diols. |
 Xindong Mu | Xindong Mu is a professor of chemical engineering at QIBEBT, Chinese Academy of Sciences, where he leads the Green Chemical Engineering Center. He obtained his PhD from Peking University in 2005, and then worked as a research associate at the University of Tokyo, Japan. He joined QIBEBT in 2008 and was promoted to full professor in 2010. He has filed over 40 patents and published over 50 peer-reviewed papers. His current research interests include green chemistry and engineering with a focus on catalysis, biomass utilization and green materials. |
1. Introduction
The production of chemicals highly depends on non-renewable fossil resources such as petroleum, coal, and natural gas. However, global issues, such as decreasing fossil fuel reserves, the growing demand for energy in the future and the concern on greenhouse gas emissions, is motivating researchers to explore alternative energy sources that can avoid these issues.1,2 Biomass energy, which has been directly acquired via combustion to produce heat for thousands of years, is being re-used because of its sustainability and enormous reserves (Fig. 1).3,4 Cellulose, mainly obtained from agricultural and forestry residues, is one of the world's largest organic raw material resources (nature renews nearly 40 billion tonnes of it every year).5 Different from starch, which is the main food source for humans, cellulose cannot be digested by humans; thus, the utilization of cellulose will not have a negative impact on the food supply. Therefore, cellulose is regarded as a promising resource that can be transformed into fuels and chemicals in future.6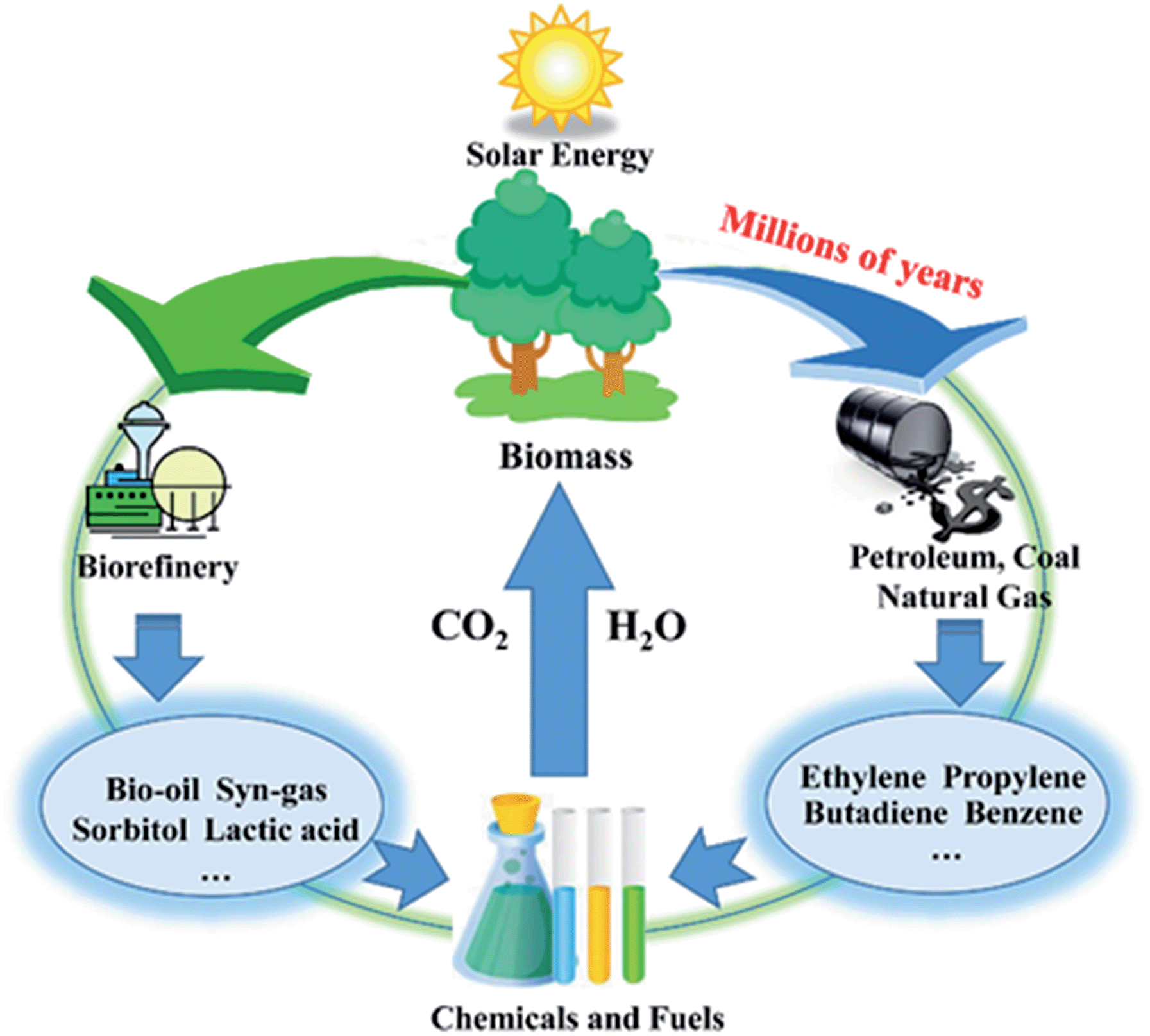 | ||
| Fig. 1 Carbon cycles in biomass and fossil resource utilization. | ||
Transformation of biomass into chemicals and fuels is generally achieved by thermal, biological, and chemical methods.6–8 Thermal techniques such as pyrolysis and gasification can take full advantage of the entire organic substance and transform biomass into liquid fuel and synthesis gas.7 Nevertheless, thermal techniques still suffer from disadvantages such as low selectivity and high energy consumption, which are unacceptable at the present stage.9 On the other hand, the biological conversion of biomass often unavoidably loses carbon and oxygen, which lead to the low selectivity and low yield of target products. In general, unlike traditional hydrocarbon raw materials, biomass possesses high oxygen content (∼50%).10,11 Thus, reasonable routes to promote biomass conversion into value-added chemicals, which have analogous carbon skeleton and maintain the oxygen atoms under relatively mild conditions in the liquid phase with a high selectivity are required.10 In order to transform the polyhydroxy compounds into high value-added chemicals via C–C and/or C–O cleavage, selective hydrogenolysis is crucial (Fig. 2).
 | ||
| Fig. 2 Comparation of O/C ratios in hydrocarbons, biomass, biomass-derived compounds and commodity polyols. | ||
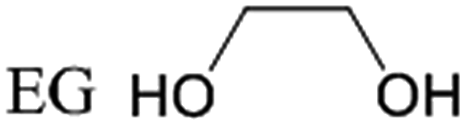
Polyols, including sorbitol, mannitol, xylitol, ethylene glycol (EG) and propylene glycol (PG) are all important chemicals. Currently, sorbitol and mannitol can be manufactured from the hydrogenation of glucose and fructose;8 xylitol is prepared by xylose hydrogenation;9 EG and PG are produced from hydration of ethylene oxide and propylene oxide, which are derived by cracking petroleum.10
Traditionally, sorbitol, mannitol, xylitol, EG and PG are important materials in the food industry, intermediates in the pharmaceutical industry, monomers in the polymer industry and additives in the cosmetics industry.6 In the new energy strategies based on biomass, valorization of these platform polyols has also been developed. For example, glycols can be used as feedstocks for fuel cells to generate electricity,11 and hydrogen12 can be produced through aqueous-phase reforming and steam reforming processes. The molecular structures and the primary applications of polyols discussed herein are listed in Table 1.
| Polyols | Primary applications | Applications |
|---|---|---|
 |
1, 2, 3, 4, 6, 10, 11, 12, 13, 14 | 1. Additive in the food industry |
| 2. Intermediate in the pharmaceutical industry | ||
| 3. Monomer in the polymer industry | ||
 |
1, 2, 3, 4, 6, 10, 11, 12, 13, 14 | 4. Additive in the cosmetic industry |
| 5. Solvent | ||
| 6. Sweetening agent | ||
 |
1, 2, 3, 4, 6, 10, 11, 12, 13, 14 | 7. Antifreeze |
| 8. Coolant | ||
 |
1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 | 9. Heat transfer fluids |
| 10. Synthesis of surfactants | ||
| 11. Production of syngas | ||
 |
1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 | 12. Production of hydrogen |
| 13. Fuel cells | ||
| 14. Production of glycols |
Catalytic upgrading of lignocellulosic biomass for the synthesis of industrial products and fuels has been reviewed recently.13–21 In this review, we focus specifically on the latest advances in the transformation of cellulose and biomass-derived compounds into high value-added polyols over heterogeneous catalysts with focus on the formation of glycols, directly obtained from cellulose, as well as polyhydroxy compounds such as sugars and sugar alcohols.
2. Hydrolytic hydrogenation of cellulose
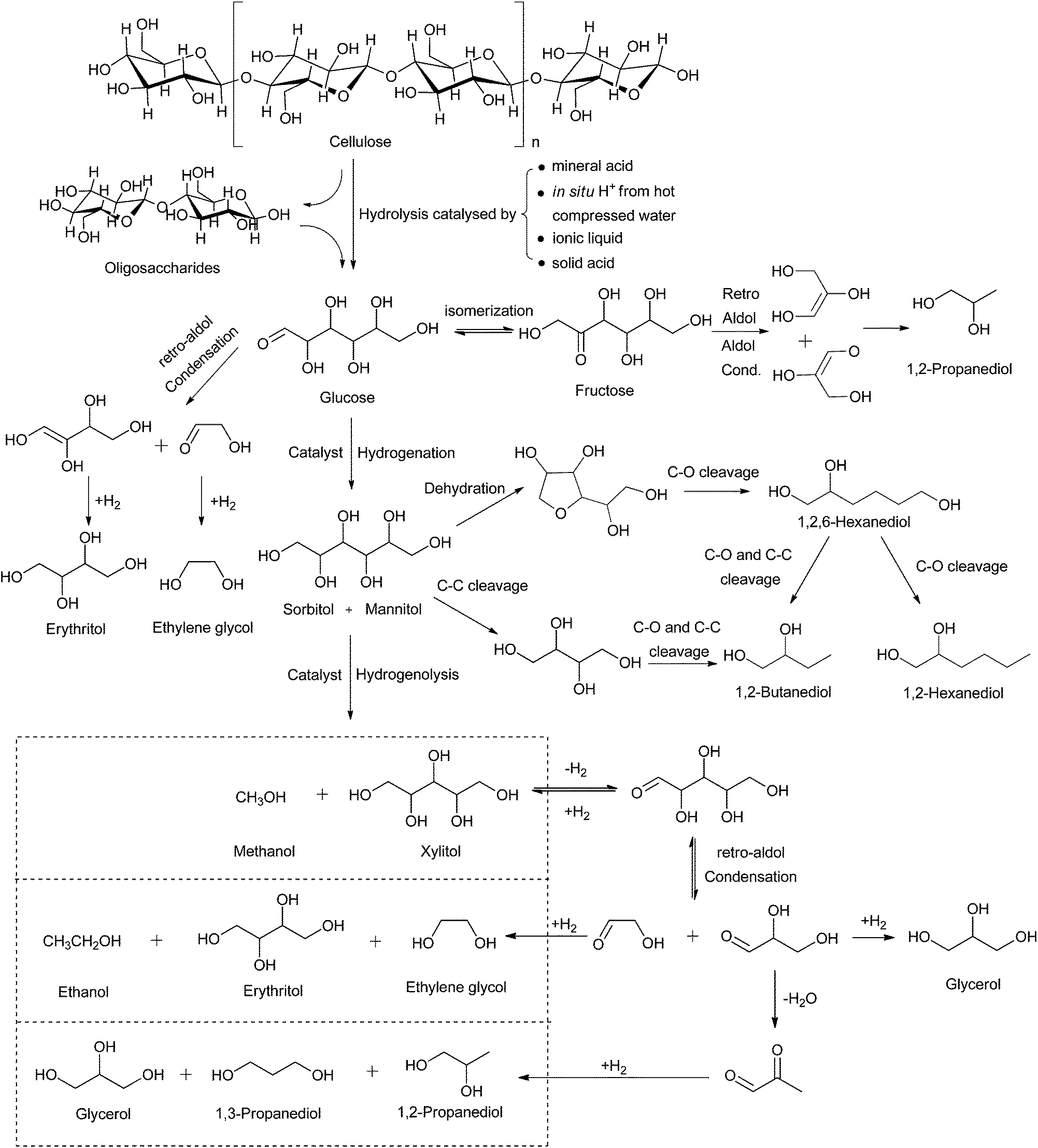
As mentioned above, lignocellulose is the most abundant form of biomass found in nature.22 However, cellulose is water insoluble because of its highly crystalline structure, which is composed of β-1,4-glycosidic bonds.23,24 Although a lot of effort has been focused on the degradation of cellulose by enzymes,23 mineral acids25 and supercritical water,26 drawbacks still exist in these processes such as the enzymatic saccharification of cellulose suffers from high cost, low reaction rates and subsequent low space-time yields and problems in enzyme recovery; mineral acid hydrolysis suffers from challenges related to waste water disposal, equipment corrosion and the recovery of acid; and the utilization of supercritical water faces problems related to energy consumption.27To date, the one-pot chemical transformation of cellulose to polyols over heterogeneous catalysts is one of the most promising routes for the effective utilization of cellulose in a more efficient and convenient manner.28 Several catalytic reaction systems were successfully established using acid catalysed hydrolysis coupled with metal-catalysed hydrogenation/hydrogenolysis;14 however, challenges still exist in this process. For example, both the substrate and the catalysts are present in the solid state in this reaction, leading to problems related to interaction between the catalysts and cellulose. Mass transfer between the substrates and the catalysts is the main barrier in the chemical conversion of cellulose.29 In the one-step hydrolytic hydrogenation/hydrogenolysis of cellulose, several independent reactions take place, including the hydrolysis of cellulose to glucose, the hydrogenation of glucose to sorbitol, the hydrogenolysis of glucose and sorbitol to low carbon glycols. The hydrolysis of cellulose is generally considered as the rate-determining step in this reaction (Fig. 3).30–32 The product distribution is controlled by the balance between the rates of cellulose hydrolysis and glucose hydrogenation/hydrogenolysis. In this part, we will review the recent progress on coupled hydrolytic hydrogenation/hydrogenolysis of cellulose and discuss the process based on the acid used in the hydrolysis step (Scheme 1).
 | ||
| Fig. 3 Typical pathway for the hydrolytic hydrogenation of cellulose. | ||
 | ||
| Scheme 1 Acid promoted hydrolysis of cellulose. | ||
2.1 Acid combined with metal catalysts in the hydrolytic hydrogenation/hydrogenolysis of cellulose
Liquid acid is utilized to promote the hydrolysis of cellulose to glucose because it is inexpensive and widely applied in industry. In this section, we mainly focus on the application of liquid acid combined with the hydrogenation catalyst in the one-pot conversion of cellulose to hexitols and glycols. Table 2 shows the hydrolytic hydrogenation/hydrogenolysis of cellulose over different catalysts promoted by an acid. Mineral acids like phosphoric acid and sulfuric acid were introduced into cellulose hydrogenation under catalysis provided by supported noble metal catalysts based on Pt, Ru and Pd at relatively low temperature with short reaction times.33 After 5 h of reaction, sugar and sugar alcohols were produced in 60% yield with 72% cellulose conversion at 433 K and 5 MPa of H2.| Starting material | Catalyst | Conditions | Products and yield/% | Ref. |
|---|---|---|---|---|
| a SOR: sorbitol, XYL: xylitol, SUA: sugar alcohols, ISO: isosorbide, SOT: sorbitan, BM: ball-milled, MBM: mixed ball milled, ZPA: Zirconium phosphate. | ||||
| 5 wt% cellulose | Ru/C | 433 K, 5 MPa H2, 1 h, H2SO4 (2.5 wt%) | SOR (33.2%), SOT (13.6%), XYL (11.3%) | 33 |
| 10 wt% cellulose-BM | Ru/H-USY | 463 K, 5 MPa H2, 13 h, HCl (177 ppm) | SUA (60%), SOT (33%) | 34 |
| 2 wt% cellulose | Ru/C | 508 K, 6 MPa H2, 1 h, HCl (0.1 M) | ISO (41.1%) | 35 |
| 2 wt% cellulose-BM | Ru/C | 463 K, 5 MPa H2, 1 h, H4SiW12O40 (1.22 × 10−2 M) | SUA (68%), SOT (19%) | 36 |
| 5 wt% cellulose | Ru/C | 433 K, 5 MPa H2, 7 h, H4SiW12O40 (55.1 mM) | C4–C6 (80.6%) | 37 |
| 2 wt% cellulose | Ru/C | 463 K, 5 MPa H2, 13 h, Cs3.5SiW ([H+] = 1.5 mM) | SUA (56%), SOT (14%) | 39 |
| 2 wt% cellulose-MBM | Ru/C + ZPA | 463 K, 6 MPa H2, 2.5 h | SUA (90%) | 41 |
| 1 wt% cellulose | Ru/C + H2WO4 | 518 K, 6 MPa H2, 0.5 h | EG (54.4%), 1,2-PG (7.3%) | 43 |
| 1 wt% cellulose-pretreated with 0.06% H3PO4 | Ni4.63Cu1Al1.82Fe0.79 | 488 K, 4 MPa H2, 3 h, H3PO4 (0.08 wt%) | SOR (68.07%), MAN (11.96%) | 44 |
Sels et al.34 introduced trace amounts of mineral acid (ppm grade) into the hydrogenation reaction system with noble metal loaded zeolites acting as hydrogenation catalysts to yield hexitols. The yield of hexitols (sugar alcohol and sorbitan) was as high as 90% under the catalysis of Ru/H-USY promoted by hydrochloric acid (177 ppm) at 463 K and 5.0 MPa of H2. Isosorbide was obtained as the main product when Ru/C and hydrochloric acid were employed to catalyse the hydrogenation of concentrated microcrystalline cellulose under 508 K as demonstrated by Zhao.35 Hydrochloric acid could accelerate both the cellulose hydrolysis and the dehydration of sorbitol. The balance between catalytic hydrolysis catalysed by hydrochloric acid and the hydrogenation of glucose promoted by Ru/C was ascribed to the high yield of isosorbide.
Apart from mineral acid, heteropolyacid (HPA) was also applied to facilitate the one-pot conversion of cellulose. Sels et al.36 found that the combination of heteropolyacid and Ru supported on carbon could promote the concentrated ball-milled cellulose feed into hexitols with notable yield (68% hexitol yield and 99% cellulose conversion) at 463 K and 5.0 MPa of H2 after 1 h of reaction. Heteropolyacids such as H3PW12O40 and H4SiW12O40 were selected in their study not only because of their high efficiency in the hydrolysis of cellulose to glucose, but also due to their ease of recovery via extraction and recrystallization processes after the reaction was completed. Palkovits et al. found that cellulose could be converted into C4–C6 sugar alcohols with a yield of 81% under the catalysis of Ru/C combined with heteropolyacid (H4SiW12O40) under the conditions of 433 K and 5 MPa of H2.37 The main drawback that restricts the utilization of liquid acid in cellulose hydrolysis lies in the recovery of the acid. In order to solve this problem, solid acids were introduced into the reaction.
Compared to liquid acid, solid acid possesses a series of advantages. For example, solid acid is easily separated from reaction system and some solid acids have a relatively strong acidity, even stronger than H2SO4.38 Therefore, solid acid is an appropriate substitute to liquid acid, and it will avoid a great number of problems such as equipment corrosion, neutralization and the waste of water resources, which is caused by use of liquid acid in industrialized applications.
Non-stoichiometric caesium salts, CsPW and CsSiW coupled with Ru/C were found to afford a much higher yield of hexitols than formerly reported HPAs and Ru/C under relatively milder reaction conditions.39 CsHPAs exhibited much higher surface acidity and hydrophobicity if the calcination temperature increased from 253 K to 873 K during the synthesis process, leading to much higher activity and selectivity. More importantly, the CsPW salts could be recovered via a procedure of recrystallization after reaction, showing its great potential for industrialized applications. Ma et al.40 presented that direct conversion of cellulose could be achieved over zirconium phosphate catalysis combined with Ru/C. Ball-milled cellulose could be transformed into sorbitol/mannitol in a yield of 81%. Mixed ball-milling of cellulose and solid acid catalysts enhanced the interaction between cellulose and solid acid catalysts and thus promoted the hydrolytic hydrogenation of cellulose.41 The yield of sugar alcohols could be 90.3% when zirconium phosphate (ZPA) was employed as the solid acid under reaction conditions of 463 K, 6 MPa of H2 and 2.5 h of reaction.
The combination of MCM-41-n-SO3H and Ru/C showed high selectivity to alkanediols or γ-valerolactone (GVL) for the hydrolytic hydrogenation of cellulose when both catalysts were added in the reaction at the beginning.42 However, due to the changes in the mesoporous structure and the loss of the acid group of MCM-41-n-SO3H, the catalyst deactivated rapidly. The sequential process yielded hexitol and GVL as the main products. Moreover, it is easy to reuse MCM-41-n-SO3H because it can be separated via the simple process of filtration. Zhang et al. employed a phase-transfer catalytic system controlled by temperature for the hydrolytic hydrogenolysis of cellulose.43 Tungsten acid, a yellow solid and water insoluble at ambient temperature, coupled with Ru/C catalysts, was utilized to promote the catalytic conversion of cellulose to EG. This reaction system could be repeated for more than 20 times with no obvious deactivation. A low concentration H3PO4 combined with Ni4.63Cu1Al1.82Fe0.79 was utilized in the hydrolytic hydrogenation of cellulose to sorbitol.44 The pretreatment of cellulose with acid with different concentrations of H3PO4 resulted in the destruction of the cellulose structure. Compared to the catalysis of Ni4.63Cu1Al1.82Fe0.79, the cellulose pretreated by 0.06% H3PO4 could be transformed to sorbitol and mannitol effectively.
2.2 Hydrolytic hydrogenation of cellulose promoted by acid formed in situ
Eckert et al.45 reported that an environmentally friendly in situ formed acid was obtained when the temperature of liquid water was above 473 K. These H+ ions disappear when the liquid water cools down to room temperature, indicating that this is a green process with possible industrial applications. In situ generated acid promoted catalytic conversion of cellulose over various catalysts are listed in Table 3.| Starting material | Catalyst | Conditions | Products and yield/% | Ref. |
|---|---|---|---|---|
| a SOR: sorbitol, MAN: mannitol, XYL: xylitol, SUA: sugar alcohols, ISO: isosorbide, SOT: sorbitan, MeOH: methanol. | ||||
| 2 wt% cellulose | Ru/C | 518 K, 6 MPa H2, 0.5 h | SOR (34.6%), MAN (11.4%), SOT (13.4) | 32 |
| 1 wt% cellulose | Ni–W2C/AC | 518 K, 6 MPa H2, 0.5 h | EG (61%), SOR (3.9%), MAN (1.9%) | 47 |
| 1 wt% cellulose | WCx/MC | 518 K, 6 MPa H2, 0.5 h | EG (72.9%), SOR (1.2%), MAN (1.4%) | 49 |
| 1 wt% cellulose | Ni5–W25/SBA-15 | 518 K, 6 MPa H2, 0.5 h | EG (72.9%), 1,2-PG (4.1%), SOR (3.1%), MAN (1.3%) | 50 |
| 1 wt% cellulose | Raney Ni + H2WO4 | 518 K, 6 MPa H2, 0.5 h | EG (65.4%), 1,2-PG (3.3%), SOR (7.5%), MAN (3.3%) | 51 |
| 1 wt% cellulose | 20% Ni/ZnO | 518 K, 6 MPa H2, 2 h | 1,2-PG (34.4%), EG (19.1%), 1,2-BDO (10.1%), 1,2-HDO (4.7%) | 31 |
| 1 wt% cellulose | 2Ni–3Cu–5ZnO | 518 K, 4 MPa H2, 0.5 h | 1,2-PG (24.3%), EG (16.9%), 1,2-BDO (7.1%), 1,2-HDO (5.3%) | 52 |
| 1.6 wt% cellulose | Ni/W/SiO2 | 518 K, 6 MPa H2, 1 h | EG (26.8%), SOR (2.3%) | 54 |
| 5 wt% cellulose | CuO/ZnO/Al2O3 | 518 K, 5 MPa H2, 2 h | MeOH (21%), EG (11.8%) | 53 |
The hydrolysis of cellulose catalysed by in situ generated reversible protons produced by high temperature liquid water was first employed for the production of sugar alcohols in Liu et al.'s research.32 Ru/C was chosen as the hydrogenation catalyst for its superior performance in glucose hydrogenation. After 30 min, the yield of hexitols was 39.3% at a conversion of 85.5% at 518 K and 6 MPa of H2. Moreover, small amounts of low carbon glycols such as PG and EG were produced because glucose was more active than the corresponding hexitols under the above mentioned reaction conditions. Once glucose was formed via the hydrolysis of cellulose, there existed two different approaches: if the rate of glucose hydrogenation was greater than that of cellulose hydrolysis, hexitols would be the main products, and on the contrary, glycols like EG and PG would be the major products. XRD patterns showed that the cellulose crystal structure did not change before and after the reaction, revealing that hydrolysis reaction mainly occurred on the surface of cellulose, which was also confirmed by subsequent investigations.46
Zhang and co-workers47 performed the hydrogenation of cellulose using tungsten carbide catalysis in order to replace the noble metal catalysts used previously. Tungsten carbide was found to give a higher yield of EG than platinum and ruthenium catalysts at the reaction condition of 518 K, 6 MPa H2, 30 min. Moreover, the yield of EG increased significantly from 27% to 61% with the promotion of a small amount of nickel. The reason that low molecular weight polyols were obtained as the main product over W2C/AC catalysts lay in the superior activity of glucose hydrogenolysis and inferior activity of glucose hydrogenation. Furthermore, the synergistic effect between nickel and W2C was another motivation that facilitated the remarkable increase in the yield of EG. Their further corresponding surface scientific studies48 indicated that the higher EG yield was due to weaker bonding between EG and Ni-promoted tungsten carbides. When 3D mesoporous carbon supported tungsten carbide nanoparticles were introduced in this reaction,49 the selectivity towards EG could be improved further to 72.9%. Zhang's group also developed a series of transition metal-W bimetallic catalysts capable of the production of EG from cellulose in one step.50 The tungsten component was found to be responsible for the C–C cleavage of glucose. On the other hand, it could also efficiently catalyse the hydrogenation of unsaturated intermediates. Thus, the final production distribution could be tuned by changing the weight ratio of transition metal to tungsten. Among the catalysts employed, Ni5–W15/SBA-15 catalysts gave a yield of EG as high as 75% at 518 K and 6.0 MPa of H2. To improve the reusability, the tungsten acid and Ru/C were combined and showed significant improvement in catalyst reusability.43,51 In their following studies,18 they found that the homogeneous tungsten bronze generated from the charged tungsten compounds functioned as uniquely active species for the C–C scission of glucose towards the formation of glycolaldehyde, which was further hydrogenated to EG catalysed by a hydrogenation active site.
In order to explore a catalyst that can effectively convert cellulose at low cost, we evaluated Ni based catalysts supported on various supports, including hydrothermally stable oxides with varying surface properties (Al2O3, kieselguhr, TiO2, SiO2, ZnO, ZrO2 and MgO) in previous investigations.31 It was found that a 20% Ni/ZnO catalyst could convert cellulose completely and give a 70.4% yield of 1,2-alkanediols composed of 1,2-PG, EG, 1,2-BDO and 1,2-HDO.
A possible mechanism of the cellulose hydrogenolysis reaction was also concluded by identifying the products formed during the reaction (Scheme 2). The main drawback of this catalyst lay in its poor hydrothermal stability, which resulted in the decrease of catalytic activity after repeated reaction runs, which was partially ascribed to the leaching of Ni. Further studies are needed to improve the stability and selectivity of current catalysts. We also prepared a series of Ni–Cu/ZnO bimetallic catalysts, which were successfully applied to the hydrogenolysis of cellulose for the purpose of producing 1,2-alkanediols as the major products.52 The molar ratio of Ni and Cu not only dominated the activity, selectivity and product distribution, but also played synergetic roles in the formation of 1,2-alkanediols during the reaction. The 2Ni–3Cu–5ZnO catalyst displayed the best 1,2-alkanediols selectivity (72.5%) with 74% cellulose conversion at 518 K, and 4 MPa of H2 after 0.5 h of reaction.
 | ||
| Scheme 2 Reaction pathway and main intermediates and products in the hydrolytic hydrogenolysis of cellulose. | ||
Palkovits et al.53 conducted the reaction under similar reaction conditions over the cheap CuO/ZnO/Al2O3 catalyst and small alcohols like methanol were obtained abundantly except for glycols and glycerol. By changing the average pore size of the support and the reduction temperature of the Ni/W/SiO2 catalysts, the crystalline size of W and the amount of surface acid site could be tuned, resulting in the alteration of the reaction activity.54 The product distribution was affected by valence state of Ni/W/SiO2 catalysts. Oxidized Ni and W species favored organic acids while the mixture of reduced and oxidized Ni and W species preferred low carbon polyols.
By employing a Pt–Mo2C/C catalyst, hydrogen could be substituted by CO and H2O in hydrolytic hydrogenation/hydrogenolysis of cellulose on the basis of Ma and co-workers' report.55 The hydrogen species needed in this reaction were produced via a water-gas shift reaction promoted by Pt–Mo2C domains present in Pt–Mo2C/C catalyst. Moreover, the Pt–C domain existing in this catalyst was responsible for the hydrogenation/hydrogenolysis reactions.
Recent investigations focused on the hydrolytic hydrogenation/hydrogenolysis of cellulose over bifunctional catalysts combined acid sites and metal active sites in hot compressed water. Such studies incorporated the advantages of solid acid with an in situ formed acid, as a consequence, cellulose could be upgraded to polyols such as sorbitol, mannitol, EG and PG, in a greener process efficiently.
2.3 Bifunctional catalysts
In the reaction of hydrolytic hydrogenation/hydrogenolysis of cellulose catalysed by bifunctional catalysts (solid acid and transition metal), cellulose first undergoes hydrolysis to produce glucose over acidic sites, and glucose is subsequently hydrogenated over the transition metal catalysts. Reaction products mainly include sorbitol, mannitol and some small glycol molecules such as EG and PG formed via the hydrogenolysis reaction of glucose and sorbitol hydrogenolysis. Table 4 demonstrates the catalytic conversion of cellulose over bifunctional catalysts.| Starting material | Catalyst | Conditions | Products and yield/% | Ref. |
|---|---|---|---|---|
| a BM: ball-milled, SOR: sorbitol, MAN: mannitol, GLU: glucose, HEX: hexitols, OLI: oligosaccharides, EGME: ethylene glycol monoether. | ||||
| 0.8 wt% cellulose | Pt/γ-Al2O3 | 463 K, 5 MPa H2, 24 h | SOR (25%), MAN (6%) | 30 |
| 2.5 wt% cellulose | Ru/C + WO3 | 478 K, 6 MPa H2, 0.5 h | EG (51.5%), PG (6.7%), SOR (15%) | 46 |
| 0.8 wt% cellulose-BM | Pt/BP2000 | 463 K, 5 MPa H2, 24 h | SOR (48%), MAN (9%) | 57 |
| 0.4 wt% cellulose-BM | Ni12P5 | 503 K, 5 MPa H2, 40 min | SOR (62%), MAN (4.5%) | 58 |
| 0.8 wt% cellulose-BM | Ru/CMK-3 | 503 K | GLU (27.6%), OLI (14.8%) | 59 |
| 2 wt% cellulose | 3.0 wt% Ni/CNF | 483 K, 6 MPa H2, 24 h | SOR (29.8%), MAN (5%), 1,2-PG (4.3%), EG (4.6%) | 61 |
| 2 wt% cellulose-BM | 7.5 wt% Ni/CNF | 463 K, 6 MPa H2, 24 h | HEX (75.6%) | 62 |
| 1 wt% cellulose | 16% Ni2P/AC | 498 K, 6 MPa H2, 1.5 h | SOR (48.4%), EG (8.2%), MAN (4.7%), 1,2-PG (2.2%) | 63 |
| 1 wt% cellulose | 16% Ni2P/SiO2 | 498 K, 6 MPa H2, 1.5 h | SOR (48%), EG (4%), MAN (4.5%), 1,2-PG (1.2%) | 63 |
| 1 wt% cellulose | 16% WP/AC | 498 K, 6 MPa H2, 1.5 h | SOR (1.2%), EG (26.5%), MAN (0.4%), 1,2-PG (2.3%) | 63 |
| 1 wt% cellulose | 1% Ru–5% Ni/MC | 518 K, 6 MPa H2, 0.5 h | EG (3.3%), 1,2-PG (3.0%), MAN (12.6%), SOR (41.6%) | 64 |
| 1 wt% cellulose | 1% Rh–5% Ni/MC | 518 K, 6 MPa H2, 0.5 h | EG (5.3%), 1,2-PG (3.5%), MAN (8.3%), SOR (51.5%) | 64 |
| 1 wt% cellulose | 1% Ir–5% Ni/MC | 518 K, 6 MPa H2, 0.5 h | EG (4.7%), 1,2-PG (3.4%), MAN (9.6%), SOR (47.9%) | 64 |
| 2 wt% cellulose | Ni/ZSM-5 | 513 K, 4 MPa H2, 4 h | HEX (48.6%) | 67 |
| 3 wt% cellulose | PtNi/ZSM-5 | 513 K, 4 MPa H2, 4 h | HEX (76.9%) | 69 |
| 2.8 wt% cellulose | Ir–BEA | 453 K, 1.6 MPa H2, 24 h | Selectivity SOR (89.2%) | 70 |
| 2.5 wt% cellulose | 3Ni–WO3/SBA-15 | 508 K, 6 MPa H2, 6 h | EG (70.7%), 1,2-PG (5.9%) | 71 |
| 0.8 wt% cellulose-BM | Ni/KB | 483 K, 5 MPa H2, 6 h | SOR (57%), MAN (6.8%) | 72 |
| 3.3 wt% cellulose | Ru/SiO2–SO3H | 423 K, 4 MPa H2, 10 h | SOR (61.2%), MAN (6.9%), PG (7.2%) | 73 |
| 0.8 wt% cellulose-BM | Ru/NbOPO4 | 443 K, 4 MPa H2, 24 h | SOR (69%) | 75 |
| 3 wt% cellulose-BM | Ru–Ni/NbOPO4 | 493 K, 3 MPa H2, 20 h solvent: methanol | EG (29.6%), EGME (35.5%) | 77 |
| 0.6 wt% cellulose-BM | Ru–PTA/MIL-100(Cr) | 463 K, 2 MPa H2, 8 h | SOR (57%), MAN (5.3%) | 78 |
| 0.8 wt% cellulose-BM | Pt/GRO | 463 K, 5 MPa H2, 24 h | SOR (58%), MAN (10%) | 79 |
| 10 wt% cellulose | CuCr + Ca(OH)2 | 518 K, 6 MPa H2, 5 h | 1,2-PG (42.6%), EG (31.6%) | 80 |
| 5 wt% cellulose (H2SO4-impregnated and BM) | Ru/C | 433 K, 5 MPa H2, 1 h | HEX (71.8%) | 83 |
Fukuoka and Dhepe30 pioneered the conversion of cellulose over solid acid supported noble metal catalysts. Cellulose could be directly hydrolytically hydrogenated into sugar alcohols over a bifunctional Pt/Al2O3 catalyst at 463 K in around 30% yield of hexitols after 24 h and sorbitol was the dominating product. When promoted by SnOx, as demonstrated by Liu et al.,56 the product composition could be tuned by varying the Sn/Pt atomic ratios in the catalyst. In order to reduce the crystallinity and particle size of cellulose which impeded the conversion of cellulose via heterogeneous catalytic transformation, ball-milling was applied in their investigations. By using platinum supported on a carbon black BP200 as a catalyst, sorbitol and mannitol were produced in 70% yield under the reaction conditions of 463 K, 5.0 MPa of H2 for 24 h.57 They also found that residual Cl on the catalysts induced side reactions like the cleavage of C–C and C–O bond, leading to lower yield of sugar alcohols. According to Fukuoka et al.58 an amorphous nickel phosphide phase generated from crystalline nickel phosphide crystalline upon increasing the temperature was responsible for the high yield of sorbitol. The leaching of P from the catalysts resulted in poor recyclability. A sorbitol yield of 60% could be obtained under the reaction conditions of 503 K, 5 MPa H2 after 40 min. As demonstrated by Fukuoka et al. water tolerable catalysts prepared by Ru supported on mesoporous carbon materials were introduced to promote the hydrolysis of cellulose.59 Ru/CMKs was proven to convert cellulose to glucose efficiently. The support material CMKs catalysed cellulose hydrolysis to oligosaccharides, and Ru was responsible for the conversion of oligosaccharides to glucose. A series of sulfonated silica/carbon nanocomposites for the highly efficient hydrolysis of cellulose to produce glucose were developed by Sels et al.60 The strength of the Brønsted acid density could be tuned by changing the carbon content of the sulfonated silica/carbon nanocomposites during the preparation steps, and thus influence the formation rate of glucose. A 50% yield of glucose was obtained at 423 K after 24 h.
In order to harmonize the acid sites and metal sites in the cellulose hydrogenation system to control the product distribution, bifunctional catalysts were needed, which joined the two kinds of active sites together properly. Hence, transition metals, such as nickel, copper and ruthenium, were supported on solid acids such as mesoporous carbon, active carbon, carbon nanofiber pretreated by mineral acid and zeolite like ZSM-5. These bifunctional catalysts can catalyse the hydrolytic hydrogenation of cellulose efficiently.
Liu et al.46 found that tungsten trioxide promoted ruthenium catalysts showed outstanding performance in the hydrogenolysis of cellulose to yield glycols at 478 K, and 6 MPa of H2. Tungsten trioxide was found to promote the hydrolysis of cellulose as well as C–C bond cleavage in sugars efficiently. EG was found to be derived from the C–C cleavage of glucose, while PG was produced by the degradation of fructose, which was formed by glucose isomerization.
As described above, solid catalysts promoted the hydrolytic hydrogenation of cellulose, which includes acid hydrolysis of cellulose to produce glucose, and the following hydrogenation of glucose to yield hexitols. If the glucose produced from the hydrolysis process was not be involved in the hydrogenation reaction immediately, glucose will undergo degradation, resulting in a decrease in the yield of hexitols. In order to find a catalyst, which can optimize the yield of hexitols, with an appropriate acid/metal ratio, Sels et al.61 oxidized carbon nanofibers with oxidation agents (HNO3 or a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HNO3–H2SO4) to introduce oxygen-containing surface groups. In their investigations, it was found that a low density of Brønsted acid sites combined with a high amount of Ni atom were the key point to maintain the high yield of hexitols. Ni supported at the tip of the carbon nanofiber prepared by catalytic vapour deposition of methane was introduced to promote the hydrogenation of cellulose.62 The bifunctional catalysts designed in this method improved the accessibility of active metal sites in the porous solids, thus giving a high yield of sorbitol (50.3%) at 463 K, and 6 MPa of H2 after 24 h.
:1 mixture of HNO3–H2SO4) to introduce oxygen-containing surface groups. In their investigations, it was found that a low density of Brønsted acid sites combined with a high amount of Ni atom were the key point to maintain the high yield of hexitols. Ni supported at the tip of the carbon nanofiber prepared by catalytic vapour deposition of methane was introduced to promote the hydrogenation of cellulose.62 The bifunctional catalysts designed in this method improved the accessibility of active metal sites in the porous solids, thus giving a high yield of sorbitol (50.3%) at 463 K, and 6 MPa of H2 after 24 h.
Dual-functional nickel phosphide catalysts were also used to promote the hydrogenation of cellulose, and sorbitol was the main product as demonstrated by Zhang et al.63 The results suggested that the hydrolysis of cellulose was promoted by homogeneous phosphoric acid generated by the leaching of P from nickel phosphide catalysts. As a consequence, this catalyst cannot sustain hydrothermal reaction conditions. Mesoporous carbon supported Ni-noble metal bimetallic catalysts were also introduced in the hydrolytic hydrogenation of cellulose.64 The mesoporous carbon prepared by a nanocasting method using commercial silica fume as the hard template not only facilitated the dispersion of metal active sites, but also played an important role in the reactant and product adsorption. The excellent performance of this catalyst largely relied on its superior ability to couple the cellulose hydrolysis and glucose hydrogenation reactions.
Nitric acid pretreated carbon nanotube (CNT) supported ruthenium catalysts were used in the direct hydrogenation of cellulose into sugar alcohol.65 The acidity of the catalyst played an important role in the hydrolysis of cellulose. The concentration of nitric acid used during CNT pretreatment and Ru particle size all exerted influence on the acid strength of the catalysts. A higher concentration of HNO3 and larger mean size of Ru particles resulted in higher hydrogenation acidity, thus leading to a high yield of sorbitol. The best sorbitol yield was obtained by catalysts with larger mean size of Ru particles and higher acidity (60% sorbitol and 98% cellulose conversion at 458 K, 8 MPa of H2 after 3 h). The research group also conducted the hydrolytic hydrogenation of ball-milled cellulose to sorbitol over Keggin-type polyoxometalate Cs3PW12O40 supported Ru catalysts in neutral water under mild conditions.66 The strong intrinsic acidity, which was related to the formation of sorbitol, was found to be generated in situ from H2 rather than Cs3PW12O40.
Zhao et al.67 prepared a series of ZSM-5 zeolite supported Ni catalysts to promote the hydrolytic hydrogenation of microcrystalline cellulose. They found that Ni/ZSM-5 catalysts prepared using an impregnation method afforded a high yield of hexitols. The Ni (1,1,1) crystal face, existing in these catalysts, was demonstrated as the main active agent in the cellulose hydrogenation reaction. The superior activity for glucose hydrogenation and inferior activity for sorbitol hydrogenolysis observed with these catalysts resulted in a high yield of hexitols. The influence of hydrogenation/dehydrogenation ability of Ni catalysts on the yield of hexitol during cellulose hydrogenation was also investigated.68 Over Ni/ZSM-5 catalyst, hexitols were produced with a selectivity of over 82%. However, the other Ni catalysts tested tended to produce small molecule glycols instead of hexitols. It was found that the high hydrogenation activity and inferior dehydrogenation activity of the Ni supported catalysts were the key points to gain high hexitol yield. The lower hexitol yield was attributed to the synergistic effect of Ni active species and acid-base sites, which accelerated the hydrogenolysis of sorbitol. Ni based bimetallic catalysts PtNi/ZSM-5 showed a hexitol yield of 76.9% under the reaction conditions of 513 K, 4 MPa of H2 after 4 h, and they could be reused 4 times.69 The high dispersion of PtNi alloy particles coupled with the superior ability of hydrogen spill over from the surface of the alloy was reasonable for the enhancement of the hydrogenation activity and the excellent hydrothermal stability of the catalysts. A BEA zeolite supported noble metal (Ir, Pd, Ru, Rh) was introduced to promote the conversion of cellulose to glycols.70 Among the selected noble metal catalysts, Ru/BEA encouraged the catalytic conversion of cellulose with the highest yield (22% conversion of cellulose, 72.8% conversion of glucose). The acidity and concentrated adsorbed hydrogen of the zeolite (are both high on Ru/BEA) were attributed to the formation of sorbitol. The selectivity of the product was related to the d-band width of the metal constituent, which was responsible for the higher selectivity of Ir than Ru. The status of the formation of sorbitol could be ameliorated by adding an additive such as pure nanoscopic hydroxylated SnF4. Nevertheless, this technique was hard for industrial applications since it was difficult to recover the material.
Wang et al.71 managed the hydrolytic hydrogenolysis of cellulose over Ni-WO3/SBA-15 catalysts in an aqueous phase. There existed a strong electronic interaction between NiO and WO3, which impacted the reduction of WO3 and NiO. WO3−x was found to be the active species responsible for the C–C cleavage in cellulose. Over the catalyst of 3% Ni–15% WO3/SBA-15, EG was produced in 70.7% yield when the conversion of cellulose was 100% at 503 K, 6 MPa of H2 after 6 h of reaction. Ni catalysts supported on metal oxide and carbons were also introduced to promote the hydrolytic hydrogenation of cellulose to hexitols.72 A high water tolerance, no basicity of active carbon and the large Ni particle size made Ni/KB an efficient catalyst for the production of hexitols.
The hydrogenation of cellulose to sorbitol catalysed by Ru/SiO2–SO3H bifunctional catalysts was conducted at low temperature.73 A high sorbitol yield was obtained because Ru nanoparticles and sulfonic groups interacted with each other through electron transfer, preventing sorbitol from further degradation. Sorbitol was produced at a yield of 61.2% at a temperature of 423 K, 4 MPa of H2 after 10 h of reaction.
The reactivity of hydrolytic hydrogenation/hydrogenolysis of cellulose over Pt/AlW and AlW was investigated by Rataboul et al.74 A comparison of the performances of Pt/AlW and AlW showed that Pt promoted the conversion of cellulose and changed the product distribution. Pyruvaldehyde was found to be the key intermediate in this reaction in the presence of Lewis acid sites. When AlW was employed as the catalyst, pyruvaldehyde could be transformed into lactic acid. With the promotion of Pt, pyruvaldehyde was hydrogenated to acetol and PG.
Mesoporous niobium phosphate supported ruthenium bifunctional catalysts were employed by Wang et al.75 in the catalytic hydrogenation of cellulose to sorbitol. NbOPO4 promoted the hydrolysis of cellulose to glucose, and Ru nanoparticles catalysed the hydrogenation of glucose to sorbitol. Sorbitol was generated in a yield of 59–69% with 90% cellulose conversion under the reaction conditions of 443 K, 4 MPa of H2 after 24 h. A two-step sequential process using the Ru/NbOPO4-pH2 and NbOPO4-pH2 catalysis was presented by the same research group for the conversion of cellulose to isosorbide.76 In this process, the hydrolytic hydrogenation of cellulose to sorbitol was promoted by Ru/NbOPO4-pH2, and the dehydration of sorbitol was catalysed by NbOPO4-pH2 to yield isosorbide. The acid amount of solid acid catalyst played a key role in the dehydration of sorbitol. When the reaction was performed in methanol, EG and ethylene glycol monoether (EGME) were generated as the main products over Ru/NbOPO4-pH2.77 Methanol was responsible for this product distribution because the acetalization of glucose with methanol protected the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of glucose, which resulted in the C–C cleavage of methyl glucoside to EG and EGME instead of glucose hydrogenation. Furthermore, the addition of Ni to the Ru/NbOPO4-pH2 catalyst prevented the degradation of EG and EGME, giving a high yield of EG and EGME. A total yield of 64% for EG and EGME was obtained over Ru–Ni/NbOPO4-pH2 under the condition of 493 K, 3 MPa of H2 after 20 h.
O bond of glucose, which resulted in the C–C cleavage of methyl glucoside to EG and EGME instead of glucose hydrogenation. Furthermore, the addition of Ni to the Ru/NbOPO4-pH2 catalyst prevented the degradation of EG and EGME, giving a high yield of EG and EGME. A total yield of 64% for EG and EGME was obtained over Ru–Ni/NbOPO4-pH2 under the condition of 493 K, 3 MPa of H2 after 20 h.
Ru–PTA/MIL-100 (Cr) was employed to catalyse the hydrolytic hydrogenation of cellulose because the ratio of acidity to the number of Ru surface atoms in this catalyst could be monitored.78 The balance between acid promoted hydrolysis and metal catalysed hydrogenation changed the product distribution. A sorbitol yield of 57% was produced under the condition of 463 K, 2 MPa of H2 after 8 h.
Wu et al.79 reported that Pt/GRO prepared by microwave-assisted EG reduction gave a high yield of sorbitol when it was employed in hydrolytic hydrogenation of cellulose. Graphene oxide was an excellent stabilizer for the Pt nanoparticles, which was related to the synergistic effects between the support and Pt nanoparticles. The hydrogen spillover of the Pt/GRO catalysts was responsible for the improvement in catalytic activity. The sorbitol yield was 58.9% under the conditions of 463 K, 5 MPa of H2 after 4 h.
Except for nickel, non-noble metal copper was also employed in the hydrogenolysis of cellulose. One of the aspects that prevent the industrialization of catalytic hydrogenation of cellulose to glycols is the low feedstock concentration (about 1–3%). It was because once the cellulose degraded into glucose through hydrolysis, the glucose produced must be hydrogenated into the corresponding sugar alcohols immediately or else coke-like precipitates will be generated and impede the reaction.
However, Liang et al.80 recently reported that concentrated cellulose (up to 15 wt%) could be converted into 1,2-PG and EG using hydrogenolysis over CuCr catalysts promoted by Ca(OH)2, with no coke-like precipitates formed. The authors believed that the addition of alkali in the reaction system promoted the C–C cleavage, and thus hampered the formation of coke-like precipitates. EG and 1,2-PG were obtained in yields of 42.6% and 31.6%, respectively, under the reaction conditions of 518 K, and 6.0 MPa of H2.
Alkaline pretreatment was utilized in the hydrolytic hydrogenolysis of cellulose by Wang et al.81 The pretreatment of cellulose with NaOH enhanced the cleavage of the cellulose chains, resulting in the promotion of the hydrolysis of cellulose. The main products of the hydrogenolysis of the alkaline pretreated cellulose were EG and PG, which was attributed to the basic conditions that alkaline pretreatment produced.
In order to degrade the robust cellulose, Beltramini et al.82 employed mechanical depolymerisation of cellulose to accelerate the production of water soluble oligomers. In the presence of bi-metallic Ni–Pt/alumina catalyst, a high yield of sorbitol and mannitol (90%) were produced under the condition of 473 K, 5 MPa of H2 after 1 h. Mechanocatalytic depolymerization was also introduced by Schüeth et al.83 to efficiently convert cellulose into low carbon glycols. The process was combined with Ru/C to promote cellulose hydrogenolysis reaction, and a high yield of hexitols (94%) was produced at 423 K and 5 MPa of H2.
2.4 Utilization of ionic liquids
Apart from water, ionic liquid, which is another green solvent, was also employed in the hydrolytic hydrogenation of cellulose. Difficulties in heterogeneous hydrolytic hydrogenation of cellulose lay in the problematic mass transfer of solid catalysts and water insoluble cellulose. However, cellulose can be dissolved in an ionic liquid,84 improving the contact between the solid catalysts and cellulose. As a result, the hydrolysis of cellulose can be improved when ionic liquid is employed as the solvent. There are some reports on the depolymerization of lignocellulose in ionic liquid.85–87 Nonetheless, few studies have focused on the one-pot transformation of lignocellulose into glycols. The utilization of an ionic liquid in the hydrolytic hydrogenation/hydrogenolysis of cellulose is shown in Table 5.| Starting material | Ionic liquid | Catalyst | Conditions | Products and yield/% | Ref. |
|---|---|---|---|---|---|
| a GLU: glucose, SOR: sorbitol, MAN: mannitol. | |||||
| 5 wt% cellulose | [Bmim]Cl | Ru/C | 423 K, 3.5 MPa H2, 24 h 0.17 wt% KOH | Selectivity: GLU (43%), SOR (29%), MAN (5%) | 89 |
| 5 wt% cellulose | [Bmim]Cl | HRuCl (CO) (PPh3)3 | 423 K, 3.5 MPa H2, 24 h 0.17 wt% KOH | Selectivity: GLU (14%), SOR (74%) | 89 |
| Cellulose | [Bmim]Cl | Ru nanoparticles | 353 K, sodium formate as hydrogen source, 5 h | SOR (94%) | 90 |
| 5 wt% cellulose | Ru/[Bmim]3PW12O40 | 433 K, 5 MPa H2, 24 h | Selectivity: SOR (70.3%) | 91 | |
Kou and Liu reported the one-pot catalytic conversion of cellulose and cellobiose over metal nanoclusters in ionic liquid.88 In their research, cellulose could be converted (16% conversion) to hexitols over the catalysis of Ru nanoclusters in an ionic liquid such as [Bmim] Cl. Cellulose unfolded in the ionic liquid ([Bmim] Cl) could be converted into hexitols using heterogeneous or homogenous catalysts.89 When promoted by Ru/C, cellulose could be successfully transformed to glucose, sorbitol and mannitol (cellulose conversion 57%, selectivity of the products: glucose 43%, sorbitol 29%, and mannitol 5%). Sorbitol could be obtained at a selectivity of 74% over the catalysis of HRuCl(CO) (PPh3)3 at a cellulose conversion of 86% under the condition of 423 K, 3.5 MPa of H2 after 24 h. Zhu et al.90 reported that an ionic liquid stabilized ruthenium nanoparticle catalyst could be employed in the hydrogenation of cellulose to hexitols. When the ionic liquid, i.e. [Bmim] Cl, was utilized in this system with sodium formate as the hydrogen source, complete cellulose conversion could be obtained with a sorbitol yield of 94% under the reaction conditions of 353 K after 5 h. Ru/[Bmim]3PW12O40 was synthesized by dispersion of Ru on ionic liquid (BmimPF6) heteropolyacid (H3W12O40 nH2O) hybrid as a support and was introduced into the hydrolytic hydrogenation of cellulose by Ge et al.91 The catalyst combined the function of hydrogenation (Ru sites) and hydrolysis (both Lewis and Brønsted acidic sites). Under the conditions of 433 K and 5 MPa of H2, microcrystalline cellulose could be converted into sorbitol with a conversion of 63.7% and a selectivity of 70.3% after 24 h of reaction. Brønsted acid generated through hydrogen spillover coupled with the supported Ru can promote the hydrolytic hydrogenation of cellulose to glycols.
2.5 Conversion of cellulose raw material to glycols
Considerable progress has been achieved in the hydrolytic hydrogenation/hydrogenolysis of cellulose to date. In order to accelerate the process of industrialization of biomass based chemicals and fuels, the emphasis of the researchers has been turned towards employing real biomass as the feedstock in this reaction. However, the composition of the real biomass feedstocks exerted a notable influence on the catalytic activity, and three major components of lignocellulose, namely, cellulose, hemicellulose and lignin, were tangled together and caused the separation of these compositions difficult to realize. Moreover, the presence of inorganic salts (CaCO3, NaHCO3 and Na2SO4)92 in lignocellulose is another obstacle, and the presence of these salts shifted the product distribution from sorbitol to by-products. Consequently, investigations on the hydrolytic hydrogenation/hydrogenolysis of raw lignocellulose are worth conducting. Catalytic conversion of raw lignocellulose is demonstrated in Table 6.| Starting material | Catalyst | Conditions | Products and yield/% | Ref. |
|---|---|---|---|---|
| a SOR: sorbitol, ISO: isosorbide, SUA: sugar alcohols, XYL: xylitol, JAS: Jerusalem artichoke stalk. | ||||
| 1 wt% corn stalks | 2% Ni–W2C | 518 K, 6 MPa H2, 2 h | EG (18.3%), PG (13.9%) | 93 |
| 1 wt% Birch | 4% Ni–30% W2C/AC | 508 K, 6 MPa H2, 4 h | EG (51.4%), 1,2-PG (14.2%) | 94 |
| 1 wt% Poplar | 4% Ni–30% W2C/AC | 508 K, 6 MPa H2, 4 h | EG (48.6%), 1,2-PG (12.8%) | 94 |
| 1 wt% Basswood | 4% Ni–30% W2C/AC | 508 K, 6 MPa H2, 4 h | EG (49.2%), 1,2-PG (11.8%) | 94 |
| 1 wt% Ashtree | 4% Ni–30% W2C/AC | 508 K, 6 MPa H2, 4 h | EG (52.7%), 1,2-PG (11.9%) | 94 |
| 1 wt% Beech | 4% Ni–30% W2C/AC | 508 K, 6 MPa H2, 4 h | EG (35.2%), 1,2-PG (11.4%) | 94 |
| 1 wt% Xylosma | 4% Ni–30% W2C/AC | 508 K, 6 MPa H2, 4 h | EG (36.4%), 1,2-PG (13.7%) | 94 |
| 5 wt% spruce | Ru/C | 433 K, 5 MPa H2, 1 h H2SO4 | SOR (36.0%), ISO (7.3%) | 33 |
| 5 wt% spruce | Ru/C | 433 K, 5 MPa H2, 1 h H3PO4 | SOR (24.3%), ISO (8.8%) | 33 |
| 5 wt% spruce | Ru/C | 433 K, 5 MPa H2, 5 h, 5 wt% H4[Si(W3O10)4] | C4–C6 SUA (64.9%) | 37 |
| 1 wt% cornstalk | 2Ni–3Cu–5ZnO | 518 K, 4 MPa H2, 0.5 h | 1,2-PG (20%), EG (15%), 1,2-BDO (4.6%), 1,2-HDO (5.4%) | 52 |
| 1 wt% corncob | 2Ni–3Cu–5ZnO | 518 K, 4 MPa H2, 0.5 h | 1,2-PG (24%), EG (17.6%), 1,2-BDO (7.4%), 1,2-HDO (4.3%) | 52 |
| 0.8 wt% silver grass | Pt/BP2000 | 463 K, 5 MPa H2, 24 h | SOR (13%), XYL (14%) | 92 |
| 1 wt% JAS | WO3 + Raney Ni | 518 K, 6 MPa H2, 2 h | EG (37.6%), 1,2-PG (6.3%) | 97 |
Spruce was employed as the lignocellulose feedstock in the hydrogenolysis reaction over Ru/C under the promotion of phosphoric acid and sulfuric acid. The yields of hexitols were 43.3% and 33.1% over Ru/C with the promotion of sulfuric acid and phosphoric acid, respectively. HPA was also utilized in hydrogenolysis of spruce, and a yield of 64.9% C4–C6 sugar alcohols was obtained with the promotion of H4[Si(W3O10)4]. In our study, the hydrogenolysis of cornstalk and corncob was conducted over Ni–Cu–ZnO catalysts.52 The selectivities of 1,2-alkanediol were 48.5% and 55.5% with cornstalk and corncob as feedstocks, respectively, under the reaction conditions of 518 K, 4 MPa of H2. Corn stalks were used as the feedstock for the hydrogenolysis reaction to yield EG and 1,2-PG.93 Cellulosic feedstocks with different chemical components and structures were prepared by introducing different pretreatment techniques in this research. The results showed that crystallinity of the cellulosic feedstock has limited effects on the formation of glycols in this reaction. The catalytic hydrogenation of cellulose pretreated by ammonia gave a total yield of 48% for both EG and 1,2-PG at 518 K, 6.0 MPa of H2. Raw woody biomass was also employed as a feed stock over supported carbide catalysts, which exhibited high activity in catalytic hydrogenation.94 Different sources of lignocellulose were introduced as feedstocks in this research and it was found that with different chemical component, different catalytic performances were observed over supported carbide catalysts. Compared to supported noble metal catalysts, low price Ni–W2C/AC showed much higher catalytic activity and gave a much higher glycol yield under the same reaction conditions.
Steam explosion and alkali were found to be an efficient pretreatment technology in the hydrogenolysis of corn stalks.95 Hemicellulose and lignin could be efficiently removed from corn stalk, showing that this is a valid approach in raw material pretreatment. Corn stalks could be converted into EG (yield 20.38%) and glycerol (yield 52.36%) over the Ni–W2C catalyst after being pretreated by steam explosion and alkali.
Kraft pulp from pulp mills was introduced as the feedstock in the hydrolytic hydrogenation reaction by Murzin et al.96 to afford sugars and sugar alcohols over metal supported on H-MCM-48 under the conditions of 458 K, 2 MPa of H2. The active sites, acidity and structure of the mesoporous material have a great influence on the yield of the sugars and sugar alcohols.
Raw Jerusalem artichoke stalks (JAS) were employed by Zhang et al.97 as a feedstock in the catalytic conversion promoted by commercial WO3 and Raney Ni. The water-soluble substances that exist in the JAS were found to cause a negative effect on the conversion of hemicellulose. By employing a simple hot water pretreatment process, the EG yield could be increased from 29.9% to 37.6% because most of the water-soluble substances could be removed.
Fukuoka et al.92 employed raw silver grass as a feedstock for the hydrolytic hydrogenation reaction using carbon supported Pt catalysts to produce polyols. It was found that lignin found in raw silver grass diminished the reaction activity of cellulose. With the existence of lignin, a small amount of sorbitol (2.8 wt%) and xylitol (7.3 wt%) was obtained. By introducing weak bases, product distribution can be tuned towards EG and PG, which was supposed to be generated from the decomposition of sugar and sorbitol. Alkali-explosion and neutralization were found to be efficient techniques that could increase the yield of sorbitol (13 wt%) and xylitol (14 wt%).
3. Hydrogenation and hydrogenolysis of sugar and sugar alcohols
The hydrogenation/hydrogenolysis of sugar and the hydrogenolysis of sugar alcohols are important steps not only in the one-pot hydrolytic hydrogenation/hydrogenolysis of lignocellulose but also in the multi-step lignocellulose transformation. Investigations on the catalytic hydrogenation/hydrogenolysis of sugar and the hydrogenolysis of sugar alcohols are essential to gain insight into the process of glycol production. In this part, we mainly focus on the recent advances in hydrogenation/hydrogenolysis of sugar and sugar alcohols with promotion by supported catalysts. To date, studies on developing catalysts utilized in the sugar and sugar alcohols hydrogenation/hydrogenolysis reactions are based on transition metal catalysts such as nickel, copper and ruthenium catalysts.3.1 Reaction mechanism of glucose and sugar alcohols hydrogenolysis
Glucose and sorbitol have similar molecular structures because sorbitol can be produced via a simple glucose hydrogenation reaction. Hydrogenolysis of these two hydroxyl compounds both need metal catalysts and base promoters under similar reaction conditions. Therefore, hydrogenolysis of glucose and sorbitol to glycols might follow a similar reaction mechanism.14 It is well known that the process of glucose and sorbitol hydrogenolysis involved the cleavage of C–C and C–O bond, as the final products comprised several low carbon polyols. As a consequence, it is necessary to verify the reaction mechanism of glucose and sorbitol hydrogenolysis so as to control the product distribution reasonably.Sohounloue et al.98 conducted the reaction of sorbitol hydrogenolysis over Ru/SiO2 catalysts by changing the reaction temperature and aqueous pH. They proposed that C–C bond cleavage during sorbitol hydrogenolysis seems to follow a retro-aldol condensation mechanism in a basic medium (Scheme 3 route B). Sorbitol first undergoes a dehydrogenation reaction to produce an intermediate product, and then an enol–ketone equilibrium could be obtained, resulting in an isomeride, and finally this isomeride comes across a retro-aldol reaction with small molecule glycols produced. Andrew et al.99 believed that C–C bond cleavage in the hydrogenolysis of sugar (glucose and fructose) also follows the same retro-aldol condensation mechanism. The distribution of reaction products was found to dependent on the competition between the reaction rate of retro-aldol cleavage and sugar hydrogenation. According to the retro-aldol condensation mechanism, hydrogenolysis of glucose and sorbitol needs a β-hydroxyl carbonyl structure generated from sorbitol dehydrogenation. The β-hydroxyl carbonyl formed undergoes the retro-aldol condensation reaction to form an aldehyde and ketone, which can be hydrogenated to low carbon glycols. In this reaction, metal catalysts promote the dehydrogenation of sugar alcohols and the hydrogenation of the intermediate products formed, while the base catalysed the retro-aldol condensation of the β-hydroxyl carbonyl.
 | ||
| Scheme 3 Reaction mechanism of sorbitol hydrogenolysis (A) C–O bond cleavage proposed by Montassier100 (B) C–C bond cleavage under the retro-aldol mechanism proposed by Sohounloue and Andrew98,99 (C) C–C bond cleavage under the retro-Michael mechanism proposed by Montassier.101 | ||
The C–O cleavage mechanism in the hydrogenolysis of glucose and sugar alcohols was proposed by Montassier et al.100 They proved that the precursor of dehydration reaction was a β-hydroxyl carbonyl, which is the same as that found in the retro-aldol condensation mechanism. After the dehydration reaction of β-hydroxyl carbonyl precursor, many α,β-unsaturated carbonyl species were generated directly and then hydrogenated to glycols (Scheme 3, route A). In this reaction, metal catalysts promote the dehydrogenation and hydrogenation reactions and the base catalysed dehydration of the β-hydroxyl carbonyl precursor. By conducting the reaction of sorbitol hydrogenolysis on supported ruthenium catalysts, Montassier et al.101 believed that sorbitol mainly undergoes retro-Michael reactions to produce two molecule glycerol by breaking the central C–C bond (Scheme 3 route C). Note that a diacarbonyl species was essential in retro-Michael mechanism.
The reaction mechanism of sugar and sugar alcohols hydrogenolysis was deduced using 1,3-diol model compounds as reactants by Wang et al.102 C–C and C–O bond cleavage in sugar and sugar alcohols hydrogenolysis were attributed to a retro-aldol mechanism and dehydration of β-hydroxyl carbonyl, respectively. The retro-Michael mechanism in sugar and sugar alcohols hydrogenolysis was found to be inappropriate, because the dehydrogenation of monocarbonyl species was unfavorable thermodynamically. As a consequence, the diacarbonyl species, a crucial intermediate in the retro-Michael mechanism would not be generated. Moreover, the precursor of the retro-Michael reaction was difficult to form because the dehydration reaction of the single carbonyl species easily occurred compared to the dehydrogenation reaction. Hydrogenolysis of sugar and sugar alcohols was supposed to occur as per the following mechanism: (1) dehydrogenation of sugar alcohols to the intermediate product; (2) the intermediate product undergoes two different pathways: (a) C–O cleavage reaction occurred via the dehydration of the intermediate product, followed by hydrogenation to produce glycols; and (b) C–C cleavage reaction realized through retro-aldol condensation of the intermediate product, followed by hydrogenation to produce small molecule glycols.
3.2 Hydrogenation and hydrogenolysis of sugars
Glucose (40 wt%) was hydrogenated into sorbitol over ruthenium catalysts supported on active charcoal pellets in a trickle-bed reactor by Gallezot et al.103 The catalysts were prepared by loading the metal on the supports through cationic exchange or anionic adsorption. The conversion of glucose could be 100%, while the selectivity of sorbitol was 99.2% under the reaction conditions of 373 K, 8 MPa of H2. The residence time of the reactant influence the epimerization of sorbitol to mannitol, which finally affected the sorbitol selectivity. Hydrogenation of glucose was also conducted over supported ruthenium catalysts104 with diverse ruthenium particles under the condition of 393 K and 1.9 MPa of H2 (Table 7). Sorbitol could be produced at a relatively high selectivity (87–96%) under the same reaction conditions and catalysts. It was found that the TOF of glucose hydrogenation was related to the particle size of ruthenium.| Starting materials | Catalysts | Conditions | Main product and yield/% | Ref. |
|---|---|---|---|---|
| a SOR: sorbitol, GLY: glycerol, MAN: mannitol, XYL: xylitol, GLU: glucose, FRU: fructose. | ||||
| 40 wt% GLU | Ru/C | 373 K, 8 MPa H2 | SOR (>99%) | 103 |
| 40 wt% GLU | Pt/ACC | 373 K, 8 MPa H2 | SOR (>99.5%) | 111 |
| 17 wt% GLU | (Ni, Mo and Cu)/kieselguhr | 423 K, 5 MPa H2, 2 h | GLY (28%), EG (22%), PG (13%), SOR (4%) | 112 |
| 1.8 wt% GLU | Ru/C | 393 K, 1.9 MPa H2, 2–3 h | SOR (96%) | 104 |
| 0.45 wt% GLU | Pt/γ-Al2O3 + hydrotalcite | 363 K, 1.6 MPa H2, 4 h | SOR (54%), MAN (14%), XYL (4%) | 9 |
| 1.7 wt% GLU | Ni–W2C/AC | 518 K, 6 MPa H2, 3 h | EG (36%), 1,2-PG (8%), SOR (9%) | 117 |
| 5 wt% GLU | Ni2.75Cu1Al1.49 | 398 K, 3 MPa H2, 3 h | SOR (65%) | 116 |
| 10 wt% GLU | Ru/MCM-41 | 393 K, 3 MPa H2, 2 h | SOR (80%) | 106 |
| 25 wt% GLU | Ru/ZSM-5 | 393 K, 4 MPa H2, 2 h | SOR (99.2%) | 110 |
Because of the superior mass transfer ability from micropore to liquid phase and the better desorption ability of sorbitol than the conventional carbon, woven rayon fabric derived active carbon cloths supported ruthenium showed outstanding performance in hydrogenation of glucose to sorbitol (99.5 selectivity at 99.7% conversion under the condition of 373 K, 8 MPa H2), based on the research of Besson et al.105
Lin et al.106 conducted the hydrogenation of glucose to sorbitol over Ru supported on MCM-41. Higher sorbitol yield (94%) was obtained over the Ru/MCM-41 catalyst compared to other catalysts such as Ru/C, Ni powder and Pd/C, under the reactions conditions of 393 K and 3 MPa of H2. The excellent reaction performance was attributed to the higher metal dispersion in Ru/MCM-41.
Zhao et al.107 presented that Ru nanoparticles containing carbon microfibers showed higher activity in glucose hydrogenation than Ru supported on multi-walled carbon nanotubes, alumina microfibers and active carbon. The high activity and stability of this catalyst was attributed to the high degree of Ru dispersion, appropriate Ru particle size and high crystallinity.
Yang et al.108 conducted the hydrogenation of glucose to sorbitol over Ru supported on multi-wall carbon nanotubes prepared by an impregnation method. These catalysts showed higher activity in the hydrogenation reaction compared with Ru supported on Al2O3, SiO2 and Raney Ni. The superior reaction activity of these catalysts was due to the fine dispersion of Ru on the support which was confirmed by TEM analysis. Kilpio et al.109 performed the hydrogenation of glucose to sorbitol over a Ru/C catalyst in a laboratory-scale trickle bed reactor and in a semi batch stirred reactor. Sorbitol was produced at a selectivity of over 90% at temperatures ranging from 363 K–403 K. The reaction temperature impacted the reaction rate for glucose hydrogenation while the selectivity of the sorbitol did not change when the temperature was altered.
Hydrogenation of D-glucose over Ru/ZSM-5 prepared by one-step template-free process was evaluated by our research group.110 Selectivity of D-sorbitol was 99.2% with 99.6% glucose conversion when the hydrogenation reaction was conducted under the reaction conditions of 393 K, 4 MPa of H2 after 2 h. The superior performance of Ru/ZSM-5 was attributed to the high dispersion of Ru, the strong interaction between Ru and ZSM-5 and the appropriate acidity-basicity balance of the Ru/ZSM-5 surface.
Perrard et al.111 conducted the glucose hydrogenation reaction over activated carbon cloth supported platinum. A high sorbitol selectivity was obtained because of the low probability of D-sorbitol epimerization, and the desorption rate of D-sorbitol from the catalysts was faster than that using conventional Pt/C. Dhepe et al.9 conducted the glucose hydrogenation reaction using Pt/γ-Al2O3 promoted by hydrotalcite, a basic promoter. Sugar alcohol was obtained in a high yield of 68% under the condition of 363 K, 1.6 MPa of H2. Glucose can be transformed into open chain form, which is readily hydrogenated into sorbitol and mannitol by the highly dispersed metal particles in the alkaline medium.
Using a kieselguhr supported Ni, Mo, and Cu catalyst, Saxena et al.112 successfully converted sucrose via hydrogenolysis to produce glycerol, EG, PG, and sorbitol. Mo and Cu were chosen to promote the activity of the supported nickel catalyst. By coordinating the amount of Ni, Mo, and Cu in the catalysts, the conversion of sucrose and the yield of glycerol could be optimized. Pachulski et al.113 introduced Ni supported on ZrO2 and/or TiO2 towards the hydrogenation of glucose to sorbitol. These catalysts showed outstanding performance in the hydrogenation of glucose compared to the Ni/SiO2 catalysts, which was attributed to the interaction between the metal and support. Raney-type Ni (promoted and unpromoted) was also employed in the hydrogenation of an aqueous solution of glucose (10 wt%) at 393 K and 4 MPa of H2 in a three-phase slurry reactor.114 During the catalytic reaction, the gluconic acid formed resulted in the leaching of Ni, Al and Fe from the surface of the catalysts, causing severe Raney Ni deactivation.
Li et al.115 found that a Co–B amorphous alloy demonstrated high activity in glucose hydrogenation to sorbitol because of the electronic interaction between Co and the B alloy and the superior hydrogen adsorption ability of Co active sites. Cr and W were both excellent promoters that could enhance the hydrogenation activity of Co–B alloy. Low-valent state Cr− or W− promoted polarization of CO bond, making the nucleophilic attack of hydrogen adsorbed on the Co active sites easier. Therefore, the reaction rate for hydrogenation could be enhanced. Ni/Cu/Al hydrotalcites prepared by a co-precipitation method were also tested by Liu et al.116 towards the hydrogenation of glucose to sorbitol. A sorbitol selectivity of 90% was obtained over a Ni2.75Cu1Al1.49 catalyst at 398 K and 3 MPa of H2.
Sels et al.117 found that concentrated sugar solution could be converted into low chain glycol EG via the hydrogenolysis reaction. A key intermediate product glycol aldehyde generated by the retro-aldol condensation of glucose was identified by the researchers, glycol aldehyde was regarded as the precursor for EG. Liang et al.118 performed a two-step hydrogenation of highly concentrated glucose over Cu–Cr catalysts with base promoters. During the low temperature step, sorbitol and mannitol was easily produced via the hydrogenation of glucose. The retro-aldol condensation reaction occurred when base was added into the reaction system. The formation of coke-like precipitates could be avoided in the presence of base promoters during the high-temperature step. Different base promoters were also introduced during the hydrogenation of glucose, the concentration of OH−, the metal ionic radius and the electric charge were found to be related to the conversion of glucose. Glucose hydrogenolysis conducted over copper catalysts supported on sulfated spherical carbon was found to yield 1,2-PG as the main product.119 The selectivity of 1,2-PG could be optimized by tuning the acid sites and the hydrogenolysis sites in the catalysts.
3.3 Hydrogenolysis of sugar alcohols
As described above, sugar alcohols, including sorbitol and xylitol, can be obtained using hydrogenation of the corresponding sugars under the catalysis of supported Ru, Pt, Ni and other metals. Hydrogenation of sugar to corresponding sugar alcohols could be conducted under pretty mild condition, while the hydrogenolysis of sugar and sugar alcohols requires higher temperatures, H2 pressure and base promoters.14 Among the sugar alcohols, sorbitol can be produced industrially on a large scale by the hydrogenation of glucose using Ni or Ru catalysts. Sorbitol is also an important renewable carbon source and has been considered as one of the 12 top building blocks in biorefinery by the United States Department of Energy (US DOE).6,120 The main products obtained from the hydrogenolysis of sugar alcohols are glycols with short carbon chains like glycerol, EG, 1,2-PG and 1,3-PG, which are important chemicals traditionally produced using petrochemical methods.10 In this part, we will review the recent advances in hydrogenolysis of sugar alcohols to produce glycols. Table 8 shows the hydrogenolysis of sugar alcohols using different catalysts.| Starting materials | Catalysts | Conditions | Products and yields/% | Ref. |
|---|---|---|---|---|
| a SOR: sorbitol, XYL: xylitol, GLY: glycerol. | ||||
| 20 wt% SOR | Ru/CNF | 493 K, 8 MPa H2, 4 h + CaO | EG (19.32%), PG (31.98%), GLY (9.53) | 122 |
| 10 wt% XYL | Ru/C | 473 K, 4 MPa H2, 1 h + Ca(OH)2 | Selectivity: EG (32.4%), PG (24.9%), GLY (9.6%) | 125 |
| 10 wt% XYL | Pt/C | 473 K, 4 MPa H2, 1 h + Ca(OH)2 | Selectivity: EG (25%), PG (23%), GLY (10%) | 125 |
| 10 wt% XYL | Cu–SiO2 | 473 K, 4 MPa H2, 2 h + Ca(OH)2 | Selectivity: EG (19.4%), PG (19.5%), GLY (4.4%) | 126 |
| 10 wt% XYL | Ni/C | 473 K, 4 MPa H2, 1 h + Ca(OH)2 | Selectivity: EG (32.0%), PG (33.7%) | 133 |
| 30 wt% SOR | Ce–Ni/Al2O3 | 493 K, 7 MPa H2, 8 h, + Ca(OH)2 | EG (17.7%), PG (35.6%), GLY (25%) | 127 |
| 5 wt% SOR | Ni2P/AC | 473 K, 4 MPa H2, 0.75 h, + Ba(OH)2 | EG (17%), PG (27.7%), | 128 |
| 20 wt% SOR | Ni–MgO | 473 K, 4 MPa H2, 4 h | Selectivity: EG (26.0%), PG (33.7%), GLY (21.1%) | 132 |
| 10 wt% SOR | Ni–Re/C | 523 K, 1 MPa H2 N2, 0.5 h + Ba(OH)2 | Selectivity: EG (15.8%), 1,2-PG (31%), GLY (6.8%) | 135 |
| 15 wt% SOR | Ni–NaY | 493 K, 6 MPa H2, 6 h + Ca(OH)2 | Selectivity: EG (7%), 1,2-PG (69%), GLY (4%) | 130 |
| 25 wt% SOR | Ru/Al2O3 | 493 K, 4 MPa H2, 4 h | Selectivity: glycols (19.1%) | 134 |
Gallezot et al.121 demonstrated that a sorbitol solution derived from biosustainable resources could be converted into C4–C6 polyols using catalytic hydrogenolysis over copper catalysts. The CuO–ZnO catalysts gave a 73% yield of C4 polyols under the condition of 453 K and 13 MPa of H2.
Ruthenium nanoparticles supported on carbon nanofibers were prepared and used in the hydrogenolysis reaction to produce small molecule glycols.122 The conversion of sorbitol and the selectivity of the target products over Ru/CNF were found to be superior to Ru supported on commercial activated carbon. Experimental results also indicated that glycerol was the precursor to PG. Further investigations were also conducted to explore the effect of calcination on sorbitol hydrogenolysis to glycols.123 Surface oxygen-containing groups (SOCGs) were introduced onto the surface of the carbon nanofibers via calcination. The interaction between the Ru particles and sorbitol molecules could be impeded by SOCGs, reducing the conversion of sorbitol. However, unsaturated species were more easily to be hydrogenated to give glycols in the presence of SOCGs because these species could be restrained around the Ru particles by the SOCGs. As a result, the selectivities of glycols could be obtained at a higher level.
The effect of different base promoters on the hydrogenolysis of sorbitol was investigated by employing different bases [NaOH, KOH, Mg (OH)2, Ba(OH)2 and CaO] in the reaction system.124 Among the bases utilized, CaO was proven to give the highest glycol selectivity. Ca2+ was found to be an important ion in this reaction because the complexation of intermediate aldehydes and Ca2+ maximize the selectivity towards glycols.
The hydrogenolysis of xylitol was conducted with the promotion of catalysts prepared by noble metals dispersed on various supports in the presence of base promoter by Sun and Liu.125 The dehydrogenation/hydrogenation activities combined with the surface acid-basicity of the catalysts dominated the final product distribution. The reaction pathway for xylitol hydrogenolysis was proposed to follow the retro-aldol condensation mechanism, xylitol underwent a process of dehydrogenation to xylose on the surface of the metal, and then, base promoted retro-aldol condensation of xylose occurred, resulting in two intermediate products, glycolaldehyde and glyceraldehyde, which were hydrogenated to EG and PG, respectively. Copper supported on SiO2 was also introduced into the xylitol hydrogenolysis reaction.126 The yields of EG and PG were 19.4% and 19.5%, respectively, under the reaction conditions of 473 K, 4.0 MPa of H2.
Ce was doped into the Ni/Al2O3 catalysts by Yuan et al.127 through different methods in order to promote their catalytic activity in the sorbitol hydrogenolysis. Over the Ce–Ni/Al2O3-CP catalyst, glycols were produced at a selectivity of 55–60% at a 90% sorbitol conversion under the reaction conditions of 513 K, 7 MPa of H2 after 12 h.
Metal phosphides supported on carbon were used to promote the hydrogenolysis of sorbitol, xylitol and glucose to produce EG and PG. Ni2P was regarded as the active phase, which gave the high activity observed in the hydrogenolysis of sorbitol128 under the reaction condition of 473 K, 4.0 MPa of H2. The yields of EG and PG was 17.0 and 28.5 mol% and 27.7 and 42.9 mol% when sorbitol and xylitol were employed as reactant, respectively.
An aqueous-phase hydrodeoxygenation of sorbitol was also conducted by Li and Huber129 using Pt/SiO2–Al2O3 catalysts. The main reaction pathway was identified by the identification of reaction intermediates. C–C bond cleavage was achieved via a retro-aldol condensation and decarbonylation, which took place on metal sites. C–O bond cleavage was accomplished through the dehydration reaction, which occurred on the acid sites.
Nickel and platinum were supported on NaY to catalyse the hydrogenolysis of sorbitol to produce low carbon glycols.130 1,2-PG was obtained as the main product over Ni–NaY while glycerol was the principal product when Pt–NaY was utilized. A negligible effect on the sorbitol conversion and product selectivity was observed when Pt was doped into the Ni–NaY catalyst; however, the addition of base promoter like Ca(OH)2 accelerated the conversion of sorbitol rather than product selectivity.
Besson et al.131 performed the hydrogenolysis of an alkaline aqueous solution of xylitol to EG and PG using Ru/C catalysts in a trickle-bed reactor. The influence of reaction parameters such as sodium hydroxide concentration, hydrogen pressure and temperature were explored in this research. The reaction results were consistent with the reaction pathway proposed before, and the hydrogenolysis of xylitol followed the retro-aldol condensation mechanism.
In previous studies, base promoters, such as NaOH and Ca(OH)2, were inevitably required in the hydrogenolysis of sugar alcohols because the base promoter was necessary in the C–C cleavage of the dehydrogenation intermediate products. However, the use of alkali brings about problems like difficulties in base recycling and water pollution. We employed MgO, which is a kind of solid base, to support nickel to promote the hydrogenolysis of sorbitol under mild condition.132 A high total selectivity (80.8%) of EG, PG and glycerol was obtained at a sorbitol conversion of 67.8% under the condition of 473 K and 4.0 MPa of H2 over the using Ni/MgO (3:7) catalysis.
Alkali promoted Ni/C catalysts prepared by physically mixing or a co-supported method were employed to catalyse the hydrogenolysis of biomass-derived xylitol to produce glycols.133 Alkali doped in the catalysts efficiently prevented the leaching and sintering of nanoparticles, and show quite high catalytic activity. The selectivity of EG, PG and glycerol was 69.5% while xylitol conversion was nearly 100% at 473 K, and 4.0 MPa of H2 after 3 h. Mariscal et al.134 evaluated the hydrogenolysis of sorbitol employing Ru supported on different oxide supports (Al2O3, SiO2, TiO2 and ZrO2) as the catalysts. Ru/Al2O3 gave the highest yield of glycols due to its high surface acidity and partially oxidized Ru species. The hydrogenolysis of sorbitol in the absence of hydrogen was carried out by Xu et al.135 using a Ni–Re/C catalyst. An important intermediate, acetol, was identified in the sorbitol hydrogenolysis reaction. Re acted as an efficient promoter that could not only prevent the sintering of Ni particles but also accelerate the rate of hydrogen generation on the active sites.
Starting from corn stover derived sugar mixtures from the pretreatment and saccharification process136 developed in our group, the yield of 1,2-alkanediols was as high as 68.5% using the Ni–ZrO2 catalyst in a 10 L scale batchwise reaction at 453 K and 6 MPa of H2 (unpublished results). The reaction can also be conducted using a continuous reactor, exhibiting great prospects for the conversion of cellulose to commodity chemicals.
4. Summary, challenges and prospects
Biomass is the only renewable carbon resource that can be converted into liquid chemicals and liquid fuels, and the valorization of cellulose and biomass derived compounds holds great potential in solving current problems such as global warming, food crisis and environmental problems. Recently, global efforts have successfully demonstrated the efficacy of heterogeneously catalyzed conversion of cellulose to key primary building blocks of sugar alcohols, and directly into industrially attractive commodity glycols in one step.Nevertheless, the present development of catalysts have not met the level required for commercialization. The components of the lignocellulose and crystalline texture result in the robust properties and insolubility of lignocellulose in most solvents.94 The currently available techniques concerning the direct conversion of lignocellulosic biomass usually suffer from the disadvantages of harsh reaction conditions, high energy input, low efficiency, and equipment corrosion. In order to solve the problems of the interaction between substrate and catalysts in this process, mechanical degradation methods,82,83 such as ball-milling, steam explosion,95 chemical pretreatment93 (ammonia, H2O2, NaOH, etc.) and ionic liquids,88,89,91 were introduced into this system to facilitate the transformation of lignocellulose into high value-added chemicals. The product distribution of the hydrolytic hydrogenation/hydrogenolysis of lignocellulose are dominated by the coordination of cellulose hydrolysis and glucose hydrogenation/hydrogenolysis. Investigators developed some catalysts that contain various ratios of acid sites to metal sites to harmonize the hydrolysis of lignocellulose and glucose hydrogenation/hydrogenolysis.50,61,65 The yield of sugar alcohols (sorbitol and mannitol) and glycols (EG and PG) can be optimized in this manner.
However, due to the relatively low conversion of reactant and selectivity to the products, breakthroughs in catalytic conversion of lignocellulose are still needed.
(1) Investigations on the mechanism of hydrolytic hydrogenation/hydrogenolysis of lignocellulose, hydrogenation/hydrogenolysis of sugars and the hydrogenolysis of sugar alcohols are still necessary. The influence of the feedstock (the type and concentration), the catalysts and the reaction conditions on the product distribution are required.
(2) Rational catalyst design is another challenge confronted in the catalytic conversion of lignocellulose. Bifunctional catalysts have been proven to be efficient in the hydrolytic hydrogenation/hydrogenolysis of lignocellulose. The acidity and basicity, interactions between the metal and support, chemical and mechanical stability and the stability of the catalysts all affect the catalytic activity and the product distribution.
(3) Up to now, the reactors utilized in most of the reports are batch reactors. However, continuous reactors are more appropriate in large-scale production. Nonetheless, the main obstacle on this road is still the insolubility of the lignocellulose in most solvents. Starting from saccharified liquid or sugar alcohols, the hydrogenolysis reaction could be very efficient even under quite mild reaction conditions. In this context, effective pretreatment and saccharification techniques should be integrated in the process for transformation of cellulosic biomass to chemicals (Fig. 4), which is also another focus in the biorefinery.
 | ||
| Fig. 4 Proposed route for the conversion of lignocellulosic biomass to glycols. | ||
Abbreviations
| EG | Ethylene glycol |
| EGME | Ethylene glycol monoether |
| 1,2-PG | 1,2-Propylene glycol |
| 1,2-BDO | 1,2-Butanediol |
| 1,2-HDO | 1,2-Hexanediol |
| HPAs | Heteropolyacid |
| CNT | Carbon nanotube |
| CNF | Carbon nanofiber |
| GVL | γ-Valerolactone |
| SOCGs | Surface oxygen-containing groups |
| GRO | Reduced graphene oxide |
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (no. 21273260 and no. 21003146), the Shandong Provincial Natural Science Foundation for Distinguished Young Scholar, China (no. JQ201305), the “135” Projects Fund of CAS-QIBEBT Director Innovation Foundation and the Chinese Academy of Sciences.References
- S. Chu and A. Majumdar, nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- C. Somerville, H. Youngs, C. Taylor, S. C. Davis and S. P. Long, Science, 2010, 329, 790–792 CrossRef CAS PubMed.
- S. Van De Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014 10.1039/c3cs60414d.
- J. Zhang, J. B. Li, S. B. Wu and Y. Liu, Ind. Eng. Chem. Res., 2013, 52, 11799–11815 CrossRef CAS.
- A. Tathod, T. Kane, E. S. Sanil and P. L. Dhepe, J. Mol. Catal. A: Chem., 2014, 388, 90–99 CrossRef PubMed.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- J. Xuan, M. K. H. Leung, D. Y. C. Leung and M. Ni, Renewable Sustainable Energy Rev., 2009, 13, 1301–1313 CrossRef CAS PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2013, 114, 1827–1870 CrossRef PubMed.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- Y. G. Zheng, X. L. Chen and Y. C. Shen, Chem. Rev., 2008, 108, 5253–5277 CrossRef CAS PubMed.
- Y. C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- L. Hu, G. Zhao, W. W. Hao, X. Tang, Y. Sun, L. Lin and S. J. Liu, RSC Adv., 2012, 2, 11184–11206 RSC.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- R. Rinaldi and F. Schuth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- Y. H. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004, 88, 797–824 CrossRef CAS PubMed.
- D. Yamaguchi, M. Kitano, S. Suganuma, K. Nakajima, H. Kato and M. Hara, J. Phys. Chem. C, 2009, 113, 3181–3188 CAS.
- R. W. Torget, J. S. Kim and Y. Y. Lee, Ind. Eng. Chem. Res., 2000, 39, 2817–2825 CrossRef CAS.
- M. Sasaki, Z. Fang, Y. Fukushima, T. Adschiri and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 2883–2890 CrossRef CAS.
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- H. Kobayashi, T. Komanoya, S. K. Guha, K. Hara and A. Fukuoka, Appl. Catal., A, 2011, 409, 13–20 CrossRef PubMed.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- X. C. Wang, L. Q. Meng, F. Wu, Y. J. Jiang, L. Wang and X. D. Mu, Green Chem., 2012, 14, 758–765 RSC.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- G. Liang, C. Wu, L. He, J. Ming, H. Cheng, L. Zhuo and F. Zhao, Green Chem., 2011, 13, 839–842 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2010, 47, 576–578 RSC.
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3665 CrossRef CAS PubMed.
- B. S. Jan Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- Q. Y. Liu, Y. H. Liao, T. J. Wang, C. L. Cai, Q. Zhang, N. Tsubaki and L. L. Ma, Ind. Eng. Chem. Res., 2014, 53, 12655–12664 CrossRef CAS.
- Y. Liao, Q. Liu, T. Wang, J. Long, Q. Zhang, L. Ma, Y. Liu and Y. Li, Energy Fuels, 2014, 28, 5778–5784 CrossRef CAS.
- Z. J. Wu, S. H. Ge, C. X. Ren, M. H. Zhang, A. Yip and C. M. Xu, Green Chem., 2012, 14, 3336–3343 RSC.
- Z. Tai, J. Zhang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2012, 48, 7052–7054 RSC.
- J. Zhang, S. B. Wu and Y. Liu, Energy Fuels, 2014, 28, 4242–4246 CrossRef CAS.
- T. S. Chamblee, R. R. Weikel, S. A. Nolen, C. L. Liotta and C. A. Eckert, Green Chem., 2004, 6, 382–386 RSC.
- Y. Liu, C. Luo and H. Liu, Angew. Chem., Int. Ed., 2012, 51, 3249–3253 CrossRef CAS PubMed.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS PubMed.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang, Y. Shu, A. L. Stottlemyer and J. G. Chen, Catal. Today, 2009, 147, 77–85 CrossRef CAS PubMed.
- Y. Zhang, A. Wang and T. Zhang, Chem. Commun., 2010, 862–864 RSC.
- M. Y. Zheng, A. Q. Wang, N. Ji, J. F. Pang, X. D. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS PubMed.
- Z. Tai, J. Zhang, A. Wang, J. Pang, M. Zheng and T. Zhang, ChemSusChem, 2013, 6, 652–658 CrossRef CAS PubMed.
- X. C. Wang, F. Wu, S. X. Yao, Y. J. Jiang, J. Guan and X. D. Mu, Chem. Lett., 2012, 41, 476–478 CrossRef CAS.
- K. Tajvidi, P. J. Hausoul and R. Palkovits, ChemSusChem, 2014, 7, 1311–1317 CrossRef CAS PubMed.
- S. J. You, I. G. Baek and E. D. Park, Appl. Catal., A, 2013, 466, 161–168 CrossRef CAS PubMed.
- J. Li, L. T. Liu, Y. Liu, M. Z. Li, Y. H. Zhu, H. C. Liu, Y. Kou, J. Z. Zhang, Y. Han and D. Ma, Energy Environ. Sci., 2014, 7, 393–398 CAS.
- T. Y. Deng and H. C. Liu, Green Chem., 2013, 15, 116–124 RSC.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- P. Yang, H. Kobayashi, K. Hara and A. Fukuoka, ChemSusChem, 2012, 5, 920–926 CrossRef CAS PubMed.
- H. Kobayashi, T. Komanoya, K. Hara and A. Fukuoka, ChemSusChem, 2010, 3, 440–443 CrossRef CAS PubMed.
- S. Van de Vyver, L. Peng, J. Geboers, H. Schepers, F. de Clippel, C. J. Gommes, B. Goderis, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1560–1563 RSC.
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS PubMed.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef CAS PubMed.
- L. N. Ding, A. Q. Wang, M. Y. Zheng and T. Zhang, ChemSusChem, 2010, 3, 818–821 CrossRef CAS PubMed.
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC.
- W. P. Deng, M. Liu, X. S. Tan, Q. H. Zhang and Y. Wang, J. Catal., 2010, 271, 22–32 CrossRef CAS PubMed.
- M. Liu, W. Deng, Q. Zhang, Y. Wang and Y. Wang, Chem. Commun., 2011, 47, 9717–9719 RSC.
- G. Liang, H. Cheng, W. Li, L. He, Y. Yu and F. Zhao, Green Chem., 2012, 14, 2146–2149 RSC.
- G. Liang, L. He, H. Cheng, W. Li, X. Li, C. Zhang, Y. Yu and F. Zhao, J. Catal., 2014, 309, 468–476 CrossRef CAS PubMed.
- G. Liang, L. He, M. Arai and F. Zhao, ChemSusChem, 2014, 7, 1415–1421 CrossRef CAS PubMed.
- A. Negoi, K. Triantafyllidis, V. I. Parvulescu and S. M. Coman, Catal. Today, 2014, 223, 122–128 CrossRef CAS PubMed.
- Y. L. Cao, J. W. Wang, M. Q. Kang and Y. L. Zhu, J. Mol. Catal. A: Chem., 2014, 381, 46–53 CrossRef CAS PubMed.
- H. Kobayashi, Y. Hosaka, K. Hara, B. Feng, Y. Hirosaki and A. Fukuoka, Green Chem., 2014, 16, 637–644 RSC.
- W. Zhu, H. Yang, J. Chen, C. Chen, L. Guo, H. Gan, X. Zhao and Z. Hou, Green Chem., 2014, 16, 1534–1542 RSC.
- F. Chambon, F. Rataboul, C. Pinel, A. Cabiac, E. Guillon and N. Essayem, ChemSusChem, 2013, 6, 500–507 CrossRef CAS PubMed.
- J. X. Xi, Y. Zhang, Q. N. Xia, X. H. Liu, J. W. Ren, G. Z. Lu and Y. Q. Wang, Appl. Catal., A, 2013, 459, 52–58 CrossRef CAS PubMed.
- J. X. Xi, Y. Zhang, D. Q. Ding, Q. N. Xia, J. J. Wang, X. H. Liu, G. Z. Lu and Y. Q. Wang, Appl. Catal., A, 2014, 469, 108–115 CrossRef CAS PubMed.
- J. Xi, D. Ding, Y. Shao, X. Liu, G. Lu and Y. Wang, ACS Sustainable Chem. Eng., 2014 DOI:10.1021/sc500380c.
- J. Chen, S. Wang, J. Huang, L. Chen, L. Ma and X. Huang, ChemSusChem, 2013, 6, 1545–1555 CrossRef CAS PubMed.
- D. Wang, W. Niu, M. Tan, M. Wu, X. Zheng, Y. Li and N. Tsubaki, ChemSusChem, 2014, 7, 1398–1406 CrossRef CAS PubMed.
- Z. Xiao, S. Jin, M. Pang and C. Liang, Green Chem., 2013, 15, 891–895 RSC.
- M. R. Liu, H. Wang, J. Y. Han and Y. F. Niu, Carbohydr. Polym., 2012, 89, 607–612 CrossRef CAS PubMed.
- A. Shrotri, L. K. Lambert, A. Tanksale and J. Beltramini, Green Chem., 2013, 15, 2761–2768 RSC.
- J. Hilgert, N. Meine, R. Rinaldi and F. Schuth, Energy Environ. Sci., 2013, 6, 92–96 CAS.
- A. Brandt, J. Grasvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- J. L. Song, H. L. Fan, J. Ma and B. X. Han, Green Chem., 2013, 15, 2619–2635 RSC.
- T. Vancov, A. S. Alston, T. Brown and S. McIntosh, Renewable Energy, 2012, 45, 1–6 CrossRef CAS PubMed.
- J. X. Long, X. H. Li, L. F. Wang and N. Zhang, Sci. China: Chem., 2012, 55, 1500–1508 CrossRef CAS.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. C. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714–8715 CrossRef CAS PubMed.
- I. A. Ignatyev, C. Van Doorslaer, P. G. Mertens, K. Binnemans and D. E. De Vos, ChemSusChem, 2010, 3, 91–96 CrossRef CAS PubMed.
- Y. Zhu, Z. N. Kong, L. P. Stubbs, H. Lin, S. Shen, E. V. Anslyn and J. A. Maguire, ChemSusChem, 2010, 3, 67–70 CrossRef CAS PubMed.
- X. N. Xie, J. Y. Han, H. Wang, X. L. Zhu, X. Liu, Y. F. Niu, Z. Q. Song and Q. F. Ge, Catal. Today, 2014, 233, 70–76 CrossRef CAS PubMed.
- H. Kobayashi, Y. Yamakoshi, Y. Hosaka, M. Yabushita and A. Fukuoka, Catal. Today, 2014, 226, 204–209 CrossRef CAS PubMed.
- J. F. Pang, M. Y. Zheng, A. Q. Wang and T. Zhang, Ind. Eng. Chem. Res., 2011, 50, 6601–6608 CrossRef CAS.
- C. Z. Li, M. Y. Zheng, A. Q. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS.
- Y. G. Sun, Y. Ma, Z. Wang and J. Yao, Bioresour. Technol., 2014, 158, 307–312 CrossRef CAS PubMed.
- M. Kaldstrom, N. Kumar, M. Tenho, M. V. Mokeev, Y. E. Moskalenko and D. Y. Murzin, ACS Catal., 2012, 2, 1381–1393 CrossRef CAS.
- L. K. Zhou, J. F. Pang, A. Q. Wang and T. Zhang, Chin. J. Catal., 2013, 34, 2041–2046 CrossRef CAS.
- D. Sohounloue, C. Montassier and J. Barbier, Korean J. Chem. Eng., 1983, 22, 391–397 CAS.
- M. A. Andrews and S. A. Klaeren, J. Am. Chem. Soc., 1989, 111, 4131–4133 CrossRef CAS.
- C. Montassier, D. Giraud and J. Barbier, Stud. Surf. Sci. Catal., 1988, 41, 165–170 CrossRef CAS.
- C. Montassier, J. C. Menezo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99–110 CrossRef CAS.
- K. Y. Wang, M. C. Hawley and T. D. Furney, Ind. Eng. Chem. Res., 1995, 34, 3766–3770 CrossRef CAS.
- P. Gallezot, N. Nicolaus, G. Fleche, P. Fuertes and A. Perrard, J. Catal., 1998, 180, 51–55 CrossRef CAS.
- A. Aho, S. Roggan, O. A. Simakova, T. Salmi and D. Y. Murzin, Catal. Today, 2014 DOI:10.1016/j.cattod.2013.12.031.
- M. Besson, P. Gallezot, A. Perrard and C. Pinel, Catal. Today, 2005, 102, 160–165 CrossRef PubMed.
- J. Zhang, L. Lin, J. Zhang and J. Shi, Carbohydr. Res., 2011, 346, 1327–1332 CrossRef CAS PubMed.
- J. Liu, P. Bai and X. S. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 3758–3763 RSC.
- J. X. Pan, J. H. Li, C. Wang and Z. Y. Yang, React. Kinet. Catal. Lett., 2007, 90, 233–242 CrossRef CAS PubMed.
- T. Kilpio, A. Aho, D. Murzin and T. Salmi, Ind. Eng. Chem. Res., 2013, 52, 7690–7703 CrossRef CAS.
- X. C. Guo, X. C. Wang, J. Guan, X. F. Chen, Z. F. Qin, X. D. Mu and M. Xian, Chin. J. Catal., 2014, 35, 733–740 CrossRef CAS.
- A. Perrard, P. Gallezot, J.-P. Joly, R. Durand, C. Baljou, B. Coq and P. Trens, Appl. Catal., A, 2007, 331, 100–104 CrossRef CAS PubMed.
- S. R. V. Utkarsh Saxena, Ind. Eng. Chem. Res., 2005, 44, 1466–1473 CrossRef.
- R. Geyer, P. Kraak, A. Pachulski and R. Schodel, Chem. Ing. Tech., 2012, 84, 513–516 CrossRef CAS.
- B. W. Hoffer, E. Crezee, F. Devred, P. R. M. Mooijman, W. G. Sloof, P. Kooyman, A. D. van Langeveld, F. Kapteijn and J. A. Moulijn, Appl. Catal., A, 2003, 253, 437–452 CrossRef CAS.
- H. X. Li, H. Li and M. H. Wang, Appl. Catal., A, 2001, 207, 129–137 CrossRef CAS.
- J. Zhang, S. B. Wu, Y. Liu and B. Li, Catal. Commun., 2013, 35, 23–26 CrossRef CAS PubMed.
- R. Ooms, M. Dusselier, J. A. Geboers, B. Op de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC.
- Z. H. Xiao, S. H. Jin, G. Y. Sha, C. T. Williams and C. H. Liang, Ind. Eng. Chem. Res., 2014, 53, 8735–8743 CrossRef CAS.
- D. Liang, C. W. Liu, S. P. Deng, Y. L. Zhu and C. X. Lv, Catal. Commun., 2014, 54, 108–113 CrossRef CAS PubMed.
- J. J. Bozell and a. G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- B. Blanc, A. Bourrel, P. Gallezot, T. Haas and P. Taylor, Green Chem., 2000, 2, 89–91 RSC.
- L. Zhao, J. H. Zhou, Z. J. Sui and X. G. Zhou, Chem. Eng. Sci., 2010, 65, 30–35 CrossRef CAS PubMed.
- L. Zhao, J. H. Zhou, H. Chen, M. G. Zhang, Z. J. Sui and X. G. Zhou, Korean J. Chem. Eng., 2010, 27, 1412–1418 CrossRef CAS PubMed.
- J. H. Zhou, G. C. Liu, Z. J. Sui, X. G. Zhou and W. K. Yuan, Chin. J. Catal., 2014, 35, 692–702 CrossRef CAS.
- J. Sun and H. Liu, Green Chem., 2011, 13, 135–142 RSC.
- Z. Huang, J. Chen, Y. Jia, H. Liu, C. Xia and H. Liu, Appl. Catal., B, 2014, 147, 377–386 CrossRef CAS PubMed.
- L. Ye, X. Duan, H. Lin and Y. Yuan, Catal. Today, 2012, 183, 65–71 CrossRef CAS PubMed.
- T. Soták, T. Schmidt and M. Hronec, Appl. Catal., A, 2013, 459, 26–33 CrossRef PubMed.
- N. Li and G. W. Huber, J. Catal., 2010, 270, 48–59 CrossRef CAS PubMed.
- M. Banu, S. Sivasanker, T. M. Sankaranarayanan and P. Venuvanalingam, Catal. Commun., 2011, 12, 673–677 CrossRef CAS PubMed.
- F. Auneau, M. Berchu, G. Aubert, C. Pinel, M. Besson, D. Todaro, M. Bernardi, T. Ponsetti and R. Di Felice, Catal. Today, 2014, 234, 100–106 CrossRef CAS PubMed.
- X. G. Chen, X. C. Wang, S. X. Yao and X. D. Mu, Catal. Commun., 2013, 39, 86–89 CrossRef CAS PubMed.
- J. Y. Sun and H. C. Liu, Catal. Today, 2014, 234, 75–82 CrossRef CAS PubMed.
- I. M. Leo, M. L. Granados, J. L. G. Fierro and R. Mariscal, Chin. J. Catal., 2014, 35, 614–621 CrossRef CAS.
- J. Zhang, F. Lu, W. Yu, J. Chen, S. Chen, J. Gao and J. Xu, Catal. Today, 2014, 234, 107–112 CrossRef CAS PubMed.
- C. Liu, E. van der Heide, H. S. Wang, B. Li, G. Yu and X. D. Mu, Biotechnol. Biofuels, 2013, 6, 97 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |